
Insights into the Genetic Profile of Two Siblings Affected by Unverricht-Lundborg Disease Using Patient-Derived hiPSCs
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Human Induced Pluripotent Stem Cells (hiPSCs) from Two Siblings Affected by ULD
2.2. Generation of Neurons from Human Induced Pluripotent Stem Cells
2.3. DNA Extraction
2.4. PCR Amplification
2.5. Sanger Sequencing
2.6. Fragment Length Analysis by Capillary Electrophoresis
2.7. CSTB Promoter Constructs and Cloning
2.8. Transfection and Luciferase Assay
2.9. RNA Extraction and Quantitative Real-Time PCR
2.10. Western Blotting
2.11. Immunofluorescence Assay
2.12. Statistical Analysis
3. Results
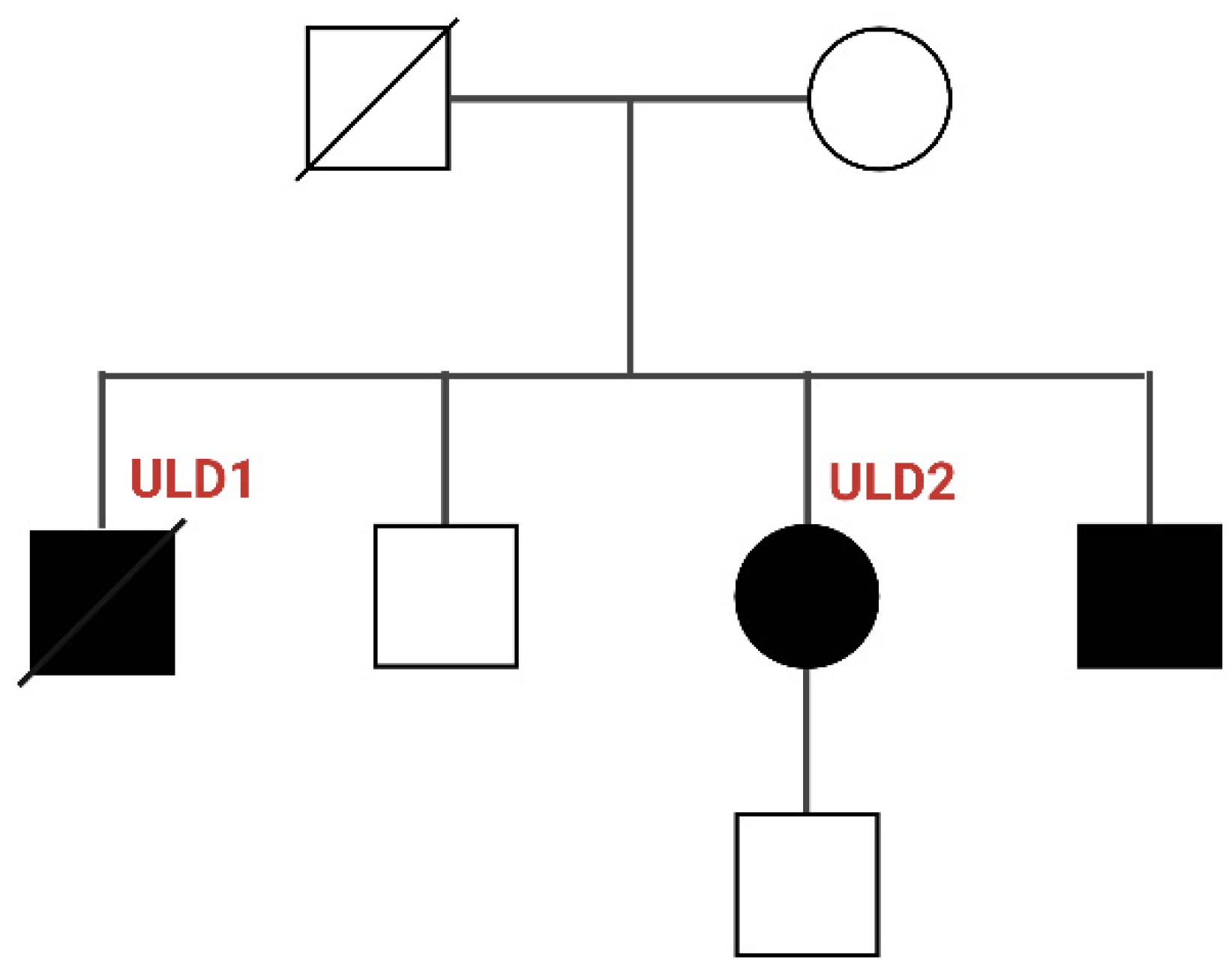
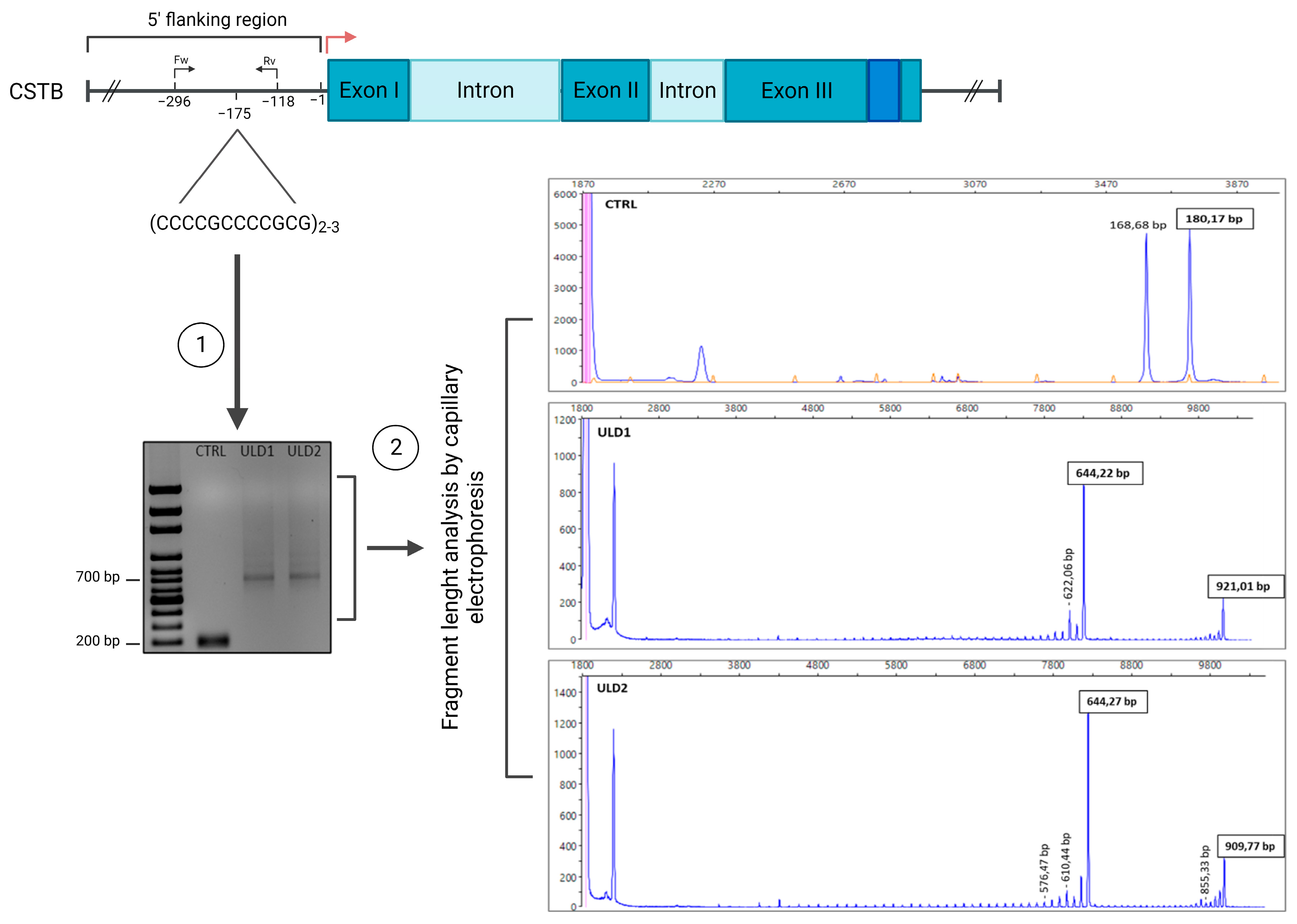
3.1. Genetic Characterization of ULD Patients
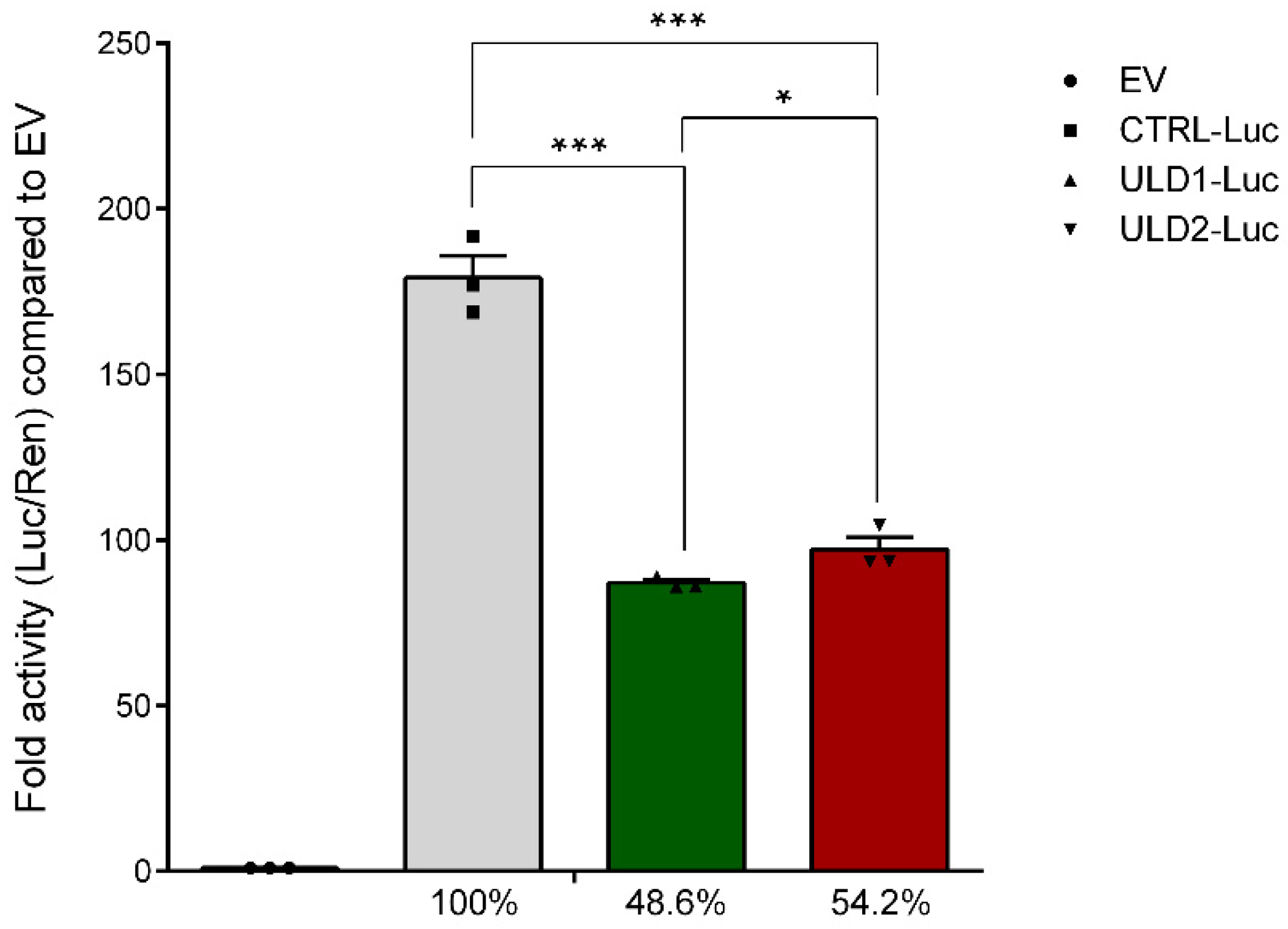
3.2. In Vitro Study of the Effect of Pathological Repeat Expansion on CSTB Promoter Activity
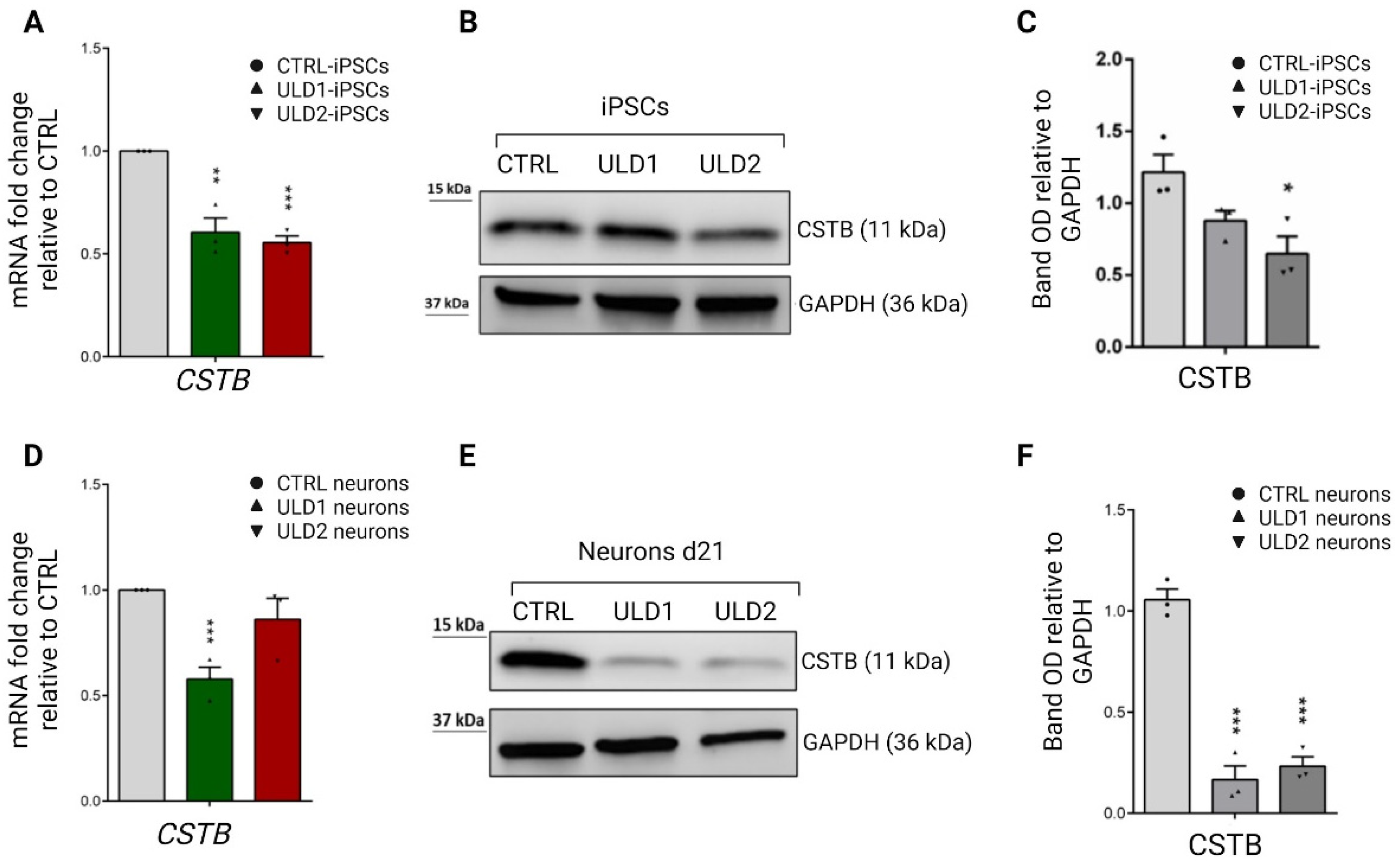
3.3. Assessment of Cystatin B Expression in ULD Patient-Derived iPSCs and Neurons
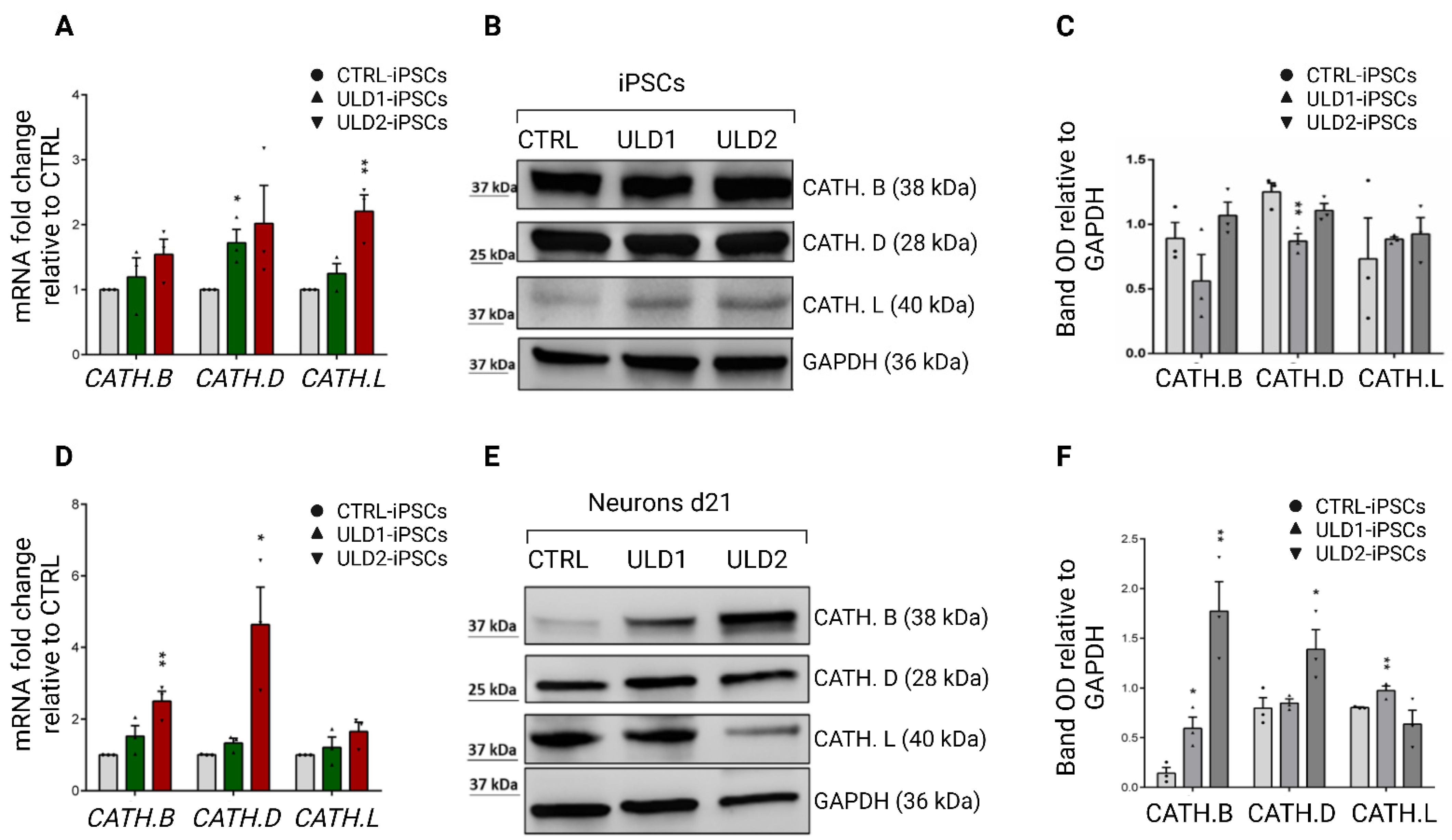
3.4. Assessment of Cathepsins Expression in ULD Patient-Derived iPSCs and Neurons
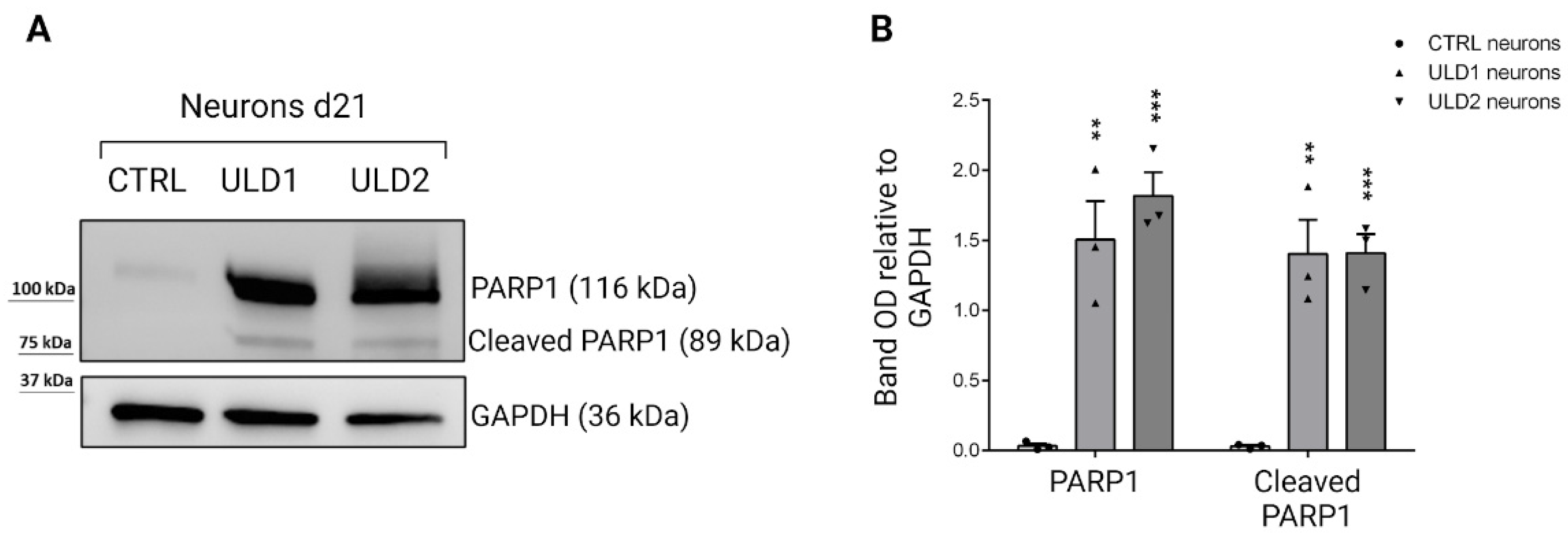
3.5. Investigation of Cathepsins-Mediated Apoptosis in ULD-Neurons
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crespel, A.; Ferlazzo, E.; Franceschetti, S.; Genton, P.; Gouider, R.; Kälviäinen, R.; Korja, M.; Lehtinen, M.K.; Mervaala, E.; Simonato, M.; et al. Unverricht-Lundborg Disease. Epileptic Disord. 2016, 18, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Genton, P.; Striano, P.; Minassian, B.A. The History of Progressive Myoclonus Epilepsies. Epileptic Disord. 2016, 18, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehesjoki, A.-E.; Tassinari, C.A.; Avanzini, G.; Michelucci, R.; Franceschetti, S.; Antonelli, A.; Rubboli, G.; de la Chapelle, A. PME of Unverricht-Lundborg Type in the Mediterranean Region: Linkage and Linkage Disequilibrium Confirm the Assignment to the EPM1 Locus. Hum. Genet. 1994, 93, 668–674. [Google Scholar] [CrossRef]
- Lehesjoki, A.E.; Kälviäinen, R. Progressive Myoclonic Epilepsy Type 1. 2004 Jun 24 [Updated 2 July 2020]. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2022. [Google Scholar]
