
Aberrant B Cell Signaling in Autoimmune Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
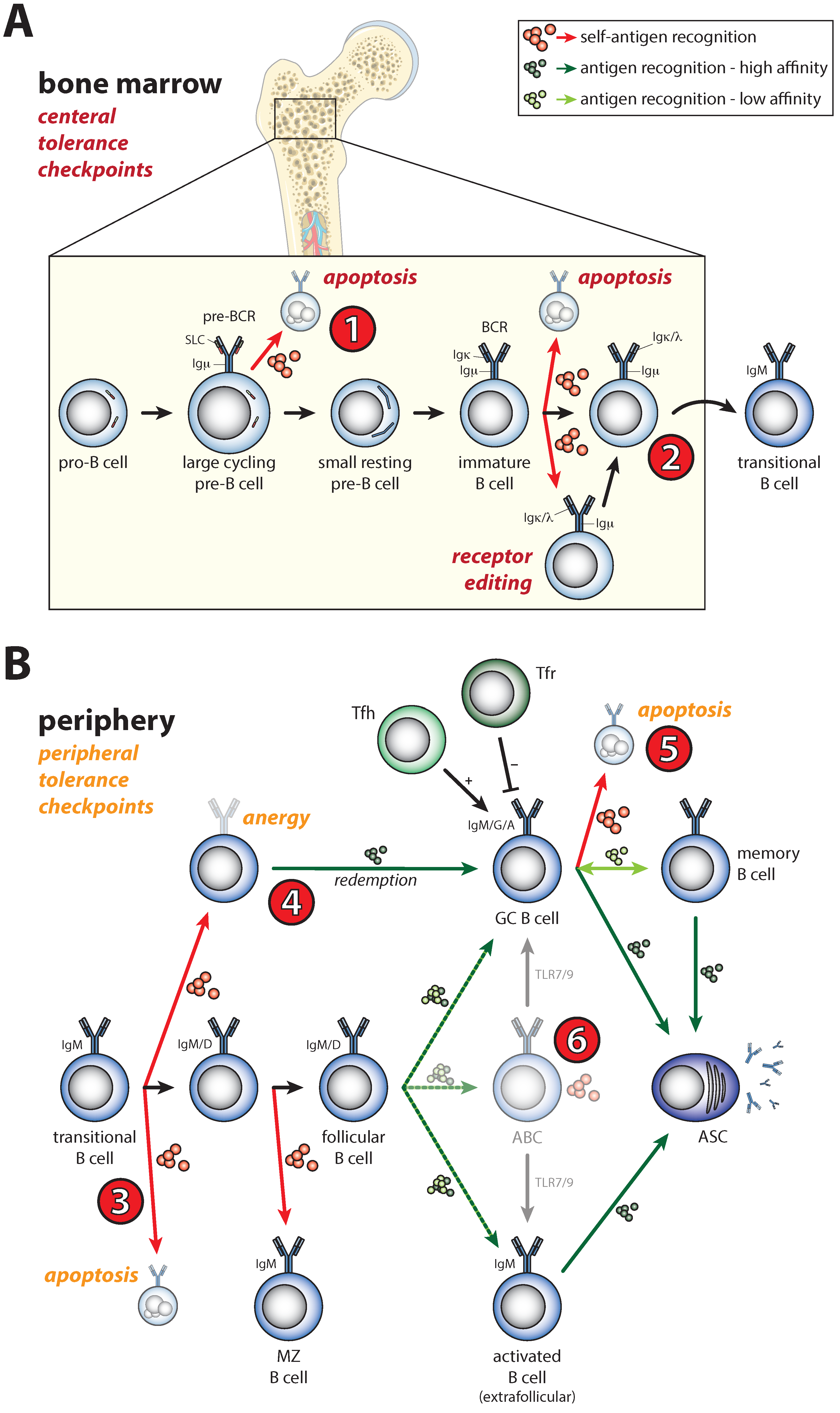
2. Shaping of the Naïve B Cell Repertoire during B Cell Development by B Cell Receptor Signals
3. Activation of Self-Reactive B Cells under Physiological Conditions
4. Defective Selection of Self-Reactive B Cells in Autoimmune Disease
5. Defective Activation of Self-Reactive B Cells in Autoimmune Disease
5.1. Enhanced Sensitivity of Naïve B Cells for Activating Signals
5.2. Activation of Anergic B Cells
5.3. GC Selection Defects and Spontaneous GC Formation
5.4. Enhanced Plasmablast Differentiation
5.5. Expansion of the Age-Associated B Cell Population
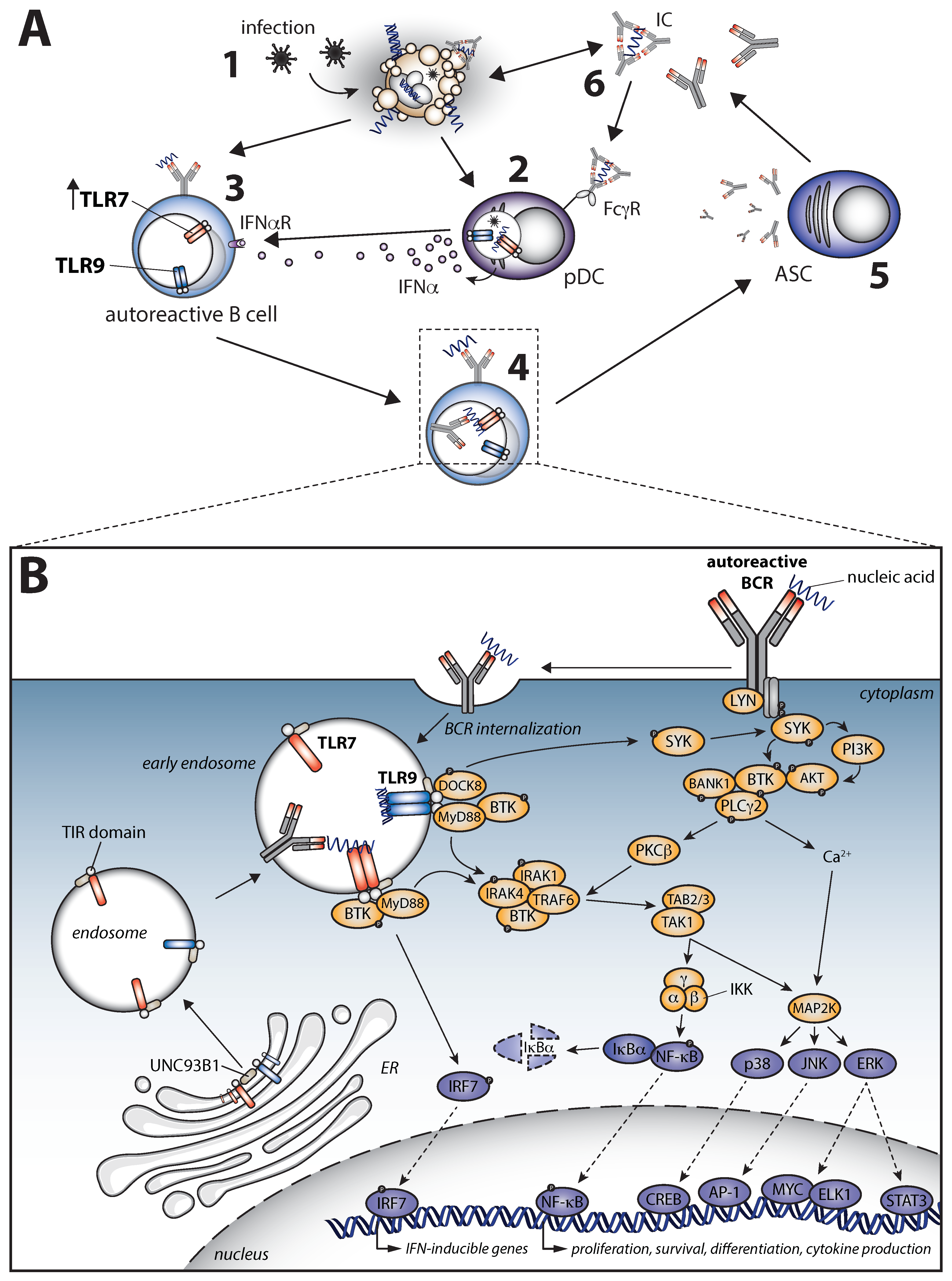
6. B Cell Receptor Signaling in Autoimmunity
6.1. Dual Role of LYN in BCR Signaling
6.2. Aberrant Levels and Activation of SYK, BTK, and PLCγ2 in Autoimmune Disease
6.3. PI3K–Akt–mTORC-Regulated Metabolism Is Essential for Normal B Cell Differentiation and Silencing of Autoreactive B Cells
7. Other Signaling Pathways Implicated in Autoimmunity
7.1. BAFF and APRIL as Drivers of B-Cell-Mediated Autoimmunity
7.2. CD40–CD40L Costimulatory Signals and PTPN22 Downregulation in Autoimmunity
7.3. Inhibitory Co-Receptors of BCR Signaling Acting through SHP-1
7.4. Inhibitory Co-Receptors of BCR Signaling Acting through SHIP-1
7.5. A Pathogenic Crosstalk between B Cell Receptor and Toll-like Receptor Signaling in Autoimmune Disease
8. Concluding Remarks and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkov, M.; van Schie, K.A.; van der Woude, D. Autoantibodies and B Cells: The ABC of rheumatoid arthritis pathophysiology. Immunol. Rev. 2020, 294, 148–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orrù, V.; Steri, M.; Sidore, C.; Marongiu, M.; Serra, V.; Olla, S.; Sole, G.; Lai, S.; Dei, M.; Mulas, A.; et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat. Genet. 2020, 52, 1036–1045. [Google Scholar] [CrossRef]
- Westra, H.-J.; Martínez-Bonet, M.; Onengut-Gumuscu, S.; Lee, A.; Luo, Y.; Teslovich, N.; Worthington, J.; Martin, J.; Huizinga, T.; Klareskog, L.; et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 2018, 50, 1366–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Tsao, B.P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat. Rev. Rheumatol. 2010, 6, 683–692. [Google Scholar] [CrossRef]
- Gómez Hernández, G.; Morell, M.; Alarcón-Riquelme, M.E. The Role of BANK1 in B Cell Signaling and Disease. Cells 2021, 10, 1184. [Google Scholar] [CrossRef]
- Tizaoui, K.; Terrazzino, S.; Cargnin, S.; Lee, K.H.; Gauckler, P.; Li, H.; Shin, J.I.; Kronbichler, A. The role of PTPN22 in the pathogenesis of autoimmune diseases: A comprehensive review. Semin. Arthritis Rheum. 2021, 51, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, J.; Xiao, F.; Zheng, L.; Zhu, X.; Wu, L.; Zhao, C.; Wang, S.; Rui, K.; Zou, H.; et al. B cell-activating factor and its targeted therapy in autoimmune diseases. Cytokine Growth Factor Rev. 2022, 64, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Isenberg, D.A. New therapies for systemic lupus erythematosus—Past imperfect, future tense. Nat. Rev. Rheumatol. 2019, 15, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Fetter, T.; Niebel, D.; Braegelmann, C.; Wenzel, J. Skin-Associated B Cells in the Pathogenesis of Cutaneous Autoimmune Diseases—Implications for Therapeutic Approaches. Cells 2020, 9, 2627. [Google Scholar] [CrossRef] [PubMed]
- Neys, S.F.H.; Rip, J.; Hendriks, R.W.; Corneth, O.B.J. Bruton’s Tyrosine Kinase Inhibition as an Emerging Therapy in Systemic Autoimmune Disease. Drugs 2021, 81, 1605–1626. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Giallourakis, C.; Mostoslavsky, R.; Alt, F.W. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu. Rev. Immunol. 2006, 24, 541–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, M.R.; Mandal, M.; Ochiai, K.; Singh, H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol. 2014, 14, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, M.; Okoreeh, M.K.; Kennedy, D.E.; Maienschein-Cline, M.; Ai, J.; McLean, K.C.; Kaverina, N.; Veselits, M.; Aifantis, I.; Gounari, F.; et al. CXCR4 signaling directs Igk recombination and the molecular mechanisms of late B lymphopoiesis. Nat. Immunol. 2019, 20, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, L.; Sasaki, Y.; Calado, D.P.; Zhang, B.; Paik, J.H.; DePinho, R.A.; Kutok, J.L.; Kearney, J.F.; Otipoby, K.L.; Rajewsky, K. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Shojaee, S.; Chan, L.N.; Buchner, M.; Cazzaniga, V.; Cosgun, K.N.; Geng, H.; Qiu, Y.H.; von Minden, M.D.; Ernst, T.; Hochhaus, A.; et al. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med. 2016, 22, 379–387. [Google Scholar] [CrossRef]
- Stein, M.; Dutting, S.; Mougiakakos, D.; Bosl, M.; Fritsch, K.; Reimer, D.; Urbanczyk, S.; Steinmetz, T.; Schuh, W.; Bozec, A.; et al. A defined metabolic state in pre B cells governs B-cell development and is counterbalanced by Swiprosin-2/EFhd1. Cell Death Differ. 2017, 24, 1239–1252. [Google Scholar] [CrossRef] [Green Version]
- Beck, T.C.; Gomes, A.C.; Cyster, J.G.; Pereira, J.P. CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. J. Exp. Med. 2014, 211, 2567–2581. [Google Scholar] [CrossRef]
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant autoantibody production by early human B cell precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamed, D.; Benschop, R.J.; Cambier, J.C.; Nemazee, D. Developmental regulation of B lymphocyte immune tolerance compartmentalizes clonal selection from receptor selection. Cell 1998, 92, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Nagaoka, H.; Gonzalez-Aseguinolaza, G.; Tsuji, M.; Nussenzweig, M.C. Immunization and infection change the number of recombination activating gene (RAG)-expressing B cells in the periphery by altering immature lymphocyte production. J. Exp. Med. 2000, 191, 2113–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giltiay, N.V.; Giordano, D.; Clark, E.A. The Plasticity of Newly Formed B Cells. J. Immunol. 2019, 203, 3095–3104. [Google Scholar] [CrossRef] [PubMed]
- Wesemann, D.R.; Portuguese, A.J.; Meyers, R.M.; Gallagher, M.P.; Cluff-Jones, K.; Magee, J.M.; Panchakshari, R.A.; Rodig, S.J.; Kepler, T.B.; Alt, F.W. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature 2013, 501, 112–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandel, P.C.; Monroe, J.G. Negative selection of immature B cells by receptor editing or deletion is determined by site of antigen encounter. Immunity 1999, 10, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Nagaoka, H.; Jankovic, M.; Misulovin, Z.; Suh, H.; Rolink, A.; Melchers, F.; Meffre, E.; Nussenzweig, M.C. Continued RAG expression in late stages of B cell development and no apparent re-induction after immunization. Nature 1999, 400, 682–687. [Google Scholar] [CrossRef]
- Monroe, R.J.; Seidl, K.J.; Gaertner, F.; Han, S.; Chen, F.; Sekiguchi, J.; Wang, J.; Ferrini, R.; Davidson, L.; Kelsoe, G.; et al. RAG2:GFP knockin mice reveal novel aspects of RAG2 expression in primary and peripheral lymphoid tissues. Immunity 1999, 11, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Su, T.T.; Rawlings, D.J. Transitional B lymphocyte subsets operate as distinct checkpoints in murine splenic B cell development. J. Immunol. 2002, 168, 2101–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.H.; Haberman, A.M.; Sant’Angelo, D.B.; Hannum, L.G.; Cancro, M.P.; Janeway, C.A., Jr.; Shlomchik, M.J. A B-cell receptor-specific selection step governs immature to mature B cell differentiation. Proc. Natl. Acad. Sci. USA 2000, 97, 2743–2748. [Google Scholar] [CrossRef]
- Goodnow, C.C.; Crosbie, J.; Adelstein, S.; Lavoie, T.B.; Smith-Gill, S.J.; Brink, R.A.; Pritchard-Briscoe, H.; Wotherspoon, J.S.; Loblay, R.H.; Raphael, K.; et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988, 334, 676–682. [Google Scholar] [CrossRef]
- Tanaka, S.; Ise, W.; Baba, Y.; Kurosaki, T. Silencing and activating anergic B cells. Immunol. Rev. 2022, 307, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lesley, R.; Xu, Y.; Kalled, S.L.; Hess, D.M.; Schwab, S.R.; Shu, H.B.; Cyster, J.G. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 2004, 20, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Thien, M.; Phan, T.G.; Gardam, S.; Amesbury, M.; Basten, A.; Mackay, F.; Brink, R. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity 2004, 20, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweighoffer, E.; Tybulewicz, V.L. BAFF signaling in health and disease. Curr. Opin. Immunol. 2021, 71, 124–131. [Google Scholar] [CrossRef]
- Depascale, R.; Gatto, M.; Zen, M.; Saccon, F.; Larosa, M.; Zanatta, E.; Bindoli, S.; Doria, A.; Iaccarino, L. Belimumab: A step forward in the treatment of systemic lupus erythematosus. Expert. Opin. Biol. Ther. 2021, 21, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.P.; Rajewsky, K. Rapid elimination of mature autoreactive B cells demonstrated by Cre-induced change in B cell antigen receptor specificity in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 13171–13175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toellner, K.M.; Gulbranson-Judge, A.; Taylor, D.R.; Sze, D.M.; MacLennan, I.C. Immunoglobulin switch transcript production in vivo related to the site and time of antigen-specific B cell activation. J. Exp. Med. 1996, 183, 2303–2312. [Google Scholar] [CrossRef] [Green Version]
- Roco, J.A.; Mesin, L.; Binder, S.C.; Nefzger, C.; Gonzalez-Figueroa, P.; Canete, P.F.; Ellyard, J.; Shen, Q.; Robert, P.A.; Cappello, J.; et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019, 51, 337–350. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Cooke, M.P.; Ho, W.Y.; Grein, J.; Townsend, S.E.; Davis, M.M.; Goodnow, C.C. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature 1995, 376, 181–184. [Google Scholar] [CrossRef]
- Breitfeld, D.; Ohl, L.; Kremmer, E.; Ellwart, J.; Sallusto, F.; Lipp, M.; Forster, R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 2000, 192, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Rott, L.S.; Clark-Lewis, I.; Campbell, D.J.; Wu, L.; Butcher, E.C. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J. Exp. Med. 2001, 193, 1373–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaerli, P.; Willimann, K.; Lang, A.B.; Lipp, M.; Loetscher, P.; Moser, B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 2000, 192, 1553–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yu, M.; Zheng, Y.; Fu, G.; Xin, G.; Zhu, W.; Luo, L.; Burns, R.; Li, Q.Z.; Dent, A.L.; et al. CXCR5(+)PD-1(+) follicular helper CD8 T cells control B cell tolerance. Nat. Commun. 2019, 10, 4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollenberg, I.; Agua-Doce, A.; Hernandez, A.; Almeida, C.; Oliveira, V.G.; Faro, J.; Graca, L. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 2011, 187, 4553–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, K.P.; Kuhn, R.; Rajewsky, K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 1997, 90, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Asano, M.; Shinton, S.A.; Gui, M.; Allman, D.; Stewart, C.L.; Silver, J.; Hardy, R.R. Positive selection of natural autoreactive B cells. Science 1999, 285, 113–116. [Google Scholar] [CrossRef]
