
Branched-Chain Amino Acids Are Linked with Alzheimer’s Disease-Related Pathology and Cognitive Deficits
 , , , , ,
, , , , ,
Abstract
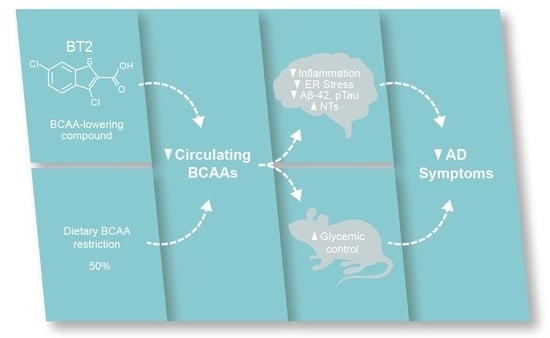
:
1. Introduction
2. Materials and Methods
2.1. Human Data Collection
2.2. Animals
2.3. Validation of BCAA Metabolism in AD Mouse Model
2.4. BCAA Restriction Experiment
2.5. BT2 Experiment
2.6. HT-22 In Vitro Experiment
2.7. Y-Maze Behavioral Test
2.8. Quantification of Brain Monoamines
2.9. Untargeted Metabolomics
2.10. BCAA Assay
2.11. Western Blots
2.12. ELISA
2.13. Real-Time Quantitative PCR (RT-qPCR)
2.14. Statistical Analysis
3. Results
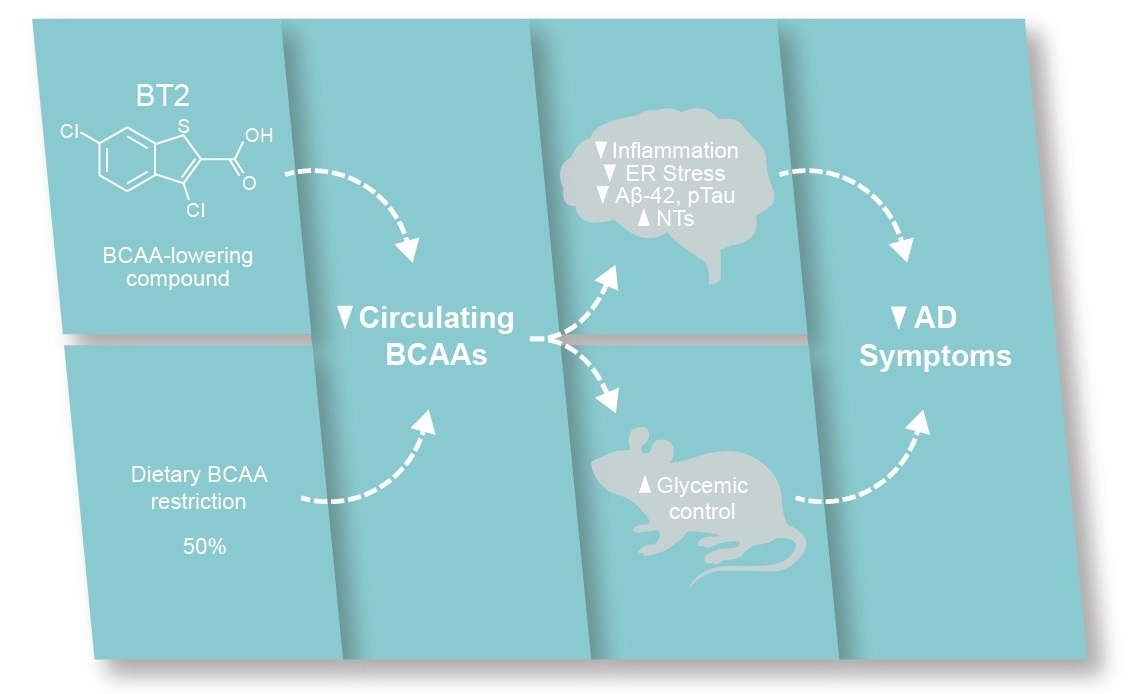
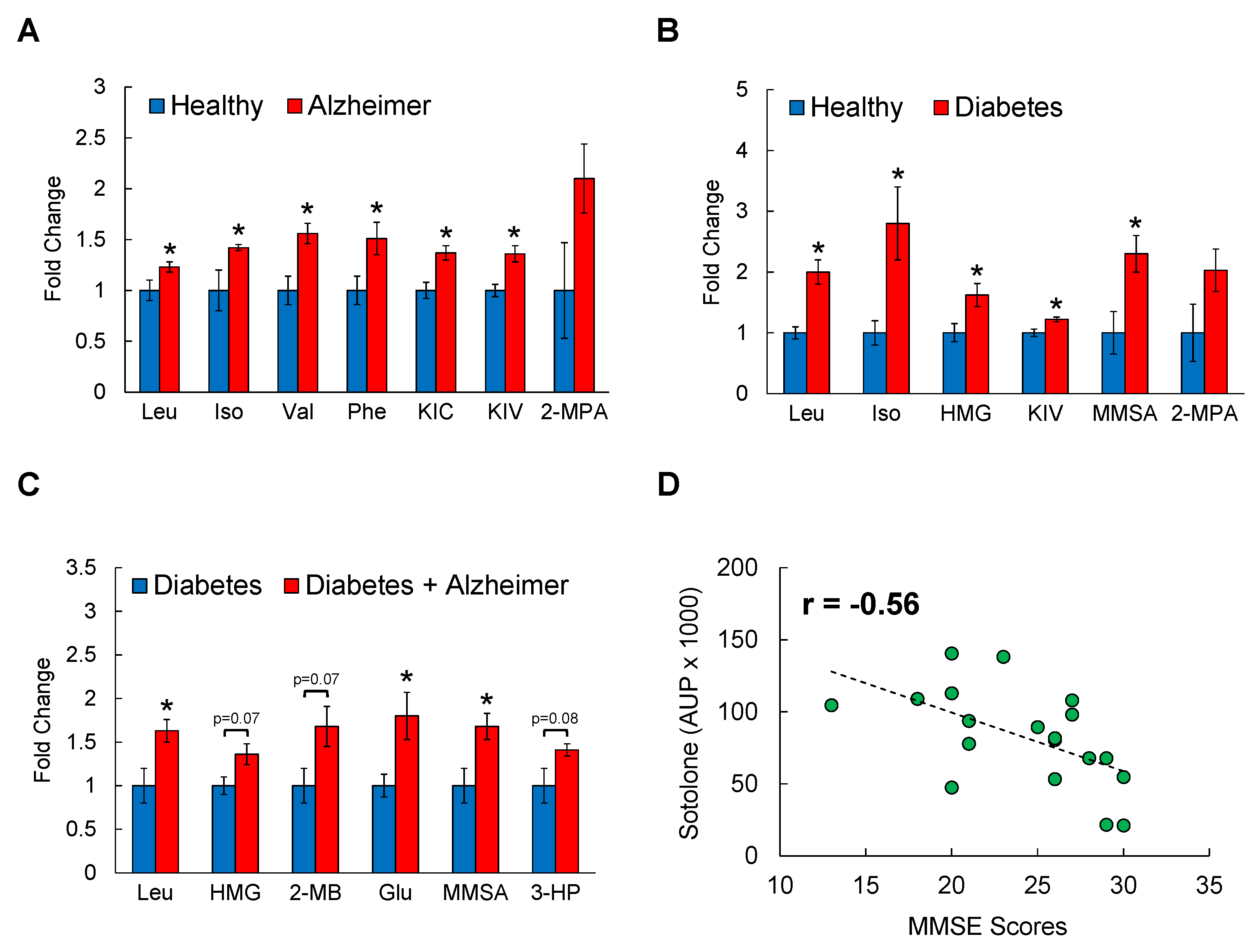
3.1. Serum BCAAs and Their Metabolites Are Associated with AD
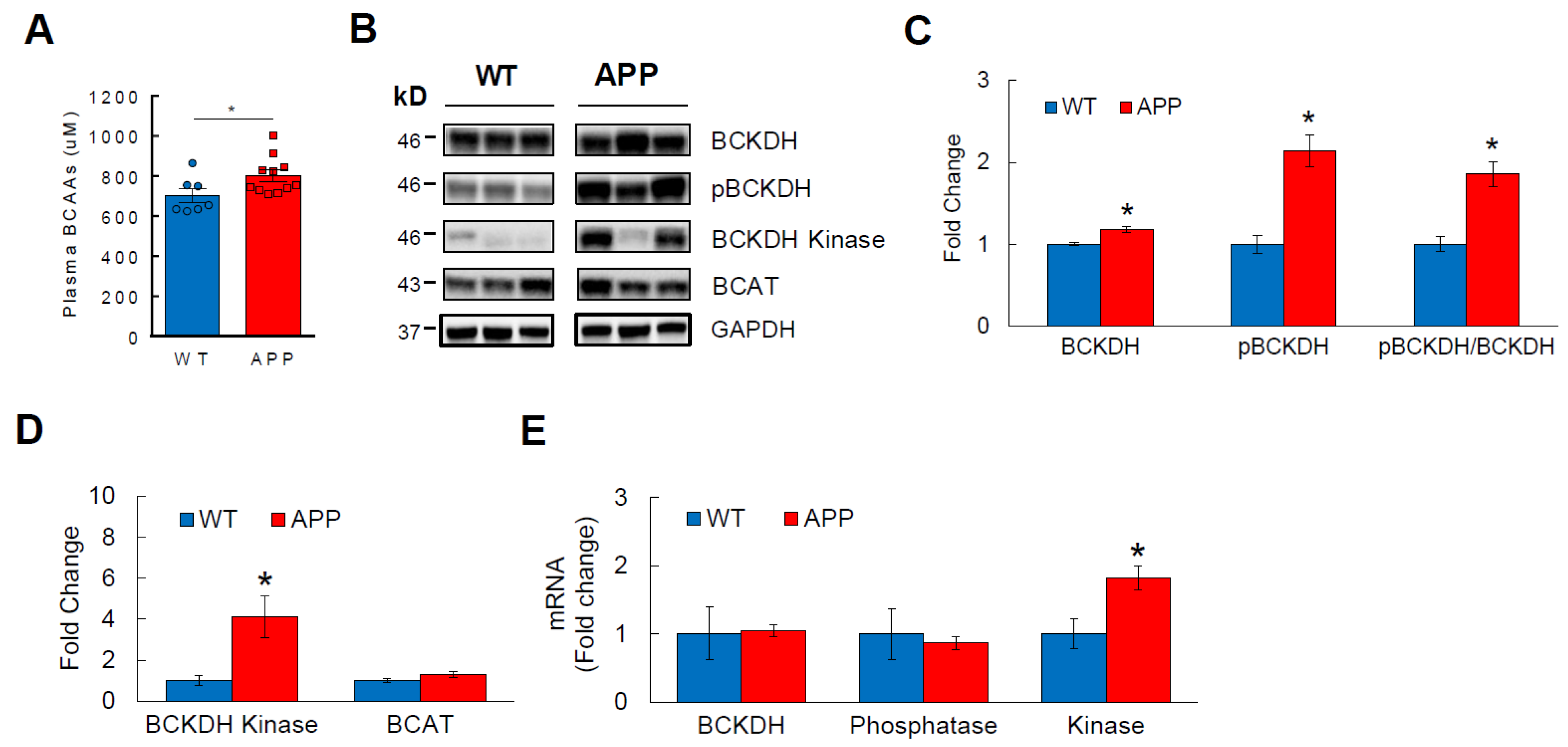