- Michelucci, R.; Pasini, E.; Riguzzi, P.; Andermann, E.; Kälviäinen, R.; Genton, P. Myoclonus and Seizures in Progressive Myoclonus Epilepsies: Pharmacology and Therapeutic Trials. Epileptic Disord. 2016, 18, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Kälviäinen, R.; Khyuppenen, J.; Koskenkorva, P.; Eriksson, K.; Vanninen, R.; Mervaala, E. Clinical Picture of EPM1-Unverricht-Lundborg Disease. Epilepsia 2008, 49, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Pennacchio, L.A.; Lehesjoki, A.E.; Stone, N.E.; Willour, V.L.; Virtaneva, K.; Miao, J.; D’Amato, E.; Ramirez, L.; Faham, M.; Koskiniemi, M.; et al. Mutations in the Gene Encoding Cystatin B in Progressive Myoclonus Epilepsy (EPM1). Science 1996, 271, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Lafrenière, R.G.; Rochefort, D.L.; Chrétien, N.; Rommens, J.M.; Cochius, J.I.; Kälviäinen, R.; Nousiainen, U.; Patry, G.; Farrell, K.; Söderfeldt, B.; et al. Unstable Insertion in the 5′ Flanking Region of the Cystatin B Gene Is the Most Common Mutation in Progressive Myoclonus Epilepsy Type 1, EPM1. Nat. Genet. 1997, 15, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Mirotsou, M.; Buresi, C.; Peitsch, M.C.; Rossier, C.; Ouazzani, R.; Baldy-Moulinier, M.; Bottani, A.; Malafosse, A.; Antonarakis, S.E. Identification of Mutations in Cystatin B, the Gene Responsible for the Unverricht-Lundborg Type of Progressive Myoclonus Epilepsy. Am. J. Hum. Genet. 1997, 60, 342–351. [Google Scholar]
- Virtaneva, K.; D’Amato, E.; Miao, J.; Koskiniemi, M.; Norio, R.; Avanzini, G.; Franceschetti, S.; Michelucci, R.; Tassinari, C.A.; Omer, S.; et al. Unstable Minisatellite Expansion Causing Recessively Inherited Myoclonus Epilepsy, EPM1. Nat. Genet. 1997, 15, 393–396. [Google Scholar] [CrossRef]
- Joensuu, T.; Kuronen, M.; Alakurtti, K.; Tegelberg, S.; Hakala, P.; Aalto, A.; Huopaniemi, L.; Aula, N.; Michellucci, R.; Eriksson, K.; et al. Cystatin B: Mutation Detection, Alternative Splicing and Expression in Progressive Myoclonus Epilepsy of Unverricht-Lundborg Type (EPM1) Patients. Eur. J. Hum. Genet. 2007, 15, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, T.; Lehesjoki, A.-E.; Kopra, O. Molecular Background of EPM1—Unverricht–Lundborg Disease. Epilepsia 2008, 49, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Scott, H.S.; Genton, P.; Grid, D.; Ouazzani, R.; M’Rabet, A.; Ibrahim, S.; Gouider, R.; Dravet, C.; Chkili, T.; et al. A PCR Amplification Method Reveals Instability of the Dodecamer Repeat in Progressive Myoclonus Epilepsy (EPM1) and No Correlation between the Size of the Repeat and Age at Onset. Am. J. Hum. Genet. 1998, 62, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Rinne, R.; Saukko, P.; Järvinen, M.; Lehesjoki, A.-E. Reduced Cystatin B Activity Correlates with Enhanced Cathepsin Activity in Progressive Myoclonus Epilepsy. Ann. Med. 2002, 34, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Riccio, M.; Di Giaimo, R.; Pianetti, S.; Palmieri, P.P.; Melli, M.; Santi, S. Nuclear Localization of Cystatin B, the Cathepsin Inhibitor Implicated in Myoclonus Epilepsy (EPM1). Exp. Cell Res. 2001, 262, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, K.; Weber, E.; Rinne, R.; Theil, G.; de Haan, G.