- Wen, L.; Brill-Dashoff, J.; Shinton, S.A.; Asano, M.; Hardy, R.R.; Hayakawa, K. Evidence of marginal-zone B cell-positive selection in spleen. Immunity 2005, 23, 297–308. [Google Scholar] [CrossRef]
- Tsiantoulas, D.; Gruber, S.; Binder, C.J. B-1 cell immunoglobulin directed against oxidation-specific epitopes. Front. Immunol. 2012, 3, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vas, J.; Grönwall, C.; Marshak-Rothstein, A.; Silverman, G.J. Natural antibody to apoptotic cell membranes inhibits the proinflammatory properties of lupus autoantibody immune complexes. Arthritis Rheum. 2012, 64, 3388–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannoor, K.; Matejuk, A.; Xu, Y.; Beardall, M.; Chen, C. Expression of natural autoantibodies in MRL-lpr mice protects from lupus nephritis and improves survival. J. Immunol. 2012, 188, 3628–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amendt, T.; Allies, G.; Nicolo, A.; El Ayoubi, O.; Young, M.; Roszer, T.; Setz, C.S.; Warnatz, K.; Jumaa, H. Autoreactive antibodies control blood glucose by regulating insulin homeostasis. Proc. Natl. Acad. Sci. USA 2022, 119, e2115695119. [Google Scholar] [CrossRef] [PubMed]
- Amendt, T.; Jumaa, H. Memory IgM protects endogenous insulin from autoimmune destruction. EMBO J. 2021, 40, e107621. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Gordon, S.M.; Waldman, M.; Edison, J.D.; Little, D.J.; Stitt, R.S.; Bailey, W.T.; Hughes, J.B.; Olson, S.W. Autoantibodies are present before the clinical diagnosis of systemic sclerosis. PLoS ONE 2019, 14, e0214202. [Google Scholar] [CrossRef] [Green Version]
- Rantapaa-Dahlqvist, S.; de Jong, B.A.; Berglin, E.; Hallmans, G.; Wadell, G.; Stenlund, H.; Sundin, U.; van Venrooij, W.J. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2741–2749. [Google Scholar] [CrossRef]
- Theander, E.; Jonsson, R.; Sjostrom, B.; Brokstad, K.; Olsson, P.; Henriksson, G. Prediction of Sjogren’s Syndrome Years Before Diagnosis and Identification of Patients With Early Onset and Severe Disease Course by Autoantibody Profiling. Arthritis Rheumatol. 2015, 67, 2427–2436. [Google Scholar] [CrossRef] [Green Version]
- Verge, C.F.; Gianani, R.; Kawasaki, E.; Yu, L.; Pietropaolo, M.; Jackson, R.A.; Chase, H.P.; Eisenbarth, G.S. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996, 45, 926–933. [Google Scholar] [CrossRef]
- Halkom, A.; Wu, H.; Lu, Q. Contribution of mouse models in our understanding of lupus. Int. Rev. Immunol. 2020, 39, 174–187. [Google Scholar] [CrossRef]
- Meehan, G.R.; Thomas, R.; Al Khabouri, S.; Wehr, P.; Hilkens, C.M.; Wraith, D.C.; Sieghart, D.; Bonelli, M.; Nagy, G.; Garside, P.; et al. Preclinical models of arthritis for studying immunotherapy and immune tolerance. Ann. Rheum. Dis. 2021, 80, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Masli, S.; Dartt, D.A. Mouse Models of Sjögren’s Syndrome with Ocular Surface Disease. Int. J. Mol. Sci. 2020, 21, 9112. [Google Scholar] [CrossRef] [PubMed]
- Bonasia, C.G.; Abdulahad, W.H.; Rutgers, A.; Heeringa, P.; Bos, N.A. B Cell Activation and Escape of Tolerance Checkpoints: Recent Insights from Studying Autoreactive B Cells. Cells 2021, 10, 1190. [Google Scholar] [CrossRef] [PubMed]
- Suurmond, J.; Atisha-Fregoso, Y.; Marasco, E.; Barlev, A.N.; Ahmed, N.; Calderon, S.A.; Wong, M.Y.; Mackay, M.C.; Aranow, C.; Diamond, B. Loss of an IgG plasma cell checkpoint in patients with lupus. J. Allergy Clin. Immunol. 2019, 143, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Dorner, T.; Lipsky, P.E. Immunoglobulin variable-region gene usage in systemic autoimmune diseases. Arthritis Rheum. 2001, 44, 2715–2727. [Google Scholar] [CrossRef]
- Yurasov, S.; Hammersen, J.; Tiller, T.; Tsuiji, M.; Wardemann, H. B-cell tolerance checkpoints in healthy humans and patients with systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2005, 1062, 165–174. [Google Scholar] [CrossRef]
- Yurasov, S.; Tiller, T.; Tsuiji, M.; Velinzon, K.; Pascual, V.; Wardemann, H.; Nussenzweig, M.C. Persistent expression of autoantibodies in SLE patients in remission. J. Exp. Med. 2006, 203, 2255–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meffre, E.; O’Connor, K.C. Impaired B-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol. Rev. 2019, 292, 90–101. [Google Scholar] [CrossRef]
- Castiello, M.C.; Bosticardo, M.; Pala, F.; Catucci, M.; Chamberlain, N.; van Zelm, M.C.; Driessen, G.J.; Pac, M.; Bernatowska, E.; Scaramuzza, S.; et al. Wiskott-Aldrich Syndrome protein deficiency perturbs the homeostasis of B-cell compartment in humans. J. Autoimmun. 2014, 50, 42–50. [Google Scholar] [CrossRef]
- Ng, Y.S.; Wardemann, H.; Chelnis, J.; Cunningham-Rundles, C.; Meffre, E. Bruton’s tyrosine kinase is essential for human B cell tolerance. J. Exp. Med. 2004, 200, 927–934. [Google Scholar] [CrossRef]
- Isnardi, I.; Ng, Y.S.; Srdanovic, I.; Motaghedi, R.; Rudchenko, S.; von Bernuth, H.; Zhang, S.Y.; Puel, A.; Jouanguy, E.; Picard, C.; et al. IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity 2008, 29, 746–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuraoka, M.; Holl, T.M.; Liao, D.; Womble, M.; Cain, D.W.; Reynolds, A.E.; Kelsoe, G. Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11560–11565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, G.; Ng, Y.S.; Bannock, J.M.; Lavoie, A.; Walter, J.E.; Notarangelo, L.D.; Kilic, S.S.; Aksu, G.; Debre, M.; Rieux-Laucat, F.; et al. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc. Natl. Acad. Sci. USA 2011, 108, 11554–11559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Jiang, L.; Melo-Jorge, M.; Puthenveetil, M.; Zhang, X.; Carroll, M.C.; Imanishi-Kari, T. T cell-independent somatic hypermutation in murine B cells with an immature phenotype. Immunity 2004, 20, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Cantaert, T.; Schickel, J.N.; Bannock, J.M.; Ng, Y.S.; Massad, C.; Oe, T.; Wu, R.; Lavoie, A.; Walter, J.E.; Notarangelo, L.D.; et al. Activation-Induced Cytidine Deaminase Expression in Human B Cell Precursors Is Essential for Central B Cell Tolerance. Immunity 2015, 43, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Kuraoka, M.; Snowden, P.B.; Nojima, T.; Verkoczy, L.; Haynes, B.F.; Kitamura, D.; Kelsoe, G. BCR and Endosomal TLR Signals Synergize to Increase AID Expression and Establish Central B Cell Tolerance. Cell Rep. 2017, 18, 1627–1635. [Google Scholar] [CrossRef]
- Tipton, C.M.; Fucile, C.F.; Darce, J.; Chida, A.; Ichikawa, T.; Gregoretti, I.; Schieferl, S.; Hom, J.; Jenks, S.; Feldman, R.J.; et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol. 2015, 16, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Scharer, C.D.; Blalock, E.L.; Barwick, B.G.; Haines, R.R.; Wei, C.; Sanz, I.; Boss, J.M. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naïve SLE B cells. Sci. Rep. 2016, 6, 27030. [Google Scholar] [CrossRef]
- Scharer, C.D.; Blalock, E.L.; Mi, T.; Barwick, B.G.; Jenks, S.A.; Deguchi, T.; Cashman, K.S.; Neary, B.E.; Patterson, D.G.; Hicks, S.L.; et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat. Immunol. 2019, 20, 1071–1082. [Google Scholar] [CrossRef]
- Joo, H.; Coquery, C.; Xue, Y.; Gayet, I.; Dillon, S.R.; Punaro, M.; Zurawski, G.; Banchereau, J.; Pascual, V.; Oh, S. Serum from patients with SLE instructs monocytes to promote IgG and IgA plasmablast differentiation. J. Exp. Med. 2012, 209, 1335–1348. [Google Scholar] [CrossRef]
- Franks, S.E.; Cambier, J.C. Putting on the Brakes: Regulatory Kinases and Phosphatases Maintaining B Cell Anergy. Front. Immunol. 2018, 9, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duty, J.A.; Szodoray, P.; Zheng, N.Y.; Koelsch, K.A.; Zhang, Q.; Swiatkowski, M.; Mathias, M.; Garman, L.; Helms, C.; Nakken, B.; et al. Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J. Exp. Med. 2009, 206, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Szodoray, P.; Stanford, S.M.; Molberg, Ø.; Munthe, L.A.; Bottini, N.; Nakken, B. T-helper signals restore B-cell receptor signaling in autoreactive anergic B cells by upregulating CD45 phosphatase activity. J. Allergy Clin. Immunol. 2016, 138, 839–851.e838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S.F.; Huang, Y.; Kaur, K.; Popova, L.I.; Ho, I.Y.; Pauli, N.T.; Henry Dunand, C.J.; Taylor, W.M.; Lim, S.; Huang, M.; et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl. Med. 2015, 7, 316ra192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthmiller, J.J.; Lan, L.Y.; Fernandez-Quintero, M.L.; Han, J.; Utset, H.A.; Bitar, D.J.; Hamel, N.J.; Stovicek, O.; Li, L.; Tepora, M.; et al. Polyreactive Broadly Neutralizing B cells Are Selected to Provide Defense against Pandemic Threat Influenza Viruses. Immunity 2020, 53, 1230–1244.e5. [Google Scholar] [CrossRef]
- Sabouri, Z.; Schofield, P.; Horikawa, K.; Spierings, E.; Kipling, D.; Randall, K.L.; Langley, D.; Roome, B.; Vazquez-Lombardi, R.; Rouet, R.; et al. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc. Natl. Acad. Sci. USA 2014, 111, E2567–E2575. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.H.; Jackson, J.; Christ, D.; Goodnow, C.C. Clonal redemption of autoantibodies by somatic hypermutation away from self-reactivity during human immunization. J. Exp. Med. 2016, 213, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Cappione, A., 3rd; Anolik, J.H.; Pugh-Bernard, A.; Barnard, J.; Dutcher, P.; Silverman, G.; Sanz, I. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J. Clin. Investig. 2005, 115, 3205–3216. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.D.; Liang, D.; Wu, X.N.; Li, Y.; Niu, J.W.; Zhou, C.; Wang, L.; Chen, H.; Zheng, W.J.; Fei, Y.Y.; et al. Contribution and underlying mechanisms of CXCR4 overexpression in patients with systemic lupus erythematosus. Cell. Mol. Immunol. 2017, 14, 842–849. [Google Scholar] [CrossRef] [Green Version]
- Simpson, N.; Gatenby, P.A.; Wilson, A.; Malik, S.; Fulcher, D.A.; Tangye, S.G.; Manku, H.; Vyse, T.J.; Roncador, G.; Huttley, G.A.; et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 234–244. [Google Scholar] [CrossRef]
- Wang, J.; Shan, Y.; Jiang, Z.; Feng, J.; Li, C.; Ma, L.; Jiang, Y. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin. Exp. Immunol. 2013, 174, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Wu, Z.B.; Ding, J.; Zheng, Z.H.; Li, X.Y.; Chen, L.N.; Zhu, P. Role of the frequency of blood CD4(+) CXCR5(+) CCR6(+) T cells in autoimmunity in patients with Sjögren’s syndrome. Biochem. Biophys. Res. Commun. 2012, 422, 238–244. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Kroese, F.G.; Meiners, P.M.; Corneth, O.B.; Huitema, M.G.; Haacke, E.A.; van der Vegt, B.; Arends, S.; Vissink, A.; Bootsma, H.; et al. B Cell Depletion Therapy Normalizes Circulating Follicular Th Cells in Primary Sjögren Syndrome. J. Rheumatol. 2017, 44, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Gago da Graça, C.; van Baarsen, L.G.M.; Mebius, R.E. Tertiary Lymphoid Structures: Diversity in Their Development, Composition, and Role. J. Immunol. 2021, 206, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Alduraibi, F.; Ponder, D.; Duck, W.L.; Morrow, C.D.; Foote, J.B.; Schoeb, T.R.; Fatima, H.; Elson, C.O., 3rd; Hsu, H.C.; et al. Host Genetics But Not Commensal Microbiota Determines the Initial Development of Systemic Autoimmune Disease in BXD2 Mice. Arthritis Rheumatol. 2022, 74, 634–640. [Google Scholar] [CrossRef]
- Domeier, P.P.; Schell, S.L.; Rahman, Z.S. Spontaneous germinal centers and autoimmunity. Autoimmunity 2017, 50, 4–18. [Google Scholar] [CrossRef] [Green Version]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Kil, L.P.; de Bruijn, M.J.; van Nimwegen, M.; Corneth, O.B.; van Hamburg, J.P.; Dingjan, G.M.; Thaiss, F.; Rimmelzwaan, G.F.; Elewaut, D.; Delsing, D.; et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 2012, 119, 3744–3756. [Google Scholar] [CrossRef]
- Becker-Herman, S.; Meyer-Bahlburg, A.; Schwartz, M.A.; Jackson, S.W.; Hudkins, K.L.; Liu, C.; Sather, B.D.; Khim, S.; Liggitt, D.; Song, W.; et al. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J. Exp. Med. 2011, 208, 2033–2042. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Silva, D.G.; Martin, J.L.; Pratama, A.; Hu, X.; Chang, P.P.; Walters, G.; Vinuesa, C.G. Interferon-γ excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity 2012, 37, 880–892. [Google Scholar] [CrossRef]
- Hsu, H.C.; Yang, P.; Wang, J.; Wu, Q.; Myers, R.; Chen, J.; Yi, J.; Guentert, T.; Tousson, A.; Stanus, A.L.; et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2008, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, L.; Xie, D.; Reddy, S.; Sleasman, J.W.; Ma, L.; Zhong, X.P. Regulation of Intrinsic and Bystander T Follicular Helper Cell Differentiation and Autoimmunity by Tsc1. Front. Immunol. 2021, 12, 620437. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.A.; Zhu, M.; Zhang, W. Spontaneous Differentiation of T Follicular Helper Cells in LATY136F Mutant Mice. Front. Immunol. 2021, 12, 656817. [Google Scholar] [CrossRef] [PubMed]
- Arkatkar, T.; Du, S.W.; Jacobs, H.M.; Dam, E.M.; Hou, B.; Buckner, J.H.; Rawlings, D.J.; Jackson, S.W. B cell-derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J. Exp. Med. 2017, 214, 3207–3217. [Google Scholar] [CrossRef]
- Corneth, O.B.; de Bruijn, M.J.; Rip, J.; Asmawidjaja, P.S.; Kil, L.P.; Hendriks, R.W. Enhanced Expression of Bruton’s Tyrosine Kinase in B Cells Drives Systemic Autoimmunity by Disrupting T Cell Homeostasis. J. Immunol. 2016, 197, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Munroe, M.E.; Lu, R.; Zhao, Y.D.; Fife, D.A.; Robertson, J.M.; Guthridge, J.M.; Niewold, T.B.; Tsokos, G.C.; Keith, M.P.; Harley, J.B.; et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis. 2016, 75, 2014–2021. [Google Scholar] [CrossRef] [Green Version]
- Zumaquero, E.; Stone, S.L.; Scharer, C.D.; Jenks, S.A.; Nellore, A.; Mousseau, B.; Rosal-Vela, A.; Botta, D.; Bradley, J.E.; Wojciechowski, W.; et al. IFNγ induces epigenetic programming of human T-bet(hi) B cells and promotes TLR7/8 and IL-21 induced differentiation. eLife 2019, 8, e41641. [Google Scholar] [CrossRef]
- Chiang, K.; Largent, A.D.; Arkatkar, T.; Thouvenel, C.D.; Du, S.W.; Shumlak, N.; Woods, J.; Li, Q.Z.; Liu, Y.; Hou, B.; et al. Cutting Edge: A Threshold of B Cell Costimulatory Signals Is Required for Spontaneous Germinal Center Formation in Autoimmunity. J. Immunol. 2021, 207, 2217–2222. [Google Scholar] [CrossRef]
- Domeier, P.P.; Chodisetti, S.B.; Soni, C.; Schell, S.L.; Elias, M.J.; Wong, E.B.; Cooper, T.K.; Kitamura, D.; Rahman, Z.S. IFN-γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J. Exp. Med. 2016, 213, 715–732. [Google Scholar] [CrossRef] [Green Version]
- Soni, C.; Wong, E.B.; Domeier, P.P.; Khan, T.N.; Satoh, T.; Akira, S.; Rahman, Z.S. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J. Immunol. 2014, 193, 4400–4414. [Google Scholar] [CrossRef]
- Sanchez, H.N.; Moroney, J.B.; Gan, H.; Shen, T.; Im, J.L.; Li, T.; Taylor, J.R.; Zan, H.; Casali, P. B cell-intrinsic epigenetic modulation of antibody responses by dietary fiber-derived short-chain fatty acids. Nat. Commun. 2020, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkerts, J.; Redegeld, F.; Folkerts, G.; Blokhuis, B.; van den Berg, M.P.; de Bruijn, M.J.; van IJcken, W.F.; Junt, T.; Tam, S.Y.; Galli, S.J.; et al. Butyrate inhibits human mast cell activation via epigenetic regulation of FcεRI-mediated signaling. Allergy 2020, 75, 1966–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malkiel, S.; Barlev, A.N.; Atisha-Fregoso, Y.; Suurmond, J.; Diamond, B. Plasma Cell Differentiation Pathways in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 427. [Google Scholar] [CrossRef] [Green Version]
- Seavey, M.M.; Lu, L.D.; Stump, K.L.; Wallace, N.H.; Ruggeri, B.A. Novel, orally active, proteasome inhibitor, delanzomib (CEP-18770), ameliorates disease symptoms and glomerulonephritis in two preclinical mouse models of SLE. Int. Immunopharmacol. 2012, 12, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.T.; Conley, T.; Muchamuel, T.; Jiang, J.; Lee, S.; Owen, T.; Barnard, J.; Nevarez, S.; Goldman, B.I.; Kirk, C.J.; et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012, 64, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.B.; Barati, M.T.; Powell, D.W.; Turbeville, H.R.; Ryan, M.J. Plasma Cell Depletion Attenuates Hypertension in an Experimental Model of Autoimmune Disease. Hypertension 2018, 71, 719–728. [Google Scholar] [CrossRef]
- Alexander, T.; Cheng, Q.; Klotsche, J.; Khodadadi, L.; Waka, A.; Biesen, R.; Hoyer, B.F.; Burmester, G.R.; Radbruch, A.; Hiepe, F. Proteasome inhibition with bortezomib induces a therapeutically relevant depletion of plasma cells in SLE but does not target their precursors. Eur. J. Immunol. 2018, 48, 1573–1579. [Google Scholar] [CrossRef] [Green Version]
- Walhelm, T.; Gunnarsson, I.; Heijke, R.; Leonard, D.; Trysberg, E.; Eriksson, P.; Sjöwall, C. Clinical Experience of Proteasome Inhibitor Bortezomib Regarding Efficacy and Safety in Severe Systemic Lupus Erythematosus: A Nationwide Study. Front. Immunol. 2021, 12, 756941. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Anolik, J.; Cappione, A.; Zheng, B.; Pugh-Bernard, A.; Brooks, J.; Lee, E.H.; Milner, E.C.; Sanz, I. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J. Immunol. 2007, 178, 6624–6633. [Google Scholar] [CrossRef]
- Phalke, S.; Rivera-Correa, J.; Jenkins, D.; Flores Castro, D.; Giannopoulou, E.; Pernis, A.B. Molecular mechanisms controlling age-associated B cells in autoimmunity. Immunol. Rev. 2022, 307, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Mouat, I.C.; Goldberg, E.; Horwitz, M.S. Age-associated B cells in autoimmune diseases. Cell. Mol. Life Sci. 2022, 79, 402. [Google Scholar] [CrossRef] [PubMed]
- Brodie, E.J.; Infantino, S.; Low, M.S.Y.; Tarlinton, D.M. Lyn, Lupus, and (B) Lymphocytes, a Lesson on the Critical Balance of Kinase Signaling in Immunity. Front. Immunol. 2018, 9, 401. [Google Scholar] [CrossRef] [Green Version]
- Oellerich, T.; Bremes, V.; Neumann, K.; Bohnenberger, H.; Dittmann, K.; Hsiao, H.H.; Engelke, M.; Schnyder, T.; Batista, F.D.; Urlaub, H.; et al. The B-cell antigen receptor signals through a preformed transducer module of SLP65 and CIN85. EMBO J. 2011, 30, 3620–3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, J.; Wong, L.E.; Pirkuliyeva, S.; Schulz, K.; Schwiegk, C.; Fünfgeld, K.G.; Keppler, S.; Batista, F.D.; Urlaub, H.; Habeck, M.; et al. The adaptor protein CIN85 assembles intracellular signaling clusters for B cell activation. Sci. Signal. 2016, 9, ra66. [Google Scholar] [CrossRef]
- Yamanashi, Y.; Kakiuchi, T.; Mizuguchi, J.; Yamamoto, T.; Toyoshima, K. Association of B cell antigen receptor with protein tyrosine kinase Lyn. Science 1991, 251, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.; Petro, J.B.; Carlesso, G.; Shinners, N.P.; Lowe, J.; Khan, W.N. B-cell antigen receptor activates transcription factors NFAT (nuclear factor of activated T-cells) and NF-kappaB (nuclear factor kappaB) via a mechanism that involves diacylglycerol. Biochem. Soc. Trans. 2004, 32, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Beitz, L.O.; Fruman, D.A.; Kurosaki, T.; Cantley, L.C.; Scharenberg, A.M. SYK is upstream of phosphoinositide 3-kinase in B cell receptor signaling. J. Biol. Chem. 1999, 274, 32662–32666. [Google Scholar] [CrossRef] [Green Version]
- Pogue, S.L.; Kurosaki, T.; Bolen, J.; Herbst, R. B cell antigen receptor-induced activation of Akt promotes B cell survival and is dependent on Syk kinase. J. Immunol. 2000, 165, 1300–1306. [Google Scholar] [CrossRef] [Green Version]
- Chan, V.W.; Lowell, C.A.; DeFranco, A.L. Defective negative regulation of antigen receptor signaling in Lyn-deficient B lymphocytes. Curr. Biol. 1998, 8, 545–553. [Google Scholar] [CrossRef]
- Smith, K.G.; Tarlinton, D.M.; Doody, G.M.; Hibbs, M.L.; Fearon, D.T. Inhibition of the B cell by CD22: A requirement for Lyn. J. Exp. Med. 1998, 187, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.; Bikah, G.; Venkataraman, C.; Bondada, S. Negative regulation of antigen receptor-mediated signaling by constitutive association of CD5 with the SHP-1 protein tyrosine phosphatase in B-1 B cells. Eur. J. Immunol. 1999, 29, 3319–3328. [Google Scholar] [CrossRef]
- Zhang, J.; Somani, A.K.; Siminovitch, K.A. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin. Immunol. 2000, 12, 361–378. [Google Scholar] [CrossRef]
- Okada, M.; Nada, S.; Yamanashi, Y.; Yamamoto, T.; Nakagawa, H. CSK: A protein-tyrosine kinase involved in regulation of src family kinases. J. Biol. Chem. 1991, 266, 24249–24252. [Google Scholar] [CrossRef]
- Ono, M.; Bolland, S.; Tempst, P.; Ravetch, J.V. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature 1996, 383, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Bolland, S.; Pearse, R.N.; Kurosaki, T.; Ravetch, J.V. SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity 1998, 8, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Saijo, K.; Schmedt, C.; Su, I.H.; Karasuyama, H.; Lowell, C.A.; Reth, M.; Adachi, T.; Patke, A.; Santana, A.; Tarakhovsky, A. Essential role of Src-family protein tyrosine kinases in NF-kappaB activation during B cell development. Nat. Immunol. 2003, 4, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.W.; Meng, F.; Soriano, P.; DeFranco, A.L.; Lowell, C.A. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 1997, 7, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Hibbs, M.L.; Tarlinton, D.M.; Armes, J.; Grail, D.; Hodgson, G.; Maglitto, R.; Stacker, S.A.; Dunn, A.R. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell 1995, 83, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Lamagna, C.; Hu, Y.; DeFranco, A.L.; Lowell, C.A. B cell-specific loss of Lyn kinase leads to autoimmunity. J. Immunol. 2014, 192, 919–928. [Google Scholar] [CrossRef]