3.2. BCAA Catabolism Is Impaired in APPSwe Transgenic Mice
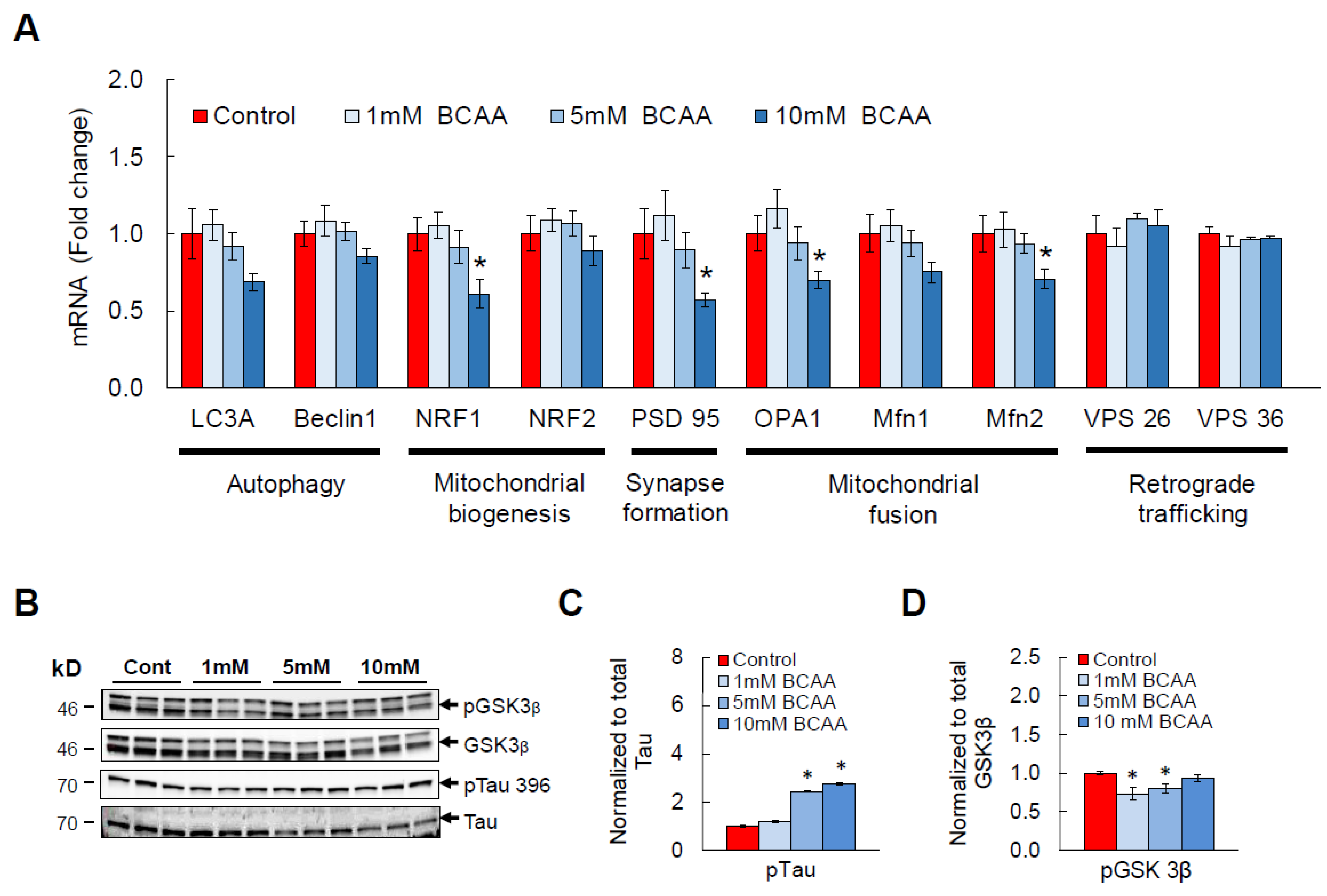
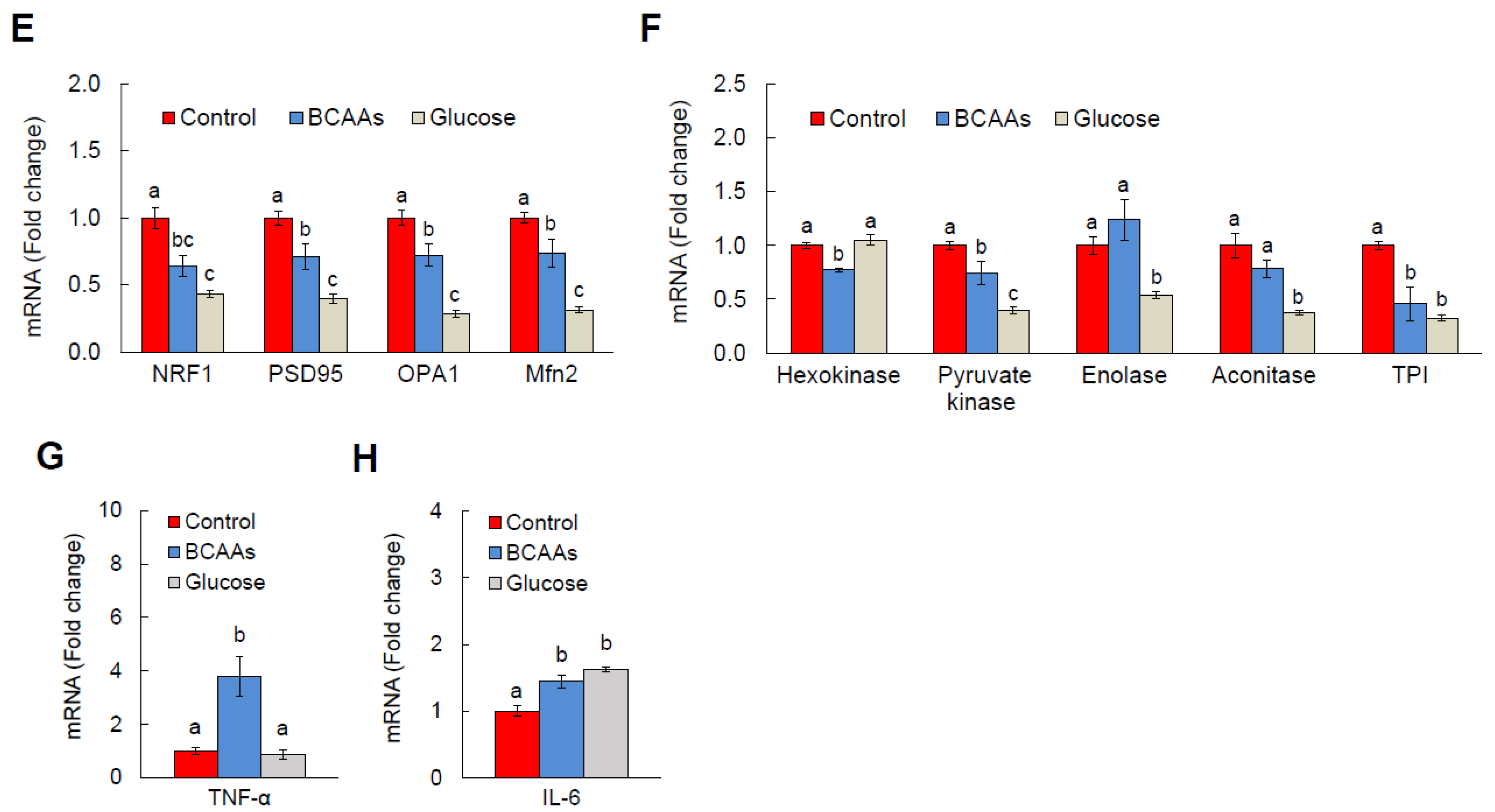
3.3. BCAA Supplementation Induces AD-Like Changes and Disrupts Cellular Functions in HT-22 Neurons
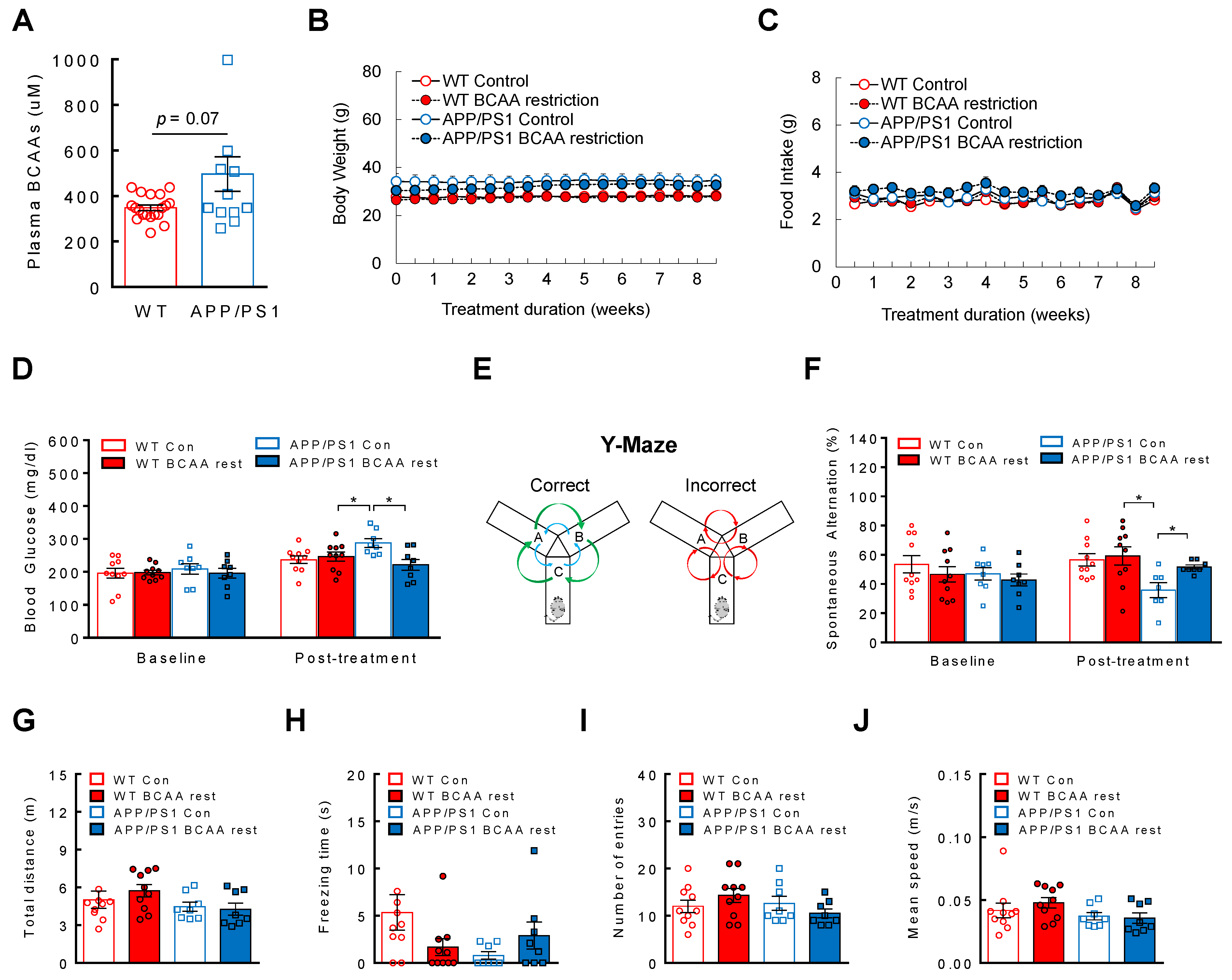
3.4. BCAA-Restriction Diet Delays Onset of Cognitive Decline in APP/PS1 Mice
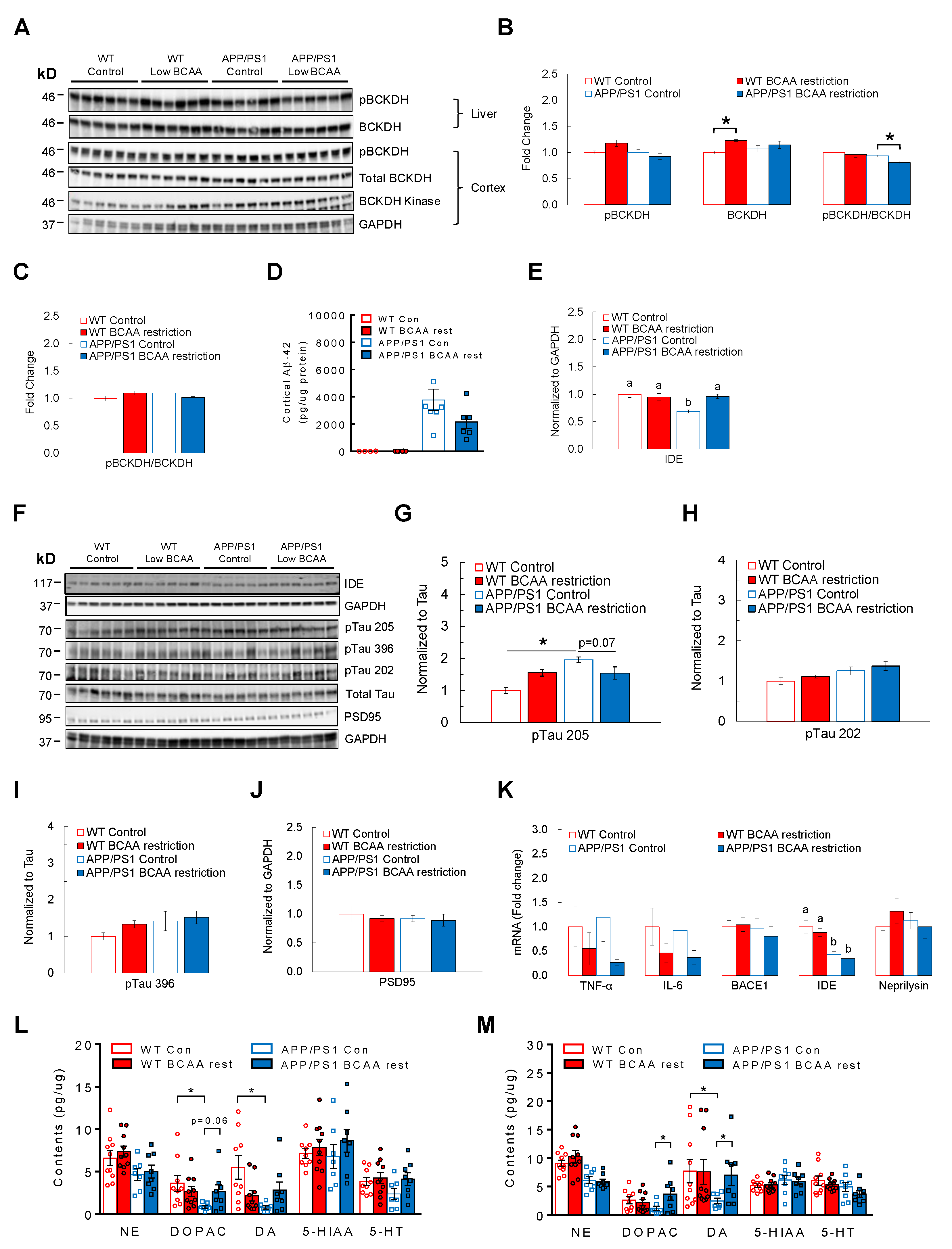