-J.; Lindhout, D.; Salmikangas, P.; Saukko, P.; Lahtinen, U.; Lehesjoki, A.-E. Loss of Lysosomal Association of Cystatin B Proteins Representing Progressive Myoclonus Epilepsy, EPM1, Mutations. Eur. J. Hum. Genet. 2005, 13, 208–215. [Google Scholar] [CrossRef]
- Joensuu, T.; Tegelberg, S.; Reinmaa, E.; Segerstråle, M.; Hakala, P.; Pehkonen, H.; Korpi, E.R.; Tyynelä, J.; Taira, T.; Hovatta, I.; et al. Gene Expression Alterations in the Cerebellum and Granule Neurons of Cstb−/− Mouse Are Associated with Early Synaptic Changes and Inflammation. PLoS ONE 2014, 9, e89321. [Google Scholar] [CrossRef] [Green Version]
- Pennacchio, L.A.; Bouley, D.M.; Higgins, K.M.; Scott, M.P.; Noebels, J.L.; Myers, R.M. Progressive Ataxia, Myoclonic Epilepsy and Cerebellar Apoptosis in Cystatin B-Deficient Mice. Nat. Genet. 1998, 20, 251–258. [Google Scholar] [CrossRef]
- Penna, E.; Cerciello, A.; Chambery, A.; Russo, R.; Cernilogar, F.M.; Pedone, E.M.; Perrone-Capano, C.; Cappello, S.; Di Giaimo, R.; Crispino, M. Cystatin B Involvement in Synapse Physiology of Rodent Brains and Human Cerebral Organoids. Front. Mol. Neurosci. 2019, 12, 195. [Google Scholar] [CrossRef] [Green Version]
- Di Matteo, F.; Pipicelli, F.; Kyrousi, C.; Tovecci, I.; Penna, E.; Crispino, M.; Chambery, A.; Russo, R.; Ayo-Martin, A.C.; Giordano, M.; et al. Cystatin B Is Essential for Proliferation and Interneuron Migration in Individuals with EPM 1 Epilepsy. EMBO Mol. Med. 2020, 12, e11419. [Google Scholar] [CrossRef]
- Shannon, P.; Pennacchio, L.A.; Houseweart, M.K.; Minassian, B.A.; Myers, R.M. Neuropathological Changes in a Mouse Model of Progressive Myoclonus Epilepsy: Cystatin B Deficiency and Unverricht-Lundborg Disease. J. Neuropathol. Exp. Neurol. 2002, 61, 1085–1091. [Google Scholar] [CrossRef]
- Franceschetti, S.; Sancini, G.; Buzzi, A.; Zucchini, S.; Paradiso, B.; Magnaghi, G.; Frassoni, C.; Chikhladze, M.; Avanzini, G.; Simonato, M. A Pathogenetic Hypothesis of Unverricht–Lundborg Disease Onset and Progression. Neurobiol. Dis. 2007, 25, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Di Giaimo, R.; Riccio, M.; Santi, S.; Galeotti, C.; Ambrosetti, D.C.; Melli, M. New Insights into the Molecular Basis of Progressive Myoclonus Epilepsy: A Multiprotein Complex with Cystatin, B. Hum. Mol. Genet. 2002, 11, 2941–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling Cardiac Disease Mechanisms Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Progress, Promises and Challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, P.; Young-Pearse, T.L. Stem Cells on the Brain: Modeling Neurodevelopmental and Neurodegenerative Diseases Using Human Induced Pluripotent Stem Cells. J. Neurogenet. 2014, 28, 5–29. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Parrotta, E.; De Angelis, M.T.; Scalise, S.; Candeloro, P.; Santamaria, G.; Paonessa, M.; Coluccio, M.L.; Perozziello, G.; De Vitis, S.; Sgura, A.; et al. Two Sides of the Same Coin? Unraveling Subtle Differences between Human Embryonic and Induced Pluripotent Stem Cells by Raman Spectroscopy. Stem Cell Res. Ther. 2017, 8, 271. [Google Scholar] [CrossRef] [Green Version]
- Moradi, S.; Mahdizadeh, H.; Šarić, T.; Kim, J.; Harati, J.; Shahsavarani, H.; Greber, B.; Moore, J.B. Research and Therapy with Induced Pluripotent Stem Cells (IPSCs): Social, Legal, and Ethical Considerations. Stem Cell Res. Ther. 2019, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced Pluripotent Stem Cells: Applications in Regenerative Medicine, Disease Modeling, and Drug Discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 Cleavage Fragments: Signatures of Cell-Death Proteases in Neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Grillone, K.; Lo Conte, M.; Esposito, C.; Aguglia, U.; Ferlazzo, E.; Perrotti, N.; Malatesta, P.; et al. Generation of Human Induced Pluripotent Stem Cell Lines (UNIMGi003-A and UNIMGi004-A) from Two Italian Siblings Affected by Unverricht-Lundborg Disease. Stem Cell Res. 2021, 53, 102329. [Google Scholar] [CrossRef]
- Parrotta, E.I.; Scalise, S.; Taverna, D.; De Angelis, M.T.; Sarro, G.; Gaspari, M.; Santamaria, G.; Cuda, G. Comprehensive Proteogenomic Analysis of Human Embryonic and Induced Pluripotent Stem Cells. J. Cell. Mol. Med. 2019, 23, 5440–5453. [Google Scholar] [CrossRef] [PubMed]
- Scalise, S.; Zannino, C.; Lucchino, V.; Lo Conte, M.; Scaramuzzino, L.; Cifelli, P.; D’Andrea, T.; Martinello, K.; Fucile, S.; Palma, E.; et al. Human IPSC Modeling of Genetic Febrile Seizure Reveals Aberrant Molecular and Physiological Features Underlying an Impaired Neuronal Activity. Biomedicines 2022, 10, 1075. [Google Scholar] [CrossRef] [PubMed]
- Canafoglia, L.; Gennaro, E.; Capovilla, G.; Gobbi, G.; Boni, A.; Beccaria, F.; Viri, M.; Michelucci, R.; Agazzi, P.; Assereto, S.; et al. Electroclinical Presentation and Genotype-Phenotype Relationships in Patients with Unverricht-Lundborg Disease Carrying Compound Heterozygous CSTB Point and Indel Mutations: Compound Heterozygous EPM1A Patients. Epilepsia 2012, 53, 2120–2127. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, K.; Virtaneva, K.; Joensuu, T.; Palvimo, J.J.; Lehesjoki, A.-E. Characterization of the Cystatin B Gene Promoter Harboring the Dodecamer Repeat Expanded in Progressive Myoclonus Epilepsy, EPM1. Gene 2000, 242, 65–73. [Google Scholar] [CrossRef]
- Lalioti, M.D.; Scott, H.S.; Antonarakis, S.E. Altered Spacing of Promoter Elements Due to the Dodecamer Repeat Expansion Contributes to Reduced Expression of the Cystatin B Gene in EPM1. Hum. Mol. Genet. 1999, 8, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Lalioti, M.D.; Scott, H.S.; Buresi, C.; Rossier, C.; Bottani, A.; Morris, M.A.; Malafosse, A.; Antonarakis, S.E. Dodecamer Repeat Expansion in Cystatin B Gene in Progressive Myoclonus Epilepsy. Nature 1997, 386, 847–851. [Google Scholar] [CrossRef]
- O’Brien, A.; Marshall, C.R.; Blaser, S.; Ray, P.N.; Yoon, G. Severe Neurodegeneration, Progressive Cerebral Volume Loss and Diffuse Hypomyelination Associated with a Homozygous Frameshift Mutation in CSTB. Eur. J. Hum. Genet. 2017, 25, 775–778. [Google Scholar] [CrossRef]
- Barrett, A.J. The Cystatins: A New Class of Peptidase Inhibitors. Trends Biochem. Sci. 1987, 12, 193–196. [Google Scholar] [CrossRef]
- Turk, V.; Bode, W. The Cystatins: Protein Inhibitors of Cysteine Proteinases. FEBS Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J. Evolution of Proteins of the Cystatin Superfamily. J. Mol. Evol. 1990, 30, 60–71. [Google Scholar] [CrossRef]
- Stubbs, M.T.; Laber, B.; Bode, W.; Huber, R.; Jerala, R.; Lenarcic, B.; Turk, V. The Refined 2.4 A X-ray Crystal Structure of Recombinant Human Stefin B in Complex with the Cysteine Proteinase Papain: A Novel Type of Proteinase Inhibitor Interaction. EMBO J. 1990, 9, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Lenarcic, B.; Krizaj, I.; Zunec, P.; Turk, V. Differences in Specificity for the Interactions of Stefins A, B and D with Cysteine Proteinases. FEBS Lett. 1996, 395, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Turk, B.; Turk, D.; Turk, V. Lysosomal Cysteine Proteases: More than Scavengers. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzymol. 2000, 1477, 98–111. [Google Scholar] [CrossRef]
- Sukharev, S.A.; Pleshakova, O.V.; Sadovnikov, V.B. Role of Proteases in Activation of Apoptosis. Cell Death Differ. 1997, 4, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Stoka, V.; Turk, B.; Turk, V. Lysosomal Cysteine Proteases: Structural Features and Their Role in Apoptosis. IUBMB Life Int. Union Biochem. Mol. Biol. Life 2005, 57, 347–353. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojič, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine Cathepsins Trigger Caspase-Dependent Cell Death through Cleavage of Bid and Antiapoptotic Bcl-2 Homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef] [Green Version]
- Gobeil, S.; Boucher, C.C.; Nadeau, D.; Poirier, G.G. Characterization of the Necrotic Cleavage of Poly(ADP-Ribose) Polymerase (PARP-1): Implication of Lysosomal Proteases. Cell Death Differ. 2001, 8, 588–594. [Google Scholar] [CrossRef] [Green Version]
- Järvinen, M.; Rinne, A. Human Spleen Cysteineproteinase Inhibitor. Purification, Fractionation into Isoelectric Variants and Some Properties of the Variants. Biochim. Biophys. Acta 1982, 708, 210–217. [Google Scholar] [CrossRef]
- Kälviäinen, R. Progressive Myoclonus Epilepsies. Semin. Neurol. 2015, 35, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.J.; Ribeiro, D.; Chaves, J.; Amaral, O. Characterization of a Rare Unverricht–Lundborg Disease Mutation. Mol. Genet. Metab. Rep. 2015, 4, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Assenza, G.; Benvenga, A.; Gennaro, E.; Tombini, M.; Campana, C.; Assenza, F.; Di Pino, G.; Di Lazzaro, V. A Novel C132-134del Mutation in Unverricht-Lundborg Disease and the Review of Literature of Heterozygous Compound Patients. Epilepsia 2017, 58, e31–e35. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H. Repeat Expansion Diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Orban, M.; Goedel, A.; Haas, J.; Sandrock-Lang, K.; Gärtner, F.; Jung, C.B.; Zieger, B.; Parrotta, E.; Kurnik, K.; Sinnecker, D.; et al. Functional Comparison of Induced Pluripotent Stem Cell- and Blood-Derived GPIIbIIIa Deficient Platelets. PLoS ONE 2015, 10, e0115978. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced Pluripotent Stem Cell Technology: A Decade of Progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- De Angelis, M.T.; Santamaria, G.; Parrotta, E.I.; Scalise, S.; Lo Conte, M.; Gasparini, S.; Ferlazzo, E.; Aguglia, U.; Ciampi, C.; Sgura, A.; et al. Establishment and Characterization of Induced Pluripotent Stem Cells (IPSCs) from Central Nervous System Lupus Erythematosus. J. Cell. Mol. Med. 2019, 23, 7382–7394. [Google Scholar] [CrossRef]
- Parrotta, E.I.; Scalise, S.; Scaramuzzino, L.; Cuda, G. Stem Cells: The Game Changers of Human Cardiac Disease Modelling and Regenerative Medicine. Int. J. Mol. Sci. 2019, 20, 5760. [Google Scholar] [CrossRef] [Green Version]
- Scalise, S.; Scaramuzzino, L.; Lucchino, V.; Esposito, C.; Malatesta, P.; Grillone, K.; Perrotti, N.; Cuda, G.; Parrotta, E.I. Generation of IPSC Lines from Two Patients Affected by Febrile Seizure Due to Inherited Missense Mutation in SCN1A Gene. Stem Cell Res. 2020, 49, 102083. [Google Scholar] [CrossRef]
- Poeta, L.; Drongitis, D.; Verrillo, L.; Miano, M.G. DNA Hypermethylation and Unstable Repeat Diseases: A Paradigm of Transcriptional Silencing to Decipher the Basis of Pathogenic Mechanisms. Genes 2020, 11, 684. [Google Scholar] [CrossRef]
- Penázová, H.; Vorlicková, M. Guanine Tetraplex Formation by Short DNA Fragments Containing Runs of Guanine and Cytosine. Biophys. J. 1997, 73, 2054–2063. [Google Scholar] [CrossRef] [Green Version]
- Kopra, O.; Joensuu, T.; Lehesjoki, A.-E. Mouse Model of Unverricht-Lundborg Disease. In Movement Disorders, 2nd ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2015; Chapter 41; pp. 671–679. [Google Scholar]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine Cathepsins: Cellular Roadmap to Different Functions. Biochimie 2008, 90, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized Roles for Cysteine Cathepsins in Health and Disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine Cathepsins: From Structure, Function and Regulation to New Frontiers. Biochim. Biophys. Acta BBA Proteins Proteomics 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases That Are Vital for Survival but Can Also Be Fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Maurer, A.; Nieke, S.; Kalbacher, H. Cathepsin D: A Cellular Roadmap. Biochem. Biophys. Res. Commun. 2008, 376, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Mohan, P.; Pawlik, M.; DeRosa, S.; Fajiculay, J.; Che, S.; Grubb, A.; Ginsberg, S.D.; Nixon, R.A.; Levy, E. Cystatin C Rescues Degenerating Neurons in a Cystatin B-Knockout Mouse Model of Progressive Myoclonus Epilepsy. Am. J. Pathol. 2010, 177, 2256–2267. [Google Scholar] [CrossRef]
- Van Der Stappen, J.W.J.; Williams, A.C.; Maciewicz, R.A.; Paraskeva, C. Activation of Cathepsin B, Secreted by a Colorectal Cancer Cell Line Requires Low PH and Is Mediated by Cathepsin, D. Int. J. Cancer 1996, 67, 547–554. [Google Scholar] [CrossRef]
- Železnik, T.Z.; Kadin, A.; Turk, V.; Dolenc, I. Aspartic Cathepsin D Degrades the Cytosolic Cysteine Cathepsin Inhibitor Stefin B in the Cells. Biochem. Biophys. Res. Commun. 2015, 465, 213–217. [Google Scholar] [CrossRef]
- Houseweart, M.K.; Vilaythong, A.; Yin, X.-M.; Turk, B.; Noebels, J.L.; Myers, R.M. Apoptosis Caused by Cathepsins Does Not Require Bid Signaling in an in Vivo Model of Progressive Myoclonus Epilepsy (EPM1). Cell Death Differ. 2003, 10, 1329–1335. [Google Scholar] [CrossRef]
- Wille, A.; Gerber, A.; Heimburg, A.; Reisenauer, A.; Peters, C.; Saftig, P.; Reinheckel, T.; Welte, T.; Bühling, F. Cathepsin L Is Involved in Cathepsin D Processing and Regulation of Apoptosis in A549 Human Lung Epithelial Cells. Biol. Chem. 2004, 385. [Google Scholar] [CrossRef]
- Chwieralski, C.E.; Welte, T.; Bühling, F. Cathepsin-Regulated Apoptosis. Apoptosis 2006, 11, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Houseweart, M.K.; Pennacchio, L.A.; Vilaythong, A.; Peters, C.; Noebels, J.L.; Myers, R.M. Cathepsin B but Not Cathepsins L or S Contributes to the Pathogenesis of Unverricht-Lundborg Progressive Myoclonus Epilepsy (EPM1). J. Neurobiol. 2003, 56, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Buzzi, A.; Chikhladze, M.; Falcicchia, C.; Paradiso, B.; Lanza, G.; Soukupova, M.; Marti, M.; Morari, M.; Franceschetti, S.; Simonato, M. Loss of Cortical GABA Terminals in Unverricht–Lundborg Disease. Neurobiol. Dis. 2012, 47, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Ishisaka, R.; Utsumi, T.; Kanno, T.; Arita, K.; Katunuma, N.; Akiyama, J.; Utsumi, K. Participation of a Cathepsin L-Type Protease in the Activation of Caspase-3. Cell Struct. Funct. 1999, 24, 465–470. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | Age | Severity | Myoclonus | Epilepsy | Dysarthria | Photosensitivity | Cognitive Impairment | Pharmacological Treatment | CSTB Mutation |
|---|---|---|---|---|---|---|---|---|---|---|
| ULD1 | M | 11 | Severe | Yes | Yes | Yes | No | Yes | VPA, LEV, PB, CLN | Homozygous for dodecamer repeat |
| ULD2 | F | 12 | Mild | Yes | Yes | No | No | No | VPA, LEV, PB | Homozygous for dodecamer repeat |
| Sample name | Fragments Size | N of Dodecamers | ||
|---|---|---|---|---|
| First allele | Second allele | First allele | Second allele | |
| CTRL-iPSCs | 180 bp | 180 bp | 2 | 2 |
| ULD1-iPSCs | 644.22 bp | 921.02 bp | 41 | 64 |
| ULD2-iPSCs | 644.27 bp | 909.77 bp | 41 | 63 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Lo Conte, M.; Zannino, C.; Benedetto, G.L.; Aguglia, U.; Ferlazzo, E.; Cuda, G.; Parrotta, E.I. Insights into the Genetic Profile of Two Siblings Affected by Unverricht-Lundborg Disease Using Patient-Derived hiPSCs. Cells 2022, 11, 3491. https://doi.org/10.3390/cells11213491
Lucchino V, Scaramuzzino L, Scalise S, Lo Conte M, Zannino C, Benedetto GL, Aguglia U, Ferlazzo E, Cuda G, Parrotta EI. Insights into the Genetic Profile of Two Siblings Affected by Unverricht-Lundborg Disease Using Patient-Derived hiPSCs. Cells. 2022; 11(21):3491. https://doi.org/10.3390/cells11213491
Chicago/Turabian StyleLucchino, Valeria, Luana Scaramuzzino, Stefania Scalise, Michela Lo Conte, Clara Zannino, Giorgia Lucia Benedetto, Umberto Aguglia, Edoardo Ferlazzo, Giovanni Cuda, and Elvira Immacolata Parrotta. 2022. "Insights into the Genetic Profile of Two Siblings Affected by Unverricht-Lundborg Disease Using Patient-Derived hiPSCs" Cells 11, no. 21: 3491. https://doi.org/10.3390/cells11213491