- Hua, Z.; Gross, A.J.; Lamagna, C.; Ramos-Hernandez, N.; Scapini, P.; Ji, M.; Shao, H.; Lowell, C.A.; Hou, B.; DeFranco, A.L. Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J. Immunol. 2014, 192, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamagna, C.; Scapini, P.; van Ziffle, J.A.; DeFranco, A.L.; Lowell, C.A. Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc. Natl. Acad. Sci. USA 2013, 110, E3311–E3320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, K.L.; Crockford, T.L.; Bouriez-Jones, T.; Milling, S.; Lambe, T.; Cornall, R.J. MyD88-dependent autoimmune disease in Lyn-deficient mice. Eur. J. Immunol. 2007, 37, 2734–2743. [Google Scholar] [CrossRef]
- Flores-Borja, F.; Kabouridis, P.S.; Jury, E.C.; Isenberg, D.A.; Mageed, R.A. Decreased Lyn expression and translocation to lipid raft signaling domains in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2005, 52, 3955–3965. [Google Scholar] [CrossRef] [PubMed]
- Liossis, S.N.; Solomou, E.E.; Dimopoulos, M.A.; Panayiotidis, P.; Mavrikakis, M.M.; Sfikakis, P.P. B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J. Investig. Med. 2001, 49, 157–165. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, J.; Mu, R.; Gao, Y.; Tan, X.; Li, Y.; Li, Z.; Yang, G. MicroRNA-30a promotes B cell hyperactivity in patients with systemic lupus erythematosus by direct interaction with Lyn. Arthritis Rheum. 2013, 65, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Taher, T.E.; Parikh, K.; Flores-Borja, F.; Mletzko, S.; Isenberg, D.A.; Peppelenbosch, M.P.; Mageed, R.A. Protein phosphorylation and kinome profiling reveal altered regulation of multiple signaling pathways in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 2412–2423. [Google Scholar] [CrossRef]
- Manjarrez-Orduno, N.; Marasco, E.; Chung, S.A.; Katz, M.S.; Kiridly, J.F.; Simpfendorfer, K.R.; Freudenberg, J.; Ballard, D.H.; Nashi, E.; Hopkins, T.J.; et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat. Genet. 2012, 44, 1227–1230. [Google Scholar] [CrossRef]
- Fleischer, S.J.; Giesecke, C.; Mei, H.E.; Lipsky, P.E.; Daridon, C.; Dorner, T. Increased frequency of a unique spleen tyrosine kinase bright memory B cell population in systemic lupus erythematosus. Arthritis Rheumatol. 2014, 66, 3424–3435. [Google Scholar] [CrossRef]
- Iwata, S.; Yamaoka, K.; Niiro, H.; Jabbarzadeh-Tabrizi, S.; Wang, S.P.; Kondo, M.; Yoshikawa, M.; Akashi, K.; Tanaka, Y. Increased Syk phosphorylation leads to overexpression of TRAF6 in peripheral B cells of patients with systemic lupus erythematosus. Lupus 2015, 24, 695–704. [Google Scholar] [CrossRef]
- Iwata, S.; Nakayamada, S.; Fukuyo, S.; Kubo, S.; Yunoue, N.; Wang, S.P.; Yoshikawa, M.; Saito, K.; Tanaka, Y. Activation of Syk in peripheral blood B cells in patients with rheumatoid arthritis: A potential target for abatacept therapy. Arthritis Rheumatol. 2015, 67, 63–73. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Kavanaugh, A.; Genovese, M.C.; Musser, T.K.; Grossbard, E.B.; Magilavy, D.B. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N. Engl. J. Med. 2010, 363, 1303–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Jiang, X.; Qin, D.; Wang, L.; Yang, J.; Wu, A.; Huang, F.; Ye, Y.; Wu, J. Efficacy and Safety of Multiple Dosages of Fostamatinib in Adult Patients With Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Front. Pharm. 2019, 10, 897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, H.S.; Boyle, D.L.; Inoue, T.; Schoot, R.; Tak, P.P.; Pine, P.; Firestein, G.S. A novel spleen tyrosine kinase inhibitor blocks c-Jun N-terminal kinase-mediated gene expression in synoviocytes. J Pharm. Exp. Ther. 2006, 317, 571–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, L.; Holmdahl, R. Genes on the X chromosome affect development of collagen-induced arthritis in mice. Clin. Exp. Immunol. 1993, 94, 459–465. [Google Scholar] [CrossRef]
- Nyhoff, L.E.; Barron, B.L.; Johnson, E.M.; Bonami, R.H.; Maseda, D.; Fensterheim, B.A.; Han, W.; Blackwell, T.S.; Crofford, L.J.; Kendall, P.L. Bruton’s Tyrosine Kinase Deficiency Inhibits Autoimmune Arthritis in Mice but Fails to Block Immune Complex-Mediated Inflammatory Arthritis. Arthritis Rheumatol. 2016, 68, 1856–1868. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.R.; Chused, T.M.; Steinberg, A.D. The effect of the X-linked immune deficiency gene (xid) upon the Y chromosome-related disease of BXSB mice. J. Immunol. 1983, 131, 1257–1262. [Google Scholar]
- Steinberg, B.J.; Smathers, P.A.; Frederiksen, K.; Steinberg, A.D. Ability of the xid gene to prevent autoimmunity in (NZB X NZW)F1 mice during the course of their natural history, after polyclonal stimulation, or following immunization with DNA. J. Clin. Investig. 1982, 70, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Rip, J.; Van Der Ploeg, E.K.; Hendriks, R.W.; Corneth, O.B.J. The Role of Bruton’s Tyrosine Kinase in Immune Cell Signaling and Systemic Autoimmunity. Crit. Rev. Immunol. 2018, 38, 17–62. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; Verstappen, G.M.P.; Paulissen, S.M.J.; de Bruijn, M.J.W.; Rip, J.; Lukkes, M.; van Hamburg, J.P.; Lubberts, E.; Bootsma, H.; Kroese, F.G.M.; et al. Enhanced Bruton’s Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes From Patients With Autoimmune Disease. Arthritis Rheumatol. 2017, 69, 1313–1324. [Google Scholar] [CrossRef] [Green Version]
- von Borstel, A.; Abdulahad, W.H.; Sanders, J.S.; Rip, J.; Neys, S.F.H.; Hendriks, R.W.; Stegeman, C.A.; Heeringa, P.; Rutgers, A.; Corneth, O.B.J. Evidence for enhanced Bruton’s tyrosine kinase activity in transitional and naive B cells of patients with granulomatosis with polyangiitis. Rheumatology 2019, 58, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Neys, S.F.H.; Heukels, P.; van Hulst, J.A.C.; Rip, J.; Wijsenbeek, M.S.; Hendriks, R.W.; Corneth, O.B.J. Aberrant B Cell Receptor Signaling in Naïve B Cells from Patients with Idiopathic Pulmonary Fibrosis. Cells 2021, 10, 1321. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Tuckwell, K.; Katsumoto, T.R.; Zhao, R.; Galanter, J.; Lee, C.; Rae, J.; Toth, B.; Ramamoorthi, N.; Hackney, J.A.; et al. Fenebrutinib versus Placebo or Adalimumab in Rheumatoid Arthritis: A Randomized, Double-Blind, Phase II Trial (ANDES Study). Arthritis Rheumatol. 2020, 72, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef]
- Isenberg, D.; Furie, R.; Jones, N.S.; Guibord, P.; Galanter, J.; Lee, C.; McGregor, A.; Toth, B.; Rae, J.; Hwang, O.; et al. Efficacy, Safety, and Pharmacodynamic Effects of the Bruton’s Tyrosine Kinase Inhibitor Fenebrutinib (GDC-0853) in Systemic Lupus Erythematosus: Results of a Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2021, 73, 1835–1846. [Google Scholar] [CrossRef]
- Middendorp, S.; Dingjan, G.M.; Maas, A.; Dahlenborg, K.; Hendriks, R.W. Function of Bruton’s tyrosine kinase during B cell development is partially independent of its catalytic activity. J. Immunol. 2003, 171, 5988–5996. [Google Scholar] [CrossRef] [Green Version]
- Middendorp, S.; Zijlstra, A.J.; Kersseboom, R.; Dingjan, G.M.; Jumaa, H.; Hendriks, R.W. Tumor suppressor function of Bruton tyrosine kinase is independent of its catalytic activity. Blood 2005, 105, 259–265. [Google Scholar] [CrossRef]
- Belver, L.; de Yébenes, V.G.; Ramiro, A.R. MicroRNAs prevent the generation of autoreactive antibodies. Immunity 2010, 33, 713–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Xie, X. MicroRNA-425 inhibits proliferation of chronic lymphocytic leukaemia cells through regulation of the Bruton’s tyrosine kinase/phospholipase Cγ2 signalling pathway. Exp. Ther. Med. 2020, 20, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Bottoni, A.; Rizzotto, L.; Lai, T.H.; Liu, C.; Smith, L.L.; Mantel, R.; Reiff, S.; El-Gamal, D.; Larkin, K.; Johnson, A.J.; et al. Targeting BTK through microRNA in chronic lymphocytic leukemia. Blood 2016, 128, 3101–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal-Quiros, M.; Wu, Y.Y.; Alarcon-Riquelme, M.E.; Castillejo-Lopez, C. BANK1 and BLK act through phospholipase C gamma 2 in B-cell signaling. PLoS ONE 2013, 8, e59842. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.H.; Athanasopoulos, V.; Ellyard, J.I.; Chuah, A.; Cappello, J.; Cook, A.; Prabhu, S.B.; Cardenas, J.; Gu, J.; Stanley, M.; et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat. Commun. 2019, 10, 2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Nalda, A.; Fortuny, C.; Rey, L.; Bunney, T.D.; Alsina, L.; Esteve-Sole, A.; Bull, D.; Anton, M.C.; Basagana, M.; Casals, F.; et al. Severe Autoinflammatory Manifestations and Antibody Deficiency Due to Novel Hypermorphic PLCG2 Mutations. J. Clin. Immunol. 2020, 40, 987–1000. [Google Scholar] [CrossRef]
- Ombrello, M.J.; Remmers, E.F.; Sun, G.; Freeman, A.F.; Datta, S.; Torabi-Parizi, P.; Subramanian, N.; Bunney, T.D.; Baxendale, R.W.; Martins, M.S.; et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N. Engl. J. Med. 2012, 366, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Constien, R.; Dear, N.; Katan, M.; Hanke, P.; Bunney, T.D.; Kunder, S.; Quintanilla-Martinez, L.; Huffstadt, U.; Schroder, A.; et al. Autoimmunity and inflammation due to a gain-of-function mutation in phospholipase C gamma 2 that specifically increases external Ca2+ entry. Immunity 2005, 22, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Limon, J.J.; Fruman, D.A. Akt and mTOR in B Cell Activation and Differentiation. Front. Immunol. 2012, 3, 228. [Google Scholar] [CrossRef] [Green Version]
- Harder, I.; Münchhalfen, M.; Andrieux, G.; Boerries, M.; Grimbacher, B.; Eibel, H.; Maccari, M.E.; Ehl, S.; Wienands, J.; Jellusova, J.; et al. Dysregulated PI3K Signaling in B Cells of CVID Patients. Cells 2022, 11, 464. [Google Scholar] [CrossRef]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef] [Green Version]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellusova, J.; Cato, M.H.; Apgar, J.R.; Ramezani-Rad, P.; Leung, C.R.; Chen, C.; Richardson, A.D.; Conner, E.M.; Benschop, R.J.; Woodgett, J.R.; et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 2017, 18, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Shinnakasu, R.; Kawai, C.; Ise, W.; Kawakami, E.; Sax, N.; Oki, T.; Kitamura, T.; Yamashita, K.; Fukuyama, H.; et al. Exit from germinal center to become quiescent memory B cells depends on metabolic reprograming and provision of a survival signal. J. Exp. Med. 2021, 218, e20200866. [Google Scholar] [CrossRef] [PubMed]
- Weisel, F.J.; Zuccarino-Catania, G.V.; Chikina, M.; Shlomchik, M.J. A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 2016, 44, 116–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malkiel, S.; Jeganathan, V.; Wolfson, S.; Manjarrez Orduno, N.; Marasco, E.; Aranow, C.; Mackay, M.; Gregersen, P.K.; Diamond, B. Checkpoints for Autoreactive B Cells in the Peripheral Blood of Lupus Patients Assessed by Flow Cytometry. Arthritis Rheumatol. 2016, 68, 2210–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.H.; Raybuck, A.L.; Stengel, K.; Wei, M.; Beck, T.C.; Volanakis, E.; Thomas, J.W.; Hiebert, S.; Haase, V.H.; Boothby, M.R. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 2016, 537, 234–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torigoe, M.; Iwata, S.; Nakayamada, S.; Sakata, K.; Zhang, M.; Hajime, M.; Miyazaki, Y.; Narisawa, M.; Ishii, K.; Shibata, H.; et al. Metabolic Reprogramming Commits Differentiation of Human CD27(+)IgD(+) B Cells to Plasmablasts or CD27(-)IgD(-) Cells. J. Immunol. 2017, 199, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blokland, S.L.M.; Hillen, M.R.; Wichers, C.G.K.; Zimmermann, M.; Kruize, A.A.; Radstake, T.; Broen, J.C.A.; van Roon, J.A.G. Increased mTORC1 activation in salivary gland B cells and T cells from patients with Sjogren’s syndrome: mTOR inhibition as a novel therapeutic strategy to halt immunopathology? RMD Open 2019, 5, e000701. [Google Scholar] [CrossRef] [Green Version]
- Clarke, A.J.; Ellinghaus, U.; Cortini, A.; Stranks, A.; Simon, A.K.; Botto, M.; Vyse, T.J. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann. Rheum. Dis. 2015, 74, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Raza, I.G.A.; Clarke, A.J. B Cell Metabolism and Autophagy in Autoimmunity. Front. Immunol. 2021, 12, 681105. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Adams, B.D.; Lai, M.; Shepherd, J.; Salvador-Bernaldez, M.; Salvador, J.M.; Lu, J.; Nemazee, D.; Xiao, C. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat. Immunol. 2016, 17, 433–440. [Google Scholar] [CrossRef]
- Chauhan, S.K.; Singh, V.V.; Rai, R.; Rai, M.; Rai, G. Differential microRNA profile and post-transcriptional regulation exist in systemic lupus erythematosus patients with distinct autoantibody specificities. J. Clin. Immunol. 2014, 34, 491–503. [Google Scholar] [CrossRef]
- Ma, F.; Zhan, Y.; Bartolomé-Cabrero, R.; Ying, W.; Asano, M.; Huang, Z.; Xiao, C.; González-Martín, A. Analysis of a miR-148a Targetome in B Cell Central Tolerance. Front. Immunol. 2022, 13, 861655. [Google Scholar] [CrossRef]
- Lai, M.; Gonzalez-Martin, A.; Cooper, A.B.; Oda, H.; Jin, H.Y.; Shepherd, J.; He, L.; Zhu, J.; Nemazee, D.; Xiao, C. Regulation of B-cell development and tolerance by different members of the miR-17 approximately 92 family microRNAs. Nat. Commun. 2016, 7, 12207. [Google Scholar] [CrossRef] [Green Version]
- Benhamou, D.; Labi, V.; Getahun, A.; Benchetrit, E.; Dowery, R.; Rajewsky, K.; Cambier, J.C.; Melamed, D. The c-Myc/miR17-92/PTEN Axis Tunes PI3K Activity to Control Expression of Recombination Activating Genes in Early B Cell Development. Front. Immunol. 2018, 9, 2715. [Google Scholar] [CrossRef]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Hines, M.J.; Coffre, M.; Mudianto, T.; Panduro, M.; Wigton, E.J.; Tegla, C.; Osorio-Vasquez, V.; Kageyama, R.; Benhamou, D.; Perez, O.; et al. miR-29 Sustains B Cell Survival and Controls Terminal Differentiation via Regulation of PI3K Signaling. Cell Rep. 2020, 33, 108436. [Google Scholar] [CrossRef]
- Wu, X.N.; Ye, Y.X.; Niu, J.W.; Li, Y.; Li, X.; You, X.; Chen, H.; Zhao, L.D.; Zeng, X.F.; Zhang, F.C.; et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci. Transl. Med. 2014, 6, 246ra299. [Google Scholar] [CrossRef]
- Smith, M.J.; Ford, B.R.; Rihanek, M.; Coleman, B.M.; Getahun, A.; Sarapura, V.D.; Gottlieb, P.A.; Cambier, J.C. Elevated PTEN expression maintains anergy in human B cells and reveals unexpectedly high repertoire autoreactivity. JCI Insight 2019, 4, e123384. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Chen, H.; Qiu, J.; Yang, H.X.; Zhang, C.Y.; Fei, Y.Y.; Zhao, L.D.; Zhou, J.X.; Wang, L.; Wu, Q.J.; et al. Antagonizing miR-7 suppresses B cell hyperresponsiveness and inhibits lupus development. J. Autoimmun. 2020, 109, 102440. [Google Scholar] [CrossRef]
- Xiao, C.; Nemazee, D.; Gonzalez-Martin, A. MicroRNA control of B cell tolerance, autoimmunity and cancer. Semin. Cancer Biol. 2020, 64, 102–107. [Google Scholar] [CrossRef]
- Chi, M.; Ma, K.; Li, Y.; Quan, M.; Han, Z.; Ding, Z.; Liang, X.; Zhang, Q.; Song, L.; Liu, C. Immunological Involvement of MicroRNAs in the Key Events of Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 699684. [Google Scholar] [CrossRef]
- Heinicke, F.; Zhong, X.; Flåm, S.T.; Breidenbach, J.; Leithaug, M.; Mæhlen, M.T.; Lillegraven, S.; Aga, A.B.; Norli, E.S.; Mjaavatten, M.D.; et al. MicroRNA Expression Differences in Blood-Derived CD19+ B Cells of Methotrexate Treated Rheumatoid Arthritis Patients. Front. Immunol. 2021, 12, 663736. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Lee, H.M.; Yu, I.S.; Choi, Y.S.; Huang, H.Y.; Hashemifar, S.S.; Lin, L.L.; Chen, M.C.; Afanasiev, N.D.; Khan, A.A.; et al. Differential cell-intrinsic regulations of germinal center B and T cells by miR-146a and miR-146b. Nat. Commun. 2018, 9, 2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrouche, L.; You, S.; Sauvaget, V.; Manda, V.; Lamarthee, B.; Desbuissons, G.; Tinel, C.; Rabant, M.; Nguyen, C.; Isnard, P.; et al. MicroRNA-146a-deficient mice develop immune complex glomerulonephritis. Sci. Rep. 2019, 9, 15597. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Luo, X.; Cui, H.; Ni, X.; Yuan, M.; Guo, Y.; Huang, X.; Zhou, H.; de Vries, N.; Tak, P.P.; et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009, 60, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Teng, G.; Hakimpour, P.; Landgraf, P.; Rice, A.; Tuschl, T.; Casellas, R.; Papavasiliou, F.N. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 2008, 28, 621–629. [Google Scholar] [CrossRef] [Green Version]
- de Yebenes, V.G.; Belver, L.; Pisano, D.G.; Gonzalez, S.; Villasante, A.; Croce, C.; He, L.; Ramiro, A.R. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J. Exp. Med. 2008, 205, 2199–2206. [Google Scholar] [CrossRef] [Green Version]
- de Yebenes, V.G.; Bartolome-Izquierdo, N.; Nogales-Cadenas, R.; Perez-Duran, P.; Mur, S.M.; Martinez, N.; Di Lisio, L.; Robbiani, D.F.; Pascual-Montano, A.; Canamero, M.; et al. miR-217 is an oncogene that enhances the germinal center reaction. Blood 2014, 124, 229–239. [Google Scholar] [CrossRef]
- Kaga, H.; Komatsuda, A.; Omokawa, A.; Ito, M.; Teshima, K.; Tagawa, H.; Sawada, K.; Wakui, H. Downregulated expression of miR-155, miR-17, and miR-181b, and upregulated expression of activation-induced cytidine deaminase and interferon-alpha in PBMCs from patients with SLE. Mod. Rheumatol. 2015, 25, 865–870. [Google Scholar] [CrossRef]
- Setz, C.S.; Khadour, A.; Renna, V.; Iype, J.; Gentner, E.; He, X.; Datta, M.; Young, M.; Nitschke, L.; Wienands, J.; et al. Pten controls B-cell responsiveness and germinal center reaction by regulating the expression of IgD BCR. EMBO J. 2019, 38, e100249. [Google Scholar] [CrossRef]
- Amendt, T.; Ayoubi, O.E.; Linder, A.T.; Allies, G.; Young, M.; Setz, C.S.; Jumaa, H. Primary Immune Responses and Affinity Maturation Are Controlled by IgD. Front. Immunol. 2021, 12, 709240. [Google Scholar] [CrossRef]
- Sabouri, Z.; Perotti, S.; Spierings, E.; Humburg, P.; Yabas, M.; Bergmann, H.; Horikawa, K.; Roots, C.; Lambe, S.; Young, C.; et al. IgD attenuates the IgM-induced anergy response in transitional and mature B cells. Nat. Commun. 2016, 7, 13381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.G. The therapeutic implications of activated immune responses via the enigmatic immunoglobulin D. Int. Rev. Immunol. 2022, 41, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.G.; Sutherland, A.P.; Newton, R.; Qian, F.; Cachero, T.G.; Scott, M.L.; Thompson, J.S.; Wheway, J.; Chtanova, T.; Groom, J.; et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J. Immunol. 2004, 173, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.S.; Bixler, S.A.; Qian, F.; Vora, K.; Scott, M.L.; Cachero, T.G.; Hession, C.; Schneider, P.; Sizing, I.D.; Mullen, C.; et al. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science 2001, 293, 2108–2111. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Casola, S.; Kutok, J.L.; Rajewsky, K.; Schmidt-Supprian, M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J. Immunol. 2004, 173, 2245–2252. [Google Scholar] [CrossRef] [Green Version]
- Castigli, E.; Wilson, S.A.; Scott, S.; Dedeoglu, F.; Xu, S.; Lam, K.P.; Bram, R.J.; Jabara, H.; Geha, R.S. TACI and BAFF-R mediate isotype switching in B cells. J. Exp. Med. 2005, 201, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Seshasayee, D.; Valdez, P.; Yan, M.; Dixit, V.M.; Tumas, D.; Grewal, I.S. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity 2003, 18, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Shulga-Morskaya, S.; Dobles, M.; Walsh, M.E.; Ng, L.G.; MacKay, F.; Rao, S.P.; Kalled, S.L.; Scott, M.L. B cell-activating factor belonging to the TNF family acts through separate receptors to support B cell survival and T cell-independent antibody formation. J. Immunol. 2004, 173, 2331–2341. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Wang, H.; Chan, B.; Roose-Girma, M.; Erickson, S.; Baker, T.; Tumas, D.; Grewal, I.S.; Dixit, V.M. Activation and accumulation of B cells in TACI-deficient mice. Nat. Immunol. 2001, 2, 638–643. [Google Scholar] [CrossRef]
- Avery, D.T.; Kalled, S.L.; Ellyard, J.I.; Ambrose, C.; Bixler, S.A.; Thien, M.; Brink, R.; Mackay, F.; Hodgkin, P.D.; Tangye, S.G. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Investig. 2003, 112, 286–297. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Pers, J.O.; Daridon, C.; Devauchelle, V.; Jousse, S.; Saraux, A.; Jamin, C.; Youinou, P. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann. N. Y. Acad. Sci. 2005, 1050, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.B.; Kandane-Rathnayake, R.; Koelmeyer, R.; Hoi, A.Y.; Harris, J.; Mackay, F.; Morand, E.F. Analysis of serum B cell-activating factor from the tumor necrosis factor family (BAFF) and its soluble receptors in systemic lupus erythematosus. Clin. Transl. Immunol. 2019, 8, e01047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweighoffer, E.; Vanes, L.; Nys, J.; Cantrell, D.; McCleary, S.; Smithers, N.; Tybulewicz, V.L. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity 2013, 38, 475–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinners, N.P.; Carlesso, G.; Castro, I.; Hoek, K.L.; Corn, R.A.; Woodland, R.T.; Scott, M.L.; Wang, D.; Khan, W.N. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J. Immunol. 2007, 179, 3872–3880. [Google Scholar] [CrossRef] [Green Version]
- Sevdali, E.; Block, V.; Lataretu, M.; Li, H.; Smulski, C.R.; Briem, J.S.; Heitz, Y.; Fischer, B.; Ramirez, N.J.; Grimbacher, B.; et al. BAFFR activates PI3K/AKT signaling in human naive but not in switched memory B cells through direct interactions with B cell antigen receptors. Cell Rep. 2022, 39, 111019. [Google Scholar] [CrossRef]
- Castro, I.; Wright, J.A.; Damdinsuren, B.; Hoek, K.L.; Carlesso, G.; Shinners, N.P.; Gerstein, R.M.; Woodland, R.T.; Sen, R.; Khan, W.N. B cell receptor-mediated sustained c-Rel activation facilitates late transitional B cell survival through control of B cell activating factor receptor and NF-kappaB2. J. Immunol. 2009, 182, 7729–7737. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.H.; Cancro, M.P. Cutting edge: B cell receptor signals regulate BLyS receptor levels in mature B cells and their immediate progenitors. J. Immunol. 2003, 170, 5820–5823. [Google Scholar] [CrossRef] [Green Version]
- Stadanlick, J.E.; Kaileh, M.; Karnell, F.G.; Scholz, J.L.; Miller, J.P.; Quinn, W.J., 3rd; Brezski, R.J.; Treml, L.S.; Jordan, K.A.; Monroe, J.G.; et al. Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat. Immunol. 2008, 9, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Song, S.S.; Shu, J.L.; Li, Y.; Wu, Y.J.; Wang, Q.T.; Chen, J.Y.; Chang, Y.; Wu, H.X.; Zhang, L.L.; et al. BAFF upregulates CD28/B7 and CD40/CD154 expression and promotes mouse T and B cell interaction in vitro via BAFF receptor. Acta Pharm. Sin. 2016, 37, 1101–1109. [Google Scholar] [CrossRef] [Green Version]
- von Bülow, G.U.; Bram, R.J. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science 1997, 278, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Hatzoglou, A.; Roussel, J.; Bourgeade, M.F.; Rogier, E.; Madry, C.; Inoue, J.; Devergne, O.; Tsapis, A. TNF receptor family member BCMA (B cell maturation) associates with TNF receptor-associated factor (TRAF) 1, TRAF2, and TRAF3 and activates NF-kappa B, elk-1, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsenelson, N.; Kanswal, S.; Puig, M.; Mostowski, H.; Verthelyi, D.; Akkoyunlu, M. Synthetic CpG oligodeoxynucleotides augment BAFF- and APRIL-mediated immunoglobulin secretion. Eur. J. Immunol. 2007, 37, 1785–1795. [Google Scholar] [CrossRef]
- Sintes, J.; Gentile, M.; Zhang, S.; Garcia-Carmona, Y.; Magri, G.; Cassis, L.; Segura-Garzón, D.; Ciociola, A.; Grasset, E.K.; Bascones, S.; et al. mTOR intersects antibody-inducing signals from TACI in marginal zone B cells. Nat. Commun. 2017, 8, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, F.S.; Kuhn, P.H.; Laurent, S.A.; Hauck, S.M.; Berer, K.; Wendlinger, S.A.; Krumbholz, M.; Khademi, M.; Olsson, T.; Dreyling, M.; et al. The immunoregulator soluble TACI is released by ADAM10 and reflects B cell activation in autoimmunity. J. Immunol. 2015, 194, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Camarena, D.C.; Ortiz-Lazareno, P.C.; Cruz, A.; Oregon-Romero, E.; Machado-Contreras, J.R.; Muñoz-Valle, J.F.; Orozco-López, M.; Marín-Rosales, M.; Palafox-Sánchez, C.A. Association of BAFF, APRIL serum levels, BAFF-R, TACI and BCMA expression on peripheral B-cell subsets with clinical manifestations in systemic lupus erythematosus. Lupus 2016, 25, 582–592. [Google Scholar] [CrossRef]
- Castigli, E.; Scott, S.; Dedeoglu, F.; Bryce, P.; Jabara, H.; Bhan, A.K.; Mizoguchi, E.; Geha, R.S. Impaired IgA class switching in APRIL-deficient mice. Proc. Natl. Acad. Sci. USA 2004, 101, 3903–3908. [Google Scholar] [CrossRef] [Green Version]
- Chu, V.T.; Enghard, P.; Schurer, S.; Steinhauser, G.; Rudolph, B.; Riemekasten, G.; Berek, C. Systemic activation of the immune system induces aberrant BAFF and APRIL expression in B cells in patients with systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 2083–2093. [Google Scholar] [CrossRef]
- Koyama, T.; Tsukamoto, H.; Masumoto, K.; Himeji, D.; Hayashi, K.; Harada, M.; Horiuchi, T. A novel polymorphism of the human APRIL gene is associated with systemic lupus erythematosus. Rheumatology 2003, 42, 980–985. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Ota, F.; Kim-Howard, X.; Kaufman, K.M.; Nath, S.K. APRIL polymorphism and systemic lupus erythematosus (SLE) susceptibility. Rheumatology 2007, 46, 1274–1276. [Google Scholar] [CrossRef] [Green Version]
- Dall’Era, M.; Chakravarty, E.; Wallace, D.; Genovese, M.; Weisman, M.; Kavanaugh, A.; Kalunian, K.; Dhar, P.; Vincent, E.; Pena-Rossi, C.; et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: Results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007, 56, 4142–4150. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.; Gordon, C.; Licu, D.; Copt, S.; Rossi, C.P.; Wofsy, D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann. Rheum. Dis. 2015, 74, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Kaegi, C.; Steiner, U.C.; Wuest, B.; Crowley, C.; Boyman, O. Systematic Review of Safety and Efficacy of Atacicept in Treating Immune-Mediated Disorders. Front. Immunol. 2020, 11, 433. [Google Scholar] [CrossRef]
- Vazgiourakis, V.M.; Zervou, M.I.; Choulaki, C.; Bertsias, G.; Melissourgaki, M.; Yilmaz, N.; Sidiropoulos, P.; Plant, D.; Trouw, L.A.; Toes, R.E.; et al. A common SNP in the CD40 region is associated with systemic lupus erythematosus and correlates with altered CD40 expression: Implications for the pathogenesis. Ann. Rheum. Dis. 2011, 70, 2184–2190. [Google Scholar] [CrossRef] [Green Version]
- Ramanujam, M.; Steffgen, J.; Visvanathan, S.; Mohan, C.; Fine, J.S.; Putterman, C. Phoenix from the flames: Rediscovering the role of the CD40-CD40L pathway in systemic lupus erythematosus and lupus nephritis. Autoimmun. Rev. 2020, 19, 102668. [Google Scholar] [CrossRef] [PubMed]
- Weißenberg, S.Y.; Szelinski, F.; Schrezenmeier, E.; Stefanski, A.L.; Wiedemann, A.; Rincon-Arevalo, H.; Welle, A.; Jungmann, A.; Nordström, K.; Walter, J.; et al. Identification and Characterization of Post-activated B Cells in Systemic Autoimmune Diseases. Front. Immunol. 2019, 10, 2136. [Google Scholar] [CrossRef] [Green Version]
- Armitage, L.H.; Wallet, M.A.; Mathews, C.E. Influence of PTPN22 Allotypes on Innate and Adaptive Immune Function in Health and Disease. Front. Immunol. 2021, 12, 636618. [Google Scholar] [CrossRef] [PubMed]
- Tsubata, T. Role of inhibitory B cell co-receptors in B cell self-tolerance to non-protein antigens. Immunol. Rev. 2022, 307, 53–65. [Google Scholar] [CrossRef]
- Tsubata, T. Ligand Recognition Determines the Role of Inhibitory B Cell Co-receptors in the Regulation of B Cell Homeostasis and Autoimmunity. Front. Immunol. 2018, 9, 2276. [Google Scholar] [CrossRef]
- Akatsu, C.; Shinagawa, K.; Numoto, N.; Liu, Z.; Ucar, A.K.; Aslam, M.; Phoon, S.; Adachi, T.; Furukawa, K.; Ito, N.; et al. CD72 negatively regulates B lymphocyte responses to the lupus-related endogenous toll-like receptor 7 ligand Sm/RNP. J. Exp. Med. 2016, 213, 2691–2706. [Google Scholar] [CrossRef] [Green Version]
- Li, D.H.; Winslow, M.M.; Cao, T.M.; Chen, A.H.; Davis, C.R.; Mellins, E.D.; Utz, P.J.; Crabtree, G.R.; Parnes, J.R. Modulation of peripheral B cell tolerance by CD72 in a murine model. Arthritis Rheum. 2008, 58, 3192–3204. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Hou, R.; Sato-Hayashizaki, A.; Man, R.; Zhu, C.; Wakabayashi, C.; Hirose, S.; Adachi, T.; Tsubata, T. Cd72(c) is a modifier gene that regulates Fas(lpr)-induced autoimmune disease. J. Immunol. 2013, 190, 5436–5445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, S.; Morimoto, S.; Suzuki, J.; Mitsuo, A.; Nakiri, Y.; Katagiri, A.; Nozawa, K.; Amano, H.; Tokano, Y.; Hashimoto, H.; et al. Down-regulation of CD72 and increased surface IgG on B cells in patients with lupus nephritis. Autoimmunity 2007, 40, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Asmiyou, A.; Bakr, A.M.; Shahin, D.A.; Wahba, Y. CD40 and CD72 expression and prognostic values among children with systemic lupus erythematosus: A case-control study. Lupus 2020, 29, 1270–1276. [Google Scholar] [CrossRef]
- Hitomi, Y.; Tsuchiya, N.; Kawasaki, A.; Ohashi, J.; Suzuki, T.; Kyogoku, C.; Fukazawa, T.; Bejrachandra, S.; Siriboonrit, U.; Chandanayingyong, D.; et al. CD72 polymorphisms associated with alternative splicing modify susceptibility to human systemic lupus erythematosus through epistatic interaction with FCGR2B. Hum. Mol. Genet. 2004, 13, 2907–2917. [Google Scholar] [CrossRef] [Green Version]
- Nitschke, L.; Carsetti, R.; Ocker, B.; Kohler, G.; Lamers, M.C. CD22 is a negative regulator of B-cell receptor signalling. Curr. Biol. 1997, 7, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Otipoby, K.L.; Andersson, K.B.; Draves, K.E.; Klaus, S.J.; Farr, A.G.; Kerner, J.D.; Perlmutter, R.M.; Law, C.L.; Clark, E.A. CD22 regulates thymus-independent responses and the lifespan of B cells. Nature 1996, 384, 634–637. [Google Scholar] [CrossRef]
- Engel, P.; Wagner, N.; Miller, A.S.; Tedder, T.F. Identification of the ligand-binding domains of CD22, a member of the immunoglobulin superfamily that uniquely binds a sialic acid-dependent ligand. J. Exp. Med. 1995, 181, 1581–1586. [Google Scholar] [CrossRef] [Green Version]
- Powell, L.D.; Sgroi, D.; Sjoberg, E.R.; Stamenkovic, I.; Varki, A. Natural ligands of the B cell adhesion molecule CD22 beta carry N-linked oligosaccharides with alpha-2,6-linked sialic acids that are required for recognition. J. Biol. Chem. 1993, 268, 7019–7027. [Google Scholar] [CrossRef]
- Chen, J.; Wang, H.; Xu, W.P.; Wei, S.S.; Li, H.J.; Mei, Y.Q.; Li, Y.G.; Wang, Y.P. Besides an ITIM/SHP-1-dependent pathway, CD22 collaborates with Grb2 and plasma membrane calcium-ATPase in an ITIM/SHP-1-independent pathway of attenuation of Ca2+i signal in B cells. Oncotarget 2016, 7, 56129–56146. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S.; Noviski, M.; Mueller, J.L.; Chuwonpad, A.; Raschke, W.C.; Weiss, A.; Zikherman, J. An extracatalytic function of CD45 in B cells is mediated by CD22. Proc. Natl. Acad. Sci. USA 2015, 112, E6515–E6524. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, T.L.; Williams, G.T.; Davies, S.L.; Neuberger, M.S. Hyperresponsive B cells in CD22-deficient mice. Science 1996, 274, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Jellusova, J.; Wellmann, U.; Amann, K.; Winkler, T.H.; Nitschke, L. CD22 x Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J. Immunol. 2010, 184, 3618–3627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatta, Y.; Tsuchiya, N.; Matsushita, M.; Shiota, M.; Hagiwara, K.; Tokunaga, K. Identification of the gene variations in human CD22. Immunogenetics 1999, 49, 280–286. [Google Scholar] [CrossRef]
- Ding, C.; Liu, Y.; Wang, Y.; Park, B.K.; Wang, C.Y.; Zheng, P.; Liu, Y. Siglecg limits the size of B1a B cell lineage by down-regulating NFkappaB activation. PLoS ONE 2007, 2, e997. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Kerr, S.; Jellusova, J.; Zhang, J.; Weisel, F.; Wellmann, U.; Winkler, T.H.; Kneitz, B.; Crocker, P.R.; Nitschke, L. Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat. Immunol. 2007, 8, 695–704. [Google Scholar] [CrossRef]
- Duong, B.H.; Tian, H.; Ota, T.; Completo, G.; Han, S.; Vela, J.L.; Ota, M.; Kubitz, M.; Bovin, N.; Paulson, J.C.; et al. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J. Exp. Med. 2010, 207, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Bökers, S.; Urbat, A.; Daniel, C.; Amann, K.; Smith, K.G.; Espéli, M.; Nitschke, L. Siglec-G deficiency leads to more severe collagen-induced arthritis and earlier onset of lupus-like symptoms in MRL/lpr mice. J. Immunol. 2014, 192, 2994–3002. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Lunz, B.; Schwab, I.; Acs, A.; Nimmerjahn, F.; Daniel, C.; Nitschke, L. Siglec-G Deficiency Leads to Autoimmunity in Aging C57BL/6 Mice. J. Immunol. 2015, 195, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Alborzian Deh Sheikh, A.; Gomaa, S.; Li, X.; Routledge, M.; Saigoh, K.; Numoto, N.; Angata, T.; Hitomi, Y.; Takematsu, H.; Tsuiji, M.; et al. A Guillain-Barre syndrome-associated SIGLEC10 rare variant impairs its recognition of gangliosides. J. Autoimmun. 2021, 116, 102571. [Google Scholar] [CrossRef]
- Bewarder, N.; Weinrich, V.; Budde, P.; Hartmann, D.; Flaswinkel, H.; Reth, M.; Frey, J. In vivo and in vitro specificity of protein tyrosine kinases for immunoglobulin G receptor (FcgammaRII) phosphorylation. Mol. Cell. Biol 1996, 16, 4735–4743. [Google Scholar] [CrossRef] [PubMed]
- Nishizumi, H.; Horikawa, K.; Mlinaric-Rascan, I.; Yamamoto, T. A double-edged kinase Lyn: A positive and negative regulator for antigen receptor-mediated signals. J. Exp. Med. 1998, 187, 1343–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolland, S.; Ravetch, J.V. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 2000, 13, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Smith, P.; Ravetch, J.V. Inhibitory Fcgamma receptor is required for the maintenance of tolerance through distinct mechanisms. J. Immunol. 2014, 192, 3021–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, T.; Ono, M.; Hikida, M.; Ohmori, H.; Ravetch, J.V. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 1996, 379, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Mackay, M.; Stanevsky, A.; Wang, T.; Aranow, C.; Li, M.; Koenig, S.; Ravetch, J.V.; Diamond, B. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J. Exp. Med. 2006, 203, 2157–2164. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.G.; Clatworthy, M.R. FcgammaRIIB in autoimmunity and infection: Evolutionary and therapeutic implications. Nat. Rev. Immunol. 2010, 10, 328–343. [Google Scholar] [CrossRef] [Green Version]
- Clatworthy, M.R.; Willcocks, L.; Urban, B.; Langhorne, J.; Williams, T.N.; Peshu, N.; Watkins, N.A.; Floto, R.A.; Smith, K.G. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcgammaRIIb reduce susceptibility to malaria. Proc. Natl. Acad. Sci. USA 2007, 104, 7169–7174. [Google Scholar] [CrossRef] [Green Version]
- Crute, B.W.; Sheraden, R.; Ott, V.L.; Harley, I.T.W.; Getahun, A.; Cambier, J.C. Inhibitory Receptor Trap: A Platform for Discovery of Inhibitory Receptors That Utilize Inositol Lipid and Phosphotyrosine Phosphatase Effectors. Front. Immunol. 2020, 11, 592329. [Google Scholar] [CrossRef]
- Losing, M.; Goldbeck, I.; Manno, B.; Oellerich, T.; Schnyder, T.; Bohnenberger, H.; Stork, B.; Urlaub, H.; Batista, F.D.; Wienands, J.; et al. The Dok-3/Grb2 protein signal module attenuates Lyn kinase-dependent activation of Syk kinase in B cell antigen receptor microclusters. J. Biol. Chem. 2013, 288, 2303–2313. [Google Scholar] [CrossRef] [Green Version]
- Manno, B.; Oellerich, T.; Schnyder, T.; Corso, J.; Losing, M.; Neumann, K.; Urlaub, H.; Batista, F.D.; Engelke, M.; Wienands, J. The Dok-3/Grb2 adaptor module promotes inducible association of the lipid phosphatase SHIP with the BCR in a coreceptor-independent manner. Eur. J. Immunol. 2016, 46, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, O.; Weingarten, L.; Padberg, I.; Pracht, C.; Sinha, R.; Hochdorfer, T.; Kuppig, S.; Backofen, R.; Reth, M.; Huber, M. The SH2-domain of SHIP1 interacts with the SHIP1 C-terminus: Impact on SHIP1/Ig-alpha interaction. Biochim. Biophys. Acta 2012, 1823, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reth, M.; Gold, M.R. What goes up must come down: A tripartite Dok-3/Grb2/SHIP1 inhibitory module limits BCR signaling. Eur. J. Immunol. 2016, 46, 2507–2511. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.; Saijo, K.; Torres, R.M.; Rajewsky, K. Ig-alpha cytoplasmic truncation renders immature B cells more sensitive to antigen contact. Immunity 1999, 11, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.M.; Hafen, K. A negative regulatory role for Ig-alpha during B cell development. Immunity 1999, 11, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Getahun, A.; Beavers, N.A.; Larson, S.R.; Shlomchik, M.J.; Cambier, J.C. Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic B cells. J. Exp. Med. 2016, 213, 751–769. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, S.K.; Getahun, A.; Gauld, S.B.; Merrell, K.T.; Tamir, I.; Smith, M.J.; Dal Porto, J.M.; Li, Q.Z.; Cambier, J.C. Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP-1 phosphatase-mediated inhibitory signaling cascade required for B cell anergy. Immunity 2011, 35, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichlin, A.; Gazumyan, A.; Nagaoka, H.; Kirsch, K.H.; Kraus, M.; Rajewsky, K.; Nussenzweig, M.C. A B cell receptor with two Igalpha cytoplasmic domains supports development of mature but anergic B cells. J. Exp. Med. 2004, 199, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Napirei, M.; Karsunky, H.; Zevnik, B.; Stephan, H.; Mannherz, H.G.; Möröy, T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat. Genet. 2000, 25, 177–181. [Google Scholar] [CrossRef]
- Yasutomo, K.; Horiuchi, T.; Kagami, S.; Tsukamoto, H.; Hashimura, C.; Urushihara, M.; Kuroda, Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001, 28, 313–314. [Google Scholar] [CrossRef]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.R.; Kozyrev, S.V.; Baechler, E.C.; Reddy, M.V.; Plenge, R.M.; Bauer, J.W.; Ortmann, W.A.; Koeuth, T.; González Escribano, M.F.; The Argentine and Spanish Collaborative Groups; et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat. Genet. 2006, 38, 550–555. [Google Scholar] [CrossRef] [PubMed]
- García-Ortiz, H.; Velázquez-Cruz, R.; Espinosa-Rosales, F.; Jiménez-Morales, S.; Baca, V.; Orozco, L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann. Rheum. Dis. 2010, 69, 1861–1865. [Google Scholar] [CrossRef]
- dos Santos, B.P.; Valverde, J.V.; Rohr, P.; Monticielo, O.A.; Brenol, J.C.; Xavier, R.M.; Chies, J.A. TLR7/8/9 polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus 2012, 21, 302–309. [Google Scholar] [CrossRef]
- Wang, C.M.; Chang, S.W.; Wu, Y.J.; Lin, J.C.; Ho, H.H.; Chou, T.C.; Yang, B.; Wu, J.; Chen, J.Y. Genetic variations in Toll-like receptors (TLRs 3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population. Sci. Rep. 2014, 4, 3792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, C.; Putterman, C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat. Rev. Nephrol. 2015, 11, 329–341. [Google Scholar] [CrossRef]
- Davis, M.L.R.; LeVan, T.D.; Yu, F.; Sayles, H.; Sokolove, J.; Robinson, W.; Michaud, K.; Thiele, G.M.; Mikuls, T.R. Associations of toll-like receptor (TLR)-4 single nucleotide polymorphisms and rheumatoid arthritis disease progression: An observational cohort study. Int. Immunopharmacol. 2015, 24, 346–352. [Google Scholar] [CrossRef]
- Lee, Y.H.; Choi, S.J.; Ji, J.D.; Song, G.G. Overall and cause-specific mortality in systemic lupus erythematosus: An updated meta-analysis. Lupus 2016, 25, 727–734. [Google Scholar] [CrossRef]
- Deng, G.M.; Nilsson, I.M.; Verdrengh, M.; Collins, L.V.; Tarkowski, A. Intra-articularly localized bacterial DNA containing CpG motifs induces arthritis. Nat. Med. 1999, 5, 702–705. [Google Scholar] [CrossRef]
- Kerfoot, S.M.; Long, E.M.; Hickey, M.J.; Andonegui, G.; Lapointe, B.M.; Zanardo, R.C.; Bonder, C.; James, W.G.; Robbins, S.M.; Kubes, P. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J. Immunol. 2004, 173, 7070–7077. [Google Scholar] [CrossRef] [Green Version]
- Pisitkun, P.; Deane, J.A.; Difilippantonio, M.J.; Tarasenko, T.; Satterthwaite, A.B.; Bolland, S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 2006, 312, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Tus, K.; Li, Q.Z.; Wang, A.; Tian, X.H.; Zhou, J.; Liang, C.; Bartov, G.; McDaniel, L.D.; Zhou, X.J.; et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. USA 2006, 103, 9970–9975. [Google Scholar] [CrossRef] [Green Version]
- Berland, R.; Fernandez, L.; Kari, E.; Han, J.H.; Lomakin, I.; Akira, S.; Wortis, H.H.; Kearney, J.F.; Ucci, A.A.; Imanishi-Kari, T. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 2006, 25, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairhurst, A.M.; Hwang, S.H.; Wang, A.; Tian, X.H.; Boudreaux, C.; Zhou, X.J.; Casco, J.; Li, Q.Z.; Connolly, J.E.; Wakeland, E.K. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur. J. Immunol. 2008, 38, 1971–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzabin, S.; Kong, P.; Medghalchi, M.; Palfreeman, A.; Williams, R.; Sacre, S. Investigation of the role of endosomal Toll-like receptors in murine collagen-induced arthritis reveals a potential role for TLR7 in disease maintenance. Arthritis Res. Ther. 2012, 14, R142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, E.R.; Pisitkun, P.; Voynova, E.; Deane, J.A.; Scott, B.L.; Caspi, R.R.; Bolland, S. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell-mediated autoimmunity. Proc. Natl. Acad. Sci. USA 2012, 109, 16276–16281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.L.; Manzin-Lorenzi, C.; Santiago-Raber, M.L. Toll-like receptor 8 deletion accelerates autoimmunity in a mouse model of lupus through a Toll-like receptor 7-dependent mechanism. Immunology 2015, 145, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.J.; Cañete, P.F.; Wang, H.; Medhavy, A.; Bones, J.; Roco, J.A.; He, Y.; Qin, Y.; Cappello, J.; Ellyard, J.I.; et al. TLR7 gain-of-function genetic variation causes human lupus. Nature 2022, 605, 349–356. [Google Scholar] [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739.e726. [Google Scholar] [CrossRef] [Green Version]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Margery-Muir, A.A.; Bundell, C.; Nelson, D.; Groth, D.M.; Wetherall, J.D. Gender balance in patients with systemic lupus erythematosus. Autoimmun. Rev. 2017, 16, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Kashgarian, M.; Alexopoulou, L.; Flavell, R.A.; Akira, S.; Shlomchik, M.J. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J. Exp. Med. 2005, 202, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desnues, B.; Macedo, A.B.; Roussel-Queval, A.; Bonnardel, J.; Henri, S.; Demaria, O.; Alexopoulou, L. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc. Natl. Acad. Sci. USA 2014, 111, 1497–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.W.; Scharping, N.E.; Kolhatkar, N.S.; Khim, S.; Schwartz, M.A.; Li, Q.Z.; Hudkins, K.L.; Alpers, C.E.; Liggitt, D.; Rawlings, D.J. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J. Immunol. 2014, 192, 4525–4532. [Google Scholar] [PubMed] [Green Version]
- Umiker, B.R.; Andersson, S.; Fernandez, L.; Korgaokar, P.; Larbi, A.; Pilichowska, M.; Weinkauf, C.C.; Wortis, H.H.; Kearney, J.F.; Imanishi-Kari, T. Dosage of X-linked Toll-like receptor 8 determines gender differences in the development of systemic lupus erythematosus. Eur. J. Immunol. 2014, 44, 1503–1516. [Google Scholar] [CrossRef] [Green Version]
- Nündel, K.; Green, N.M.; Shaffer, A.L.; Moody, K.L.; Busto, P.; Eilat, D.; Miyake, K.; Oropallo, M.A.; Cancro, M.P.; Marshak-Rothstein, A. Cell-intrinsic expression of TLR9 in autoreactive B cells constrains BCR/TLR7-dependent responses. J. Immunol. 2015, 194, 2504–2512. [Google Scholar] [CrossRef] [Green Version]
- Tilstra, J.S.; John, S.; Gordon, R.A.; Leibler, C.; Kashgarian, M.; Bastacky, S.; Nickerson, K.M.; Shlomchik, M.J. B cell-intrinsic TLR9 expression is protective in murine lupus. J. Clin. Investig. 2020, 130, 3172–3187. [Google Scholar] [CrossRef] [Green Version]
- Sieber, J.; Daridon, C.; Fleischer, S.J.; Fleischer, V.; Hiepe, F.; Alexander, T.; Heine, G.; Burmester, G.R.; Fillatreau, S.; Dörner, T. Active systemic lupus erythematosus is associated with a reduced cytokine production by B cells in response to TLR9 stimulation. Arthritis Res. Ther. 2014, 16, 477. [Google Scholar] [CrossRef] [Green Version]
- Gies, V.; Schickel, J.N.; Jung, S.; Joublin, A.; Glauzy, S.; Knapp, A.M.; Soley, A.; Poindron, V.; Guffroy, A.; Choi, J.Y.; et al. Impaired TLR9 responses in B cells from patients with systemic lupus erythematosus. JCI Insight 2018, 3, e96795. [Google Scholar]
- Kim, Y.M.; Brinkmann, M.M.; Paquet, M.E.; Ploegh, H.L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 2008, 452, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Fukui, R.; Saitoh, S.; Kanno, A.; Onji, M.; Shibata, T.; Ito, A.; Onji, M.; Matsumoto, M.; Akira, S.; Yoshida, N.; et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity 2011, 35, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Majer, O.; Liu, B.; Woo, B.J.; Kreuk, L.S.M.; Van Dis, E.; Barton, G.M. Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature 2019, 575, 371–374. [Google Scholar] [CrossRef]
- De Nardo, D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef]
- Bernasconi, N.L.; Onai, N.; Lanzavecchia, A. A role for Toll-like receptors in acquired immunity: Up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood 2003, 101, 4500–4504. [Google Scholar] [CrossRef] [PubMed]
- Busconi, L.; Bauer, J.W.; Tumang, J.R.; Laws, A.; Perkins-Mesires, K.; Tabor, A.S.; Lau, C.; Corley, R.B.; Rothstein, T.L.; Lund, F.E.; et al. Functional outcome of B cell activation by chromatin immune complex engagement of the B cell receptor and TLR9. J. Immunol. 2007, 179, 7397–7405. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, A.; Dorward, D.; Pierce, S.K. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 2008, 28, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otipoby, K.L.; Waisman, A.; Derudder, E.; Srinivasan, L.; Franklin, A.; Rajewsky, K. The B-cell antigen receptor integrates adaptive and innate immune signals. Proc. Natl. Acad. Sci. USA 2015, 112, 12145–12150. [Google Scholar] [CrossRef] [Green Version]
- Schweighoffer, E.; Nys, J.; Vanes, L.; Smithers, N.; Tybulewicz, V.L.J. TLR4 signals in B lymphocytes are transduced via the B cell antigen receptor and SYK. J. Exp. Med. 2017, 214, 1269–1280. [Google Scholar] [CrossRef] [Green Version]
- Iwata, S.; Yamaoka, K.; Niiro, H.; Nakano, K.; Wang, S.P.; Akashi, K.; Tanaka, Y. Amplification of Toll-like receptor-mediated signaling through spleen tyrosine kinase in human B-cell activation. J. Allergy Clin. Immunol. 2012, 129, 1594–1601.e1592. [Google Scholar] [CrossRef]
- Kremlitzka, M.; Mácsik-Valent, B.; Erdei, A. Syk is indispensable for CpG-induced activation and differentiation of human B cells. Cell. Mol. Life Sci. 2015, 72, 2223–2236. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, C.A.; Doyle, S.; Brunner, C.; Dunne, A.; Brint, E.; Wietek, C.; Walch, E.; Wirth, T.; O’Neill, L.A. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J. Biol. Chem. 2003, 278, 26258–26264. [Google Scholar] [CrossRef]
- Gray, P.; Dunne, A.; Brikos, C.; Jefferies, C.A.; Doyle, S.L.; O’Neill, L.A. MyD88 adapter-like (Mal) is phosphorylated by Bruton’s tyrosine kinase during TLR2 and TLR4 signal transduction. J. Biol. Chem. 2006, 281, 10489–10495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.G.; Xu, S.; Wong, E.T.; Tergaonkar, V.; Lam, K.P. Bruton’s tyrosine kinase separately regulates NFkappaB p65RelA activation and cytokine interleukin (IL)-10/IL-12 production in TLR9-stimulated B Cells. J. Biol. Chem. 2008, 283, 11189–11198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, E.F.; Quinn, S.R.; Doyle, S.L.; Vink, P.M.; van Eenennaam, H.; O’Neill, L.A. Bruton’s tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PLoS ONE 2013, 8, e74103. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.Y.; Kumar, R.; Haque, M.S.; Castillejo-López, C.; Alarcón-Riquelme, M.E. BANK1 controls CpG-induced IL-6 secretion via a p38 and MNK1/2/eIF4E translation initiation pathway. J. Immunol. 2013, 191, 6110–6116. [Google Scholar]
- Wu, Y.Y.; Kumar, R.; Iida, R.; Bagavant, H.; Alarcón-Riquelme, M.E. BANK1 Regulates IgG Production in a Lupus Model by Controlling TLR7-Dependent STAT1 Activation. PLoS ONE 2016, 11, e0156302. [Google Scholar] [CrossRef] [Green Version]
- Ni, M.; MacFarlane, A.W.t.; Toft, M.; Lowell, C.A.; Campbell, K.S.; Hamerman, J.A. B-cell adaptor for PI3K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3K. Proc. Natl. Acad. Sci. USA 2012, 109, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Troutman, T.D.; Hu, W.; Fulenchek, S.; Yamazaki, T.; Kurosaki, T.; Bazan, J.F.; Pasare, C. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc. Natl. Acad. Sci. USA 2012, 109, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Szili, D.; Bankó, Z.; Tóth, E.A.; Nagy, G.; Rojkovich, B.; Gáti, T.; Simon, M.; Hérincs, Z.; Sármay, G. TGFβ activated kinase 1 (TAK1) at the crossroad of B cell receptor and Toll-like receptor 9 signaling pathways in human B cells. PLoS ONE 2014, 9, e96381. [Google Scholar] [CrossRef] [Green Version]
- Jabara, H.H.; McDonald, D.R.; Janssen, E.; Massaad, M.J.; Ramesh, N.; Borzutzky, A.; Rauter, I.; Benson, H.; Schneider, L.; Baxi, S.; et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nat. Immunol. 2012, 13, 612–620. [Google Scholar] [CrossRef]
- Rönnblom, L.; Alm, G.V. A pivotal role for the natural interferon alpha-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J. Exp. Med. 2001, 194, F59–F63. [Google Scholar] [CrossRef]
- Bekeredjian-Ding, I.B.; Wagner, M.; Hornung, V.; Giese, T.; Schnurr, M.; Endres, S.; Hartmann, G. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J. Immunol. 2005, 174, 4043–4050. [Google Scholar] [CrossRef]
- Lau, C.M.; Broughton, C.; Tabor, A.S.; Akira, S.; Flavell, R.A.; Mamula, M.J.; Christensen, S.R.; Shlomchik, M.J.; Viglianti, G.A.; Rifkin, I.R.; et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med. 2005, 202, 1171–1177. [Google Scholar] [CrossRef]
- Poovassery, J.S.; Vanden Bush, T.J.; Bishop, G.A. Antigen receptor signals rescue B cells from TLR tolerance. J. Immunol. 2009, 183, 2974–2983. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Medetgul-Ernar, K.; Davis, M.M. Standing on the shoulders of mice. Immunity 2022, 55, 1343–1353. [Google Scholar] [CrossRef]
- Rip, J.; de Bruijn, M.J.W.; Kaptein, A.; Hendriks, R.W.; Corneth, O.B.J. Phosphoflow Protocol for Signaling Studies in Human and Murine B Cell Subpopulations. J. Immunol. 2020, 204, 2852–2863. [Google Scholar] [CrossRef]
- Marsman, C.; Jorritsma, T.; ten Brinke, A.; van Ham, S.M. Flow Cytometric Methods for the Detection of Intracellular Signaling Proteins and Transcription Factors Reveal Heterogeneity in Differentiating Human B Cell Subsets. Cells 2020, 9, 2633. [Google Scholar] [CrossRef]
- Csomos, K.; Ujhazi, B.; Blazso, P.; Herrera, J.L.; Tipton, C.M.; Kawai, T.; Gordon, S.; Ellison, M.; Wu, K.; Stowell, M.; et al. Partial RAG deficiency in humans induces dysregulated peripheral lymphocyte development and humoral tolerance defect with accumulation of T-bet(+) B cells. Nat. Immunol. 2022, 23, 1256–1272. [Google Scholar] [CrossRef]
- Hoshino, A.; Boutboul, D.; Zhang, Y.; Kuehn, H.S.; Hadjadj, J.; Özdemir, N.; Celkan, T.; Walz, C.; Picard, C.; Lenoir, C.; et al. Gain-of-function IKZF1 variants in humans cause immune dysregulation associated with abnormal T/B cell late differentiation. Sci. Immunol. 2022, 7, eabi7160. [Google Scholar] [CrossRef]
- Yazar, S.; Alquicira-Hernandez, J.; Wing, K.; Senabouth, A.; Gordon, M.G.; Andersen, S.; Lu, Q.; Rowson, A.; Taylor, T.R.P.; Clarke, L.; et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 2022, 376, eabf3041. [Google Scholar] [CrossRef]
- Chiou, J.; Geusz, R.J.; Okino, M.L.; Han, J.Y.; Miller, M.; Melton, R.; Beebe, E.; Benaglio, P.; Huang, S.; Korgaonkar, K.; et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 2021, 594, 398–402. [Google Scholar] [CrossRef]
- Farh, K.K.-H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Catalina, M.D.; Owen, K.A.; Labonte, A.C.; Grammer, A.C.; Lipsky, P.E. The pathogenesis of systemic lupus erythematosus: Harnessing big data to understand the molecular basis of lupus. J. Autoimmun. 2020, 110, 102359. [Google Scholar] [CrossRef]
- Soskic, B.; Cano-Gamez, E.; Smyth, D.J.; Ambridge, K.; Ke, Z.; Matte, J.C.; Bossini-Castillo, L.; Kaplanis, J.; Ramirez-Navarro, L.; Lorenc, A.; et al. Immune disease risk variants regulate gene expression dynamics during CD4+ T cell activation. Nat. Genet. 2022, 54, 817–826. [Google Scholar] [CrossRef]
- Foulquier, N.; Le Dantec, C.; Bettacchioli, E.; Jamin, C.; Consortium, P.C.; Consortium, P.F.C.; Alarcón-Riquelme, M.E.; Pers, J.-O. Machine learning identifies a common signature for anti-SSA/Ro60 antibody expression across autoimmune diseases. Arthritis Rheumatol. 2022, 74, 1706–1719. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corneth, O.B.J.; Neys, S.F.H.; Hendriks, R.W. Aberrant B Cell Signaling in Autoimmune Diseases. Cells 2022, 11, 3391. https://doi.org/10.3390/cells11213391
Corneth OBJ, Neys SFH, Hendriks RW. Aberrant B Cell Signaling in Autoimmune Diseases. Cells. 2022; 11(21):3391. https://doi.org/10.3390/cells11213391
Chicago/Turabian StyleCorneth, Odilia B. J., Stefan F. H. Neys, and Rudi W. Hendriks. 