3.5. BCAA-Restriction Diet Lowers AD-Related Pathology and Restores Neurotransmitter Levels in the Cortex and Hippocampus in APP/PS1 Mice
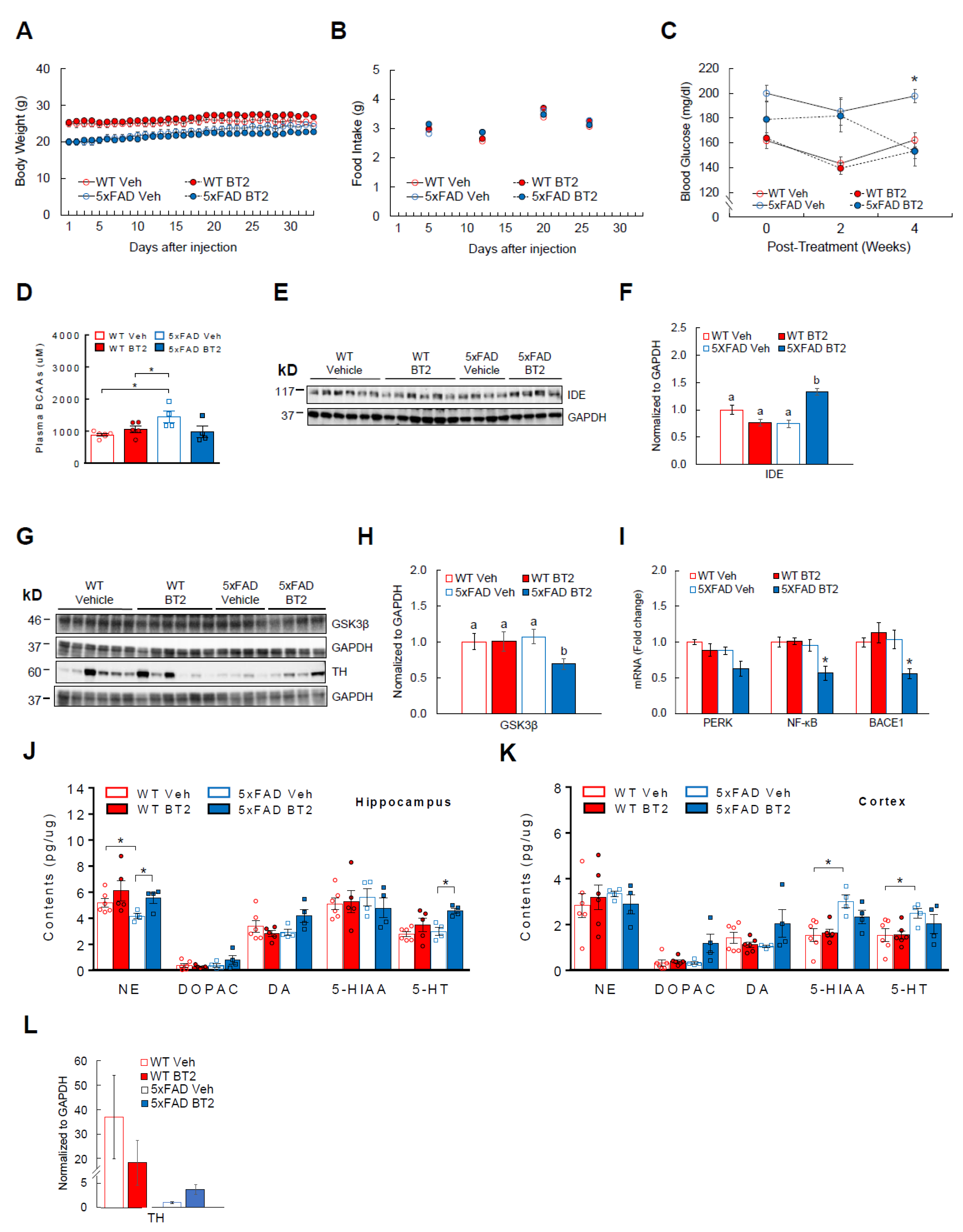
3.6. BT2, a BCAA-Lowering Compound, Effectively Reduces Aβ-42 and Enhances Cortical and Hippocampal Neurotransmitter Levels in 5xFAD Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 8 September 2022).
- Salas, I.H.; De Strooper, B. Diabetes and Alzheimer’s Disease: A Link not as Simple as it Seems. Neurochem. Res. 2019, 44, 1271–1278. [Google Scholar] [CrossRef] [Green Version]
- Umegaki, H. Type 2 diabetes as a risk factor for cognitive impairment: Current insights. Clin. Interv. Aging 2014, 9, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.H.; Hoppel, C.L.; Lok, K.H.; Zhao, L.; Wong, S.W.; Minkler, P.E.; Hwang, D.H.; Newman, J.W.; Garvey, W.T. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr. 2009, 139, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihalik, S.J.; Goodpaster, B.H.; Kelley, D.E.; Chace, D.H.; Vockley, J.; Toledo, F.G.; DeLany, J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 2010, 18, 1695–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- She, P.; Van Horn, C.; Reid, T.; Hutson, S.M.; Cooney, R.N.; Lynch, C.J. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1552–E1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Cummings, N.E.; Williams, E.M.; Kasza, I.; Konon, E.N.; Schaid, M.D.; Schmidt, B.A.; Poudel, C.; Sherman, D.S.; Yu, D.; Arriola Apelo, S.I.; et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J. Physiol. 2018, 596, 623–645. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Cummings, N.E.; Arriola Apelo, S.I.; Neuman, J.C.; Kasza, I.; Schmidt, B.A.; Cava, E.; Spelta, F.; Tosti, V.; Syed, F.A.; et al. Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health. Cell Rep. 2016, 16, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Moghei, M.; Tavajohi-Fini, P.; Beatty, B.; Adegoke, O.A. Ketoisocaproic acid, a metabolite of leucine, suppresses insulin-stimulated glucose transport in skeletal muscle cells in a BCAT2-dependent manner. Am. J. Physiol. Cell Physiol. 2016, 311, C518–C527. [Google Scholar] [CrossRef]
- White, P.J.; Lapworth, A.L.; An, J.; Wang, L.; McGarrah, R.W.; Stevens, R.D.; Ilkayeva, O.; George, T.; Muehlbauer, M.J.; Bain, J.R.; et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol. Metab. 2016, 5, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Yu, J.; Guo, Y.; Deng, J.; Li, K.; Du, Y.; Chen, S.; Zhu, J.; Sheng, H.; Guo, F. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism 2014, 63, 841–850. [Google Scholar] [CrossRef]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Shao, J.; Wu, C.Y.; Shu, L.; Dong, W.; Liu, Y.; Chen, M.; Wynn, R.M.; Wang, J.; Gui, W.J.; et al. Targeting BCAA Catabolism to Treat Obesity-Associated Insulin Resistance. Diabetes 2019, 68, 1730–1746. [Google Scholar] [CrossRef] [Green Version]
- Bridi, R.; Braun, C.A.; Zorzi, G.K.; Wannmacher, C.M.; Wajner, M.; Lissi, E.G.; Dutra-Filho, C.S. alpha-keto acids accumulating in maple syrup urine disease stimulate lipid peroxidation and reduce antioxidant defences in cerebral cortex from young rats. Metab. Brain Dis. 2005, 20, 155–167. [Google Scholar] [CrossRef]
- Lu, G.; Sun, H.; She, P.; Youn, J.Y.; Warburton, S.; Ping, P.; Vondriska, T.M.; Cai, H.; Lynch, C.J.; Wang, Y. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J. Clin. Investig. 2009, 119, 1678–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, A.U.; Leipnitz, G.; Fernandes, C.G.; Seminotti, B.; Schuck, P.F.; Wajner, M. Alpha-ketoisocaproic acid and leucine provoke mitochondrial bioenergetic dysfunction in rat brain. Brain Res. 2010, 1324, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Ren, S.; Korge, P.; Choi, J.; Dong, Y.; Weiss, J.; Koehler, C.; Chen, J.N.; Wang, Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007, 21, 784–796. [Google Scholar] [CrossRef] [Green Version]
- Haymond, M.W.; Ben-Galim, E.; Strobel, K.E. Glucose and alanine metabolism in children with maple syrup urine disease. J. Clin. Investig. 1978, 62, 398–405. [Google Scholar] [CrossRef]
- Snyderman, S.E.; Goldstein, F.; Sansaricq, C.; Norton, P.M. The relationship between the branched chain amino acids and their alpha-ketoacids in maple syrup urine disease. Pediatr. Res. 1984, 18, 851–853. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. Branched-chain amino acids: Enzyme and substrate regulation. J. Nutr. 2006, 136, 207S–211S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernstrom, J.D. Branched-chain amino acids and brain function. J. Nutr. 2005, 135, 1539S–1546S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeganathan, S.; Abdullahi, A.; Zargar, S.; Maeda, N.; Riddell, M.C.; Adegoke, O.A. Amino acid-induced impairment of insulin sensitivity in healthy and obese rats is reversible. Physiol. Rep. 2014, 2, e12067. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Pocai, A.; Wang, Y.; Sakoda, H.; Asano, T.; Backer, J.M.; Schwartz, G.J.; Rossetti, L. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J. Clin. Investig. 2008, 118, 2959–2968. [Google Scholar] [CrossRef]
- Lee, H.K.; Kwon, B.; Lemere, C.A.; de la Monte, S.; Itamura, K.; Ha, A.Y.; Querfurth, H.W. mTORC2 (Rictor) in Alzheimer’s Disease and Reversal of Amyloid-beta Expression-Induced Insulin Resistance and Toxicity in Rat Primary Cortical Neurons. J. Alzheimers Dis. 2017, 56, 1015–1036. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, A.; Belfiore, R.; Oddo, S. Genetically reducing mTOR signaling rescues central insulin dysregulation in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 68, 59–67. [Google Scholar] [CrossRef]
- Li, F.; Yin, Y.; Tan, B.; Kong, X.; Wu, G. Leucine nutrition in animals and humans: mTOR signaling and beyond. Amino Acids 2011, 41, 1185–1193. [Google Scholar] [CrossRef]
- Basun, H.; Forssell, L.G.; Almkvist, O.; Cowburn, R.F.; Eklof, R.; Winblad, B.; Wetterberg, L. Amino acid concentrations in cerebrospinal fluid and plasma in Alzheimer’s disease and healthy control subjects. J. Neural. Transm. Park. Dis Dement. Sect. 1990, 2, 295–304. [Google Scholar] [CrossRef]
- Fonteh, A.N.; Harrington, R.J.; Tsai, A.; Liao, P.; Harrington, M.G. Free amino acid and dipeptide changes in the body fluids from Alzheimer’s disease subjects. Amino Acids 2007, 32, 213–224. [Google Scholar] [CrossRef]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Metabolite profiling for the identification of altered metabolic pathways in Alzheimer’s disease. J. Pharm. Biomed. Anal. 2015, 107, 75–81. [Google Scholar] [CrossRef]
- Horgusluoglu, E.; Neff, R.; Song, W.M.; Wang, M.; Wang, Q.; Arnold, M.; Krumsiek, J.; Galindo-Prieto, B.; Ming, C.; Nho, K.; et al. Integrative metabolomics-genomics approach reveals key metabolic pathways and regulators of Alzheimer’s disease. Alzheimers Dement. 2022, 18, 1260–1278. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, C.; Simo, C.; Martin-Alvarez, P.J.; Kivipelto, M.; Winblad, B.; Cedazo-Minguez, A.; Cifuentes, A. Toward a predictive model of Alzheimer’s disease progression using capillary electrophoresis-mass spectrometry metabolomics. Anal. Chem. 2012, 84, 8532–8540. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Markus, H.S. Branched-chain amino acids and Alzheimer’s disease: A Mendelian randomization analysis. Sci. Rep. 2017, 7, 13604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ye, D.; Xie, W.; Hua, F.; Yang, Y.; Wu, J.; Gu, A.; Ren, Y.; Mao, K. Defect of branched-chain amino acid metabolism promotes the development of Alzheimer’s disease by targeting the mTOR signaling. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, H.H.; Chi, T.; Shin, A.C.; Lindtner, C.; Hsieh, W.; Ehrlich, M.; Gandy, S.; Buettner, C. Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer’s disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimers Dement. 2016, 12, 851–861. [Google Scholar] [CrossRef] [Green Version]
- Toledo, J.B.; Arnold, M.; Kastenmuller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement. 2017, 13, 965–984. [Google Scholar] [CrossRef]
- Tondo, M.; Wasek, B.; Escola-Gil, J.C.; de Gonzalo-Calvo, D.; Harmon, C.; Arning, E.; Bottiglieri, T. Altered Brain Metabolome Is Associated with Memory Impairment in the rTg4510 Mouse Model of Tauopathy. Metabolites 2020, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E.; Dutta, T.; Persson, X.M.; Mielke, M.M.; Petersen, R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS ONE 2013, 8, e63644. [Google Scholar] [CrossRef] [Green Version]
- Tynkkynen, J.; Chouraki, V.; van der Lee, S.J.; Hernesniemi, J.; Yang, Q.; Li, S.; Beiser, A.; Larson, M.G.; Saaksjarvi, K.; Shipley, M.J.; et al. Association of branched-chain amino acids and other circulating metabolites with risk of incident dementia and Alzheimer’s disease: A prospective study in eight cohorts. Alzheimers Dement. 2018, 14, 723–733. [Google Scholar] [CrossRef]
- Hutson, S.M.; Sweatt, A.J.; Lanoue, K.F. Branched-chain amino acid metabolism: Implications for establishing safe intakes. J. Nutr. 2005, 135, 1557S–1564S. [Google Scholar] [CrossRef]
- Shin, A.C.; Fasshauer, M.; Filatova, N.; Grundell, L.A.; Zielinski, E.; Zhou, J.Y.; Scherer, T.; Lindtner, C.; White, P.J.; Lapworth, A.L.; et al. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab. 2014, 20, 898–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waring, S.; O’Bryant, S.E.; Reisch, J.S.; Diaz-Arrastia, R.; Knebel, J.; Doody, R. The Texas Alzheimer’s Research Consortium longitudinal research cohort: Study design and baseline characteristics. Tex. Public Health J. 2008, 60, 9–13. [Google Scholar]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef]
- Tso, S.C.; Gui, W.J.; Wu, C.Y.; Chuang, J.L.; Qi, X.; Skvora, K.J.; Dork, K.; Wallace, A.L.; Morlock, L.K.; Lee, B.H.; et al. Benzothiophene carboxylate derivatives as novel allosteric inhibitors of branched-chain alpha-ketoacid dehydrogenase kinase. J. Biol. Chem. 2014, 289, 20583–20593. [Google Scholar] [CrossRef] [Green Version]
- Uddin, G.M.; Zhang, L.; Shah, S.; Fukushima, A.; Wagg, C.S.; Gopal, K.; Al Batran, R.; Pherwani, S.; Ho, K.L.; Boisvenue, J.; et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc. Diabetol. 2019, 18, 86. [Google Scholar] [CrossRef]
- Wessels, A.G.; Kluge, H.; Hirche, F.; Kiowski, A.; Schutkowski, A.; Corrent, E.; Bartelt, J.; Konig, B.; Stangl, G.I. High Leucine Diets Stimulate Cerebral Branched-Chain Amino Acid Degradation and Modify Serotonin and Ketone Body Concentrations in a Pig Model. PLoS ONE 2016, 11, e0150376. [Google Scholar] [CrossRef]
- Purpera, M.N.; Shen, L.; Taghavi, M.; Munzberg, H.; Martin, R.J.; Hutson, S.M.; Morrison, C.D. Impaired branched chain amino acid metabolism alters feeding behavior and increases orexigenic neuropeptide expression in the hypothalamus. J. Endocrinol. 2012, 212, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Laeger, T.; Reed, S.D.; Henagan, T.M.; Fernandez, D.H.; Taghavi, M.; Addington, A.; Munzberg, H.; Martin, R.J.; Hutson, S.M.; Morrison, C.D. Leucine acts in the brain to suppress food intake but does not function as a physiological signal of low dietary protein. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R310–R320. [Google Scholar] [CrossRef] [Green Version]
- Beckett, P.R. Spectrophotometric assay for measuring branched-chain amino acids. Methods Enzymol. 2000, 324, 40–47. [Google Scholar] [CrossRef]
- Wurtz, P.; Soininen, P.; Kangas, A.J.; Ronnemaa, T.; Lehtimaki, T.; Kahonen, M.; Viikari, J.S.; Raitakari, O.T.; Ala-Korpela, M. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 2013, 36, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Theurey, P.; Connolly, N.M.C.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 2019, 18, e12924. [Google Scholar] [CrossRef] [Green Version]
- Iwangoff, P.; Armbruster, R.; Enz, A.; Meier-Ruge, W. Glycolytic enzymes from human autoptic brain cortex: Normal aged and demented cases. Mech. Ageing Dev. 1980, 14, 203–209. [Google Scholar] [CrossRef]
- Tomlinson, D.R.; Gardiner, N.J. Glucose neurotoxicity. Nat. Rev. Neurosci. 2008, 9, 36–45. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, H.; Gu, W.; Liu, Y.; Zhang, M. Neuroprotective effects of ginsenoside Rb1 on high glucose-induced neurotoxicity in primary cultured rat hippocampal neurons. PLoS ONE 2013, 8, e79399. [Google Scholar] [CrossRef]
- Russell, J.W.; Golovoy, D.; Vincent, A.M.; Mahendru, P.; Olzmann, J.A.; Mentzer, A.; Feldman, E.L. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002, 16, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Tournissac, M.; Vandal, M.; Tremblay, C.; Bourassa, P.; Vancassel, S.; Emond, V.; Gangloff, A.; Calon, F. Dietary intake of branched-chain amino acids in a mouse model of Alzheimer’s disease: Effects on survival, behavior, and neuropathology. Alzheimers Dement. 2018, 4, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.A.; Cha, R.; Kokmen, E.; O’Brien, P.C.; Palumbo, P.J. Risk of dementia among persons with diabetes mellitus: A population-based cohort study. Am. J. Epidemiol. 1997, 145, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Ott, A.; Stolk, R.P.; Hofman, A.; van Harskamp, F.; Grobbee, D.E.; Breteler, M.M. Association of diabetes mellitus and dementia: The Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Gannon, M.; Che, P.; Chen, Y.; Jiao, K.; Roberson, E.D.; Wang, Q. Noradrenergic dysfunction in Alzheimer’s disease. Front. Neurosci. 2015, 9, 220. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yoo, M.J.; Savonenko, A.; Stirling, W.; Price, D.L.; Borchelt, D.R.; Mamounas, L.; Lyons, W.E.; Blue, M.E.; Lee, M.K. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 13805–13814. [Google Scholar] [CrossRef] [Green Version]
- Nobili, A.; Latagliata, E.C.; Viscomi, M.T.; Cavallucci, V.; Cutuli, D.; Giacovazzo, G.; Krashia, P.; Rizzo, F.R.; Marino, R.; Federici, M.; et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat. Commun. 2017, 8, 14727. [Google Scholar] [CrossRef] [PubMed]
- Rorabaugh, J.M.; Chalermpalanupap, T.; Botz-Zapp, C.A.; Fu, V.M.; Lembeck, N.A.; Cohen, R.M.; Weinshenker, D. Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain 2017, 140, 3023–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K. Where, when, and in what form does sporadic Alzheimer’s disease begin? Curr. Opin Neurol. 2012, 25, 708–714. [Google Scholar] [CrossRef]
- Mather, M.; Harley, C.W. The Locus Coeruleus: Essential for Maintaining Cognitive Function and the Aging Brain. Trends Cogn. Sci. 2016, 20, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.S.; Barrett, F.S.; Joo, J.H.; Nassery, N.; Savonenko, A.; Sodums, D.J.; Marano, C.M.; Munro, C.A.; Brandt, J.; Kraut, M.A.; et al. Molecular imaging of serotonin degeneration in mild cognitive impairment. Neurobiol. Dis. 2017, 105, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [Green Version]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Yannakoulia, M.; Kontogianni, M.; Scarmeas, N. Cognitive health and Mediterranean diet: Just diet or lifestyle pattern? Ageing Res. Rev. 2015, 20, 74–78. [Google Scholar] [CrossRef]
- De Toledo Ferraz Alves, T.C.; Ferreira, L.K.; Wajngarten, M.; Busatto, G.F. Cardiac disorders as risk factors for Alzheimer’s disease. J. Alzheimers Dis 2010, 20, 749–763. [Google Scholar] [CrossRef]
- Santos, C.Y.; Snyder, P.J.; Wu, W.C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimers Dement. 2017, 7, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, J.Y.; Kim, O.Y.; Ham, B.M.; Kim, H.J.; Kwon, D.Y.; Jang, Y.; Lee, J.H. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC-Q-TOF MS). J. Proteome Res. 2010, 9, 4368–4375. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarzar, T.G.; Lee, B.; Coughlin, R.; Kim, D.; Shen, L.; Hall, M.A. Sex Differences in the Metabolome of Alzheimer’s Disease Progression. Front. Radiol. 2022, 2. [Google Scholar] [CrossRef]
- Arnold, M.; Nho, K.; Kueider-Paisley, A.; Massaro, T.; Huynh, K.; Brauner, B.; MahmoudianDehkordi, S.; Louie, G.; Moseley, M.A.; Thompson, J.W.; et al. Sex and APOE epsilon4 genotype modify the Alzheimer’s disease serum metabolome. Nat. Commun. 2020, 11, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Nasaruddin, M.B.; Elliott, C.T.; McGuinness, B.; Passmore, A.P.; Kehoe, P.G.; Holscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Alzheimer’s disease-like pathology has transient effects on the brain and blood metabolome. Neurobiol. Aging 2016, 38, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; Kumar, A.; Keegan, R.M.; Deshmukh, R. Recent advances in the neurobiology and neuropharmacology of Alzheimer’s disease. Biomed. Pharmacother. 2018, 98, 297–307. [Google Scholar] [CrossRef]
- Zheng, H.; Zhou, Q.; Du, Y.; Li, C.; Xu, P.; Lin, L.; Xiao, J.; Gao, H. The hypothalamus as the primary brain region of metabolic abnormalities in APP/PS1 transgenic mouse model of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis 2018, 1864, 263–273. [Google Scholar] [CrossRef]
- Jakel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.A.; Joshi, M.; Jeoung, N.H.; Obayashi, M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J. Nutr. 2005, 135, 1527S–1530S. [Google Scholar] [CrossRef] [Green Version]
- Parrella, E.; Maxim, T.; Maialetti, F.; Zhang, L.; Wan, J.; Wei, M.; Cohen, P.; Fontana, L.; Longo, V.D. Protein restriction cycles reduce IGF-1 and phosphorylated Tau, and improve behavioral performance in an Alzheimer’s disease mouse model. Aging Cell 2013, 12, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Julien, J.; Morin, F.; Marette, A.; Planel, E. Differential effects of voluntary treadmill exercise and caloric restriction on tau pathogenesis in a mouse model of Alzheimer’s disease-like tau pathology fed with Western diet. Prog. Neuropsychopharmacol. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Roman, I.; Barja, G. Regulation of longevity and oxidative stress by nutritional interventions: Role of methionine restriction. Exp. Gerontol. 2013, 48, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, R.; Dong, W.; Zhao, Z. Caloric restriction can improve learning and memory in C57/BL mice probably via regulation of the AMPK signaling pathway. Exp. Gerontol. 2018, 102, 28–35. [Google Scholar] [CrossRef]
- Crane, P.K.; Walker, R.; Larson, E.B. Glucose levels and risk of dementia. N. Engl. J. Med. 2013, 369, 1863–1864. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.; Sharma, A.N.; Elased, K.M.; Guest, P.C.; Rahmoune, H.; Bahn, S. Diabetic db/db mice exhibit central nervous system and peripheral molecular alterations as seen in neurological disorders. Transl. Psychiatry 2013, 3, e263. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Lee, J.; Hong, H.J.; Jung, E.S.; Ku, Y.H.; Jeong, I.K.; Cho, Y.M.; So, I.; Park, K.S.; Mook-Jung, I. A relationship between Alzheimer’s disease and type 2 diabetes mellitus through the measurement of serum amyloid-beta autoantibodies. J. Alzheimers Dis. 2010, 19, 1371–1376. [Google Scholar] [CrossRef] [Green Version]
- Macklin, L.; Griffith, C.M.; Cai, Y.; Rose, G.M.; Yan, X.X.; Patrylo, P.R. Glucose tolerance and insulin sensitivity are impaired in APP/PS1 transgenic mice prior to amyloid plaque pathogenesis and cognitive decline. Exp. Gerontol. 2017, 88, 9–18. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Sasaki, K.; Tanizaki, Y.; Hata, J.; Fujimi, K.; Matsui, Y.; Sekita, A.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; et al. Insulin resistance is associated with the pathology of Alzheimer disease: The Hisayama study. Neurology 2010, 75, 764–770. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Ortiz, O.; Jimenez-Palomares, M.; Kay, K.R.; Berrocoso, E.; Murillo-Carretero, M.I.; Perdomo, G.; Spires-Jones, T.; Cozar-Castellano, I.; Lechuga-Sancho, A.M.; et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 2013, 38, 2462–2475. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [PubMed]
- Carrera, I.; Etcheverria, I.; Li, Y.; Fernandez-Novoa, L.; Lombardi, V.; Vigo, C.; Palacios, H.H.; Benberin, V.V.; Cacabelos, R.; Aliev, G. Immunocytochemical Characterization of Alzheimer’s Disease Hallmarks in APP/PS1 Transgenic Mice Treated with a New Anti-Amyloid-beta Vaccine. Cent. Asian J. Glob. Health 2013, 2, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Yao, L.; Li, C.; Wang, J.; Chen, S.; Zhou, X.F.; Liao, H. The blockage of the Nogo/NgR signal pathway in microglia alleviates the formation of Abeta plaques and tau phosphorylation in APP/PS1 transgenic mice. J. Neuroinflammation 2016, 13, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Ohno, M. Phospho-eIF2alpha level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS ONE 2010, 5, e12974. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.M. Role of amino acid transporters in amino acid sensing. Am. J. Clin. Nutr. 2014, 99, 223S–230S. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Diet Research Diets A11072001 | (50%) BCAA-Restricted Diet Research Diets A20112501 | |||
|---|---|---|---|---|
| Macronutrients | gm | kcal | gm | kcal |
| Protein | 34 | 33 | 34 | 33 |
| Carbohydrate | 49 | 49 | 49 | 49 |
| Fat | 8 | 18 | 8 | 18 |
| Fiber | 4 | 0 | 4 | 0 |
| Total | 91 | 100 | 91 | 100 |
| kcal/gm | 4 | 4 | ||
| Amino Acids | ||||
| Cystine | 5.5 | 22 | 6.29 | 25 |
| Isoleucine | 17.88 | 72 | 8.94 | 36 |
| Leucine | 31.63 | 127 | 15.81 | 63 |
| Lysine | 24.75 | 99 | 29.28 | 117 |
| Methionine | 6.88 | 28 | 7.86 | 31 |
| Phenylalanine | 17.88 | 72 | 17.88 | 72 |
| Threonine | 15.13 | 61 | 17.29 | 69 |
| Tryptophan | 5.5 | 22 | 5.50 | 22 |
| Valine | 23.38 | 94 | 11.69 | 47 |
| Histidine | 8.25 | 33 | 9.43 | 38 |
| Alanine | 12.38 | 50 | 14.15 | 57 |
| Arginine | 27.5 | 110 | 32.08 | 128 |
| Aspartic Acid | 20.63 | 83 | 23.58 | 94 |
| Glutamic Acid | 37.13 | 149 | 43.43 | 174 |
| Glycine | 38.5 | 154 | 45.00 | 180 |
| Proline | 17.88 | 72 | 20.43 | 82 |
| Serine | 15.13 | 61 | 17.29 | 69 |
| Tyrosine | 12.38 | 50 | 12.38 | 50 |
| Other Ingredients | ||||
| Corn Starch | 386 | 1544 | 386 | 1544 |
| Maltodextrin 10 | 100 | 400 | 100 | 400 |
| Cellulose, BW200 | 40 | 0 | 40 | 0 |
| Soybean Oil | 25 | 225 | 25 | 225 |
| Lard | 55 | 495 | 55 | 495 |
| Mineral Mix S10026 | 10 | 0 | 10 | 0 |
| Dicalcium Phosphate | 13 | 0 | 13 | 0 |
| Calcium Carbonate | 5.5 | 0 | 6 | 0 |
| Potassium Citrate, 1 H2O | 16.5 | 0 | 17 | 0 |
| Sodium Bicarbonate | 3.5 | 0 | 3.5 | 0 |
| Vitamin Mix V10001 | 10 | 40 | 10 | 40 |
| Choline Bitartrate | 2 | 0 | 2 | 0 |
| Cholesterol | 0.782 | 0 | 0.782 | 0 |
| Total | 1005.64 | 4057 | 1005.592 | 4057 |
| Gene Name | Primer Sequence (5′–3′) | |
|---|---|---|
| BCKDH | Forward | GGATGAGGAACAGGAGAAGG |
| Reverse | GGAGAAGAGGAGGCTTGG | |
| BCKDH Kinase | Forward | GACAGGTGGACTTAGATGGA |
| Reverse | CAAGAATGAGCAGAGCAGAG | |
| BCKDH Phosphatase | Forward | CCTGCTACTTCTCCACTTCA |
| Reverse | GCTCATCAATGCGGTTATCC | |
| LC3A | Forward | CCCATCGCTGACATCTATGAAC |
| Reverse | AAGGTTTCTTGGGAGGCGTA | |
| Beclin1 | Forward | ACCAGCGGGAGTATAGTGAGT |
| Reverse | CAGCTGGATCTGGGCGTAG | |
| Nrf1 | Forward | AGAAACGGAAACGGCCTCAT |
| Reverse | CATCCAACGTGGCTCTGAGT | |
| Nrf2 | Forward | ATGGAGCAAGTTTGGCAGGA |
| Reverse | GCTGGGAACAGCGGTAGTAT | |
| PSD95 | Forward | CTTCATCCTTGCTGGGGGTC |
| Reverse | TTGCGGAGGTCAACACCATT | |
| Opa1 | Forward | ACCTTGCCAGTTTAGCTCCC |
| Reverse | TTGGGACCTGCAGTGAAGAA | |
| Mfn1 | Forward | GCAGACAGCACATGGAGAGA |
| Reverse | GATCCGATTCCGAGCTTCCG | |
| Mfn2 | Forward | TGCACCGCCATATAGAGGAAG |
| Reverse | TCTGCAGTGAACTGGCAATG | |
| VPS26 | Forward | CCAGCCGAAGTGTCCATA |
| Reverse | CCATACGCCTCAGTTGTG | |
| VPS36 | Forward | ACCTCCAGACACCTTCAG |
| Reverse | CTCCATTAGTAGCCAGAATAAGTG | |
| Hexokinase | Forward | TGCCACTGAGTTGTCTGT |
| Reverse | CTACCACCACCACCATCA | |
| TPI | Forward | CAGCAGGCACAGGAAGTA |
| Reverse | CCAGTCACAGAACCTCCATAA | |
| Enolase | Forward | CACAGTTGCCACCATCTC |
| Reverse | TTCTCTTCGTCCTCTCACAT | |
| Aconitase | Forward | AGATACGGACGCTTACCATT |
| Reverse | CGGCACTTCTATGTTCTTATGTT | |
| Pyruvate Kinase | Forward | CGCAACACTGGCATCATT |
| Reverse | TGGCTTCACGGACATTCT | |
| B2M | Forward | GAAGCCGAACATACTGAACTG |
| Reverse | CTGAAGGACATATCTGACATCTCT | |
| GAPDH | Forward | GGTGAAGGTCGGTGTGAAC |
| Reverse | TGAGTGGAGTCATACTGGAACA | |
| BACE1 | Forward | CCTATGCGATGCGAATGTT |
| Reverse | TCTCCTTCCTGTCTCTATCCT | |
| PSEN1 | Forward | CACCGTTGTCCTACTTCCA |
| Reverse | CTCCTCATCTTCTTCCTCATCTT | |
| PERK | Forward | ACGGTTACTATCTGCCATACTAC |
| Reverse | CCTTCTTGCGGATGTTCTTG | |
| TNF-α | Forward | ACCACCATCAAGGACTCAA |
| Reverse | AAGGTCTGAAGGTAGGAAGG | |
| IL-6 | Forward | ACAGAAGGAGTGGCTAAG |
| Reverse | AGAGAACAACATAAGTCAGATAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddik, M.A.B.; Mullins, C.A.; Kramer, A.; Shah, H.; Gannaban, R.B.; Zabet-Moghaddam, M.; Huebinger, R.M.; Hegde, V.K.; MohanKumar, S.M.J.; MohanKumar, P.S.; et al. Branched-Chain Amino Acids Are Linked with Alzheimer’s Disease-Related Pathology and Cognitive Deficits. Cells 2022, 11, 3523. https://doi.org/10.3390/cells11213523
Siddik MAB, Mullins CA, Kramer A, Shah H, Gannaban RB, Zabet-Moghaddam M, Huebinger RM, Hegde VK, MohanKumar SMJ, MohanKumar PS, et al. Branched-Chain Amino Acids Are Linked with Alzheimer’s Disease-Related Pathology and Cognitive Deficits. Cells. 2022; 11(21):3523. https://doi.org/10.3390/cells11213523
Chicago/Turabian StyleSiddik, Md Abu Bakkar, Caitlyn A. Mullins, Alyssa Kramer, Harsh Shah, Ritchel B. Gannaban, Masoud Zabet-Moghaddam, Ryan M. Huebinger, Vijay K. Hegde, Sheba M. J. MohanKumar, Puliyur S. MohanKumar, and et al. 2022. "Branched-Chain Amino Acids Are Linked with Alzheimer’s Disease-Related Pathology and Cognitive Deficits" Cells 11, no. 21: 3523. https://doi.org/10.3390/cells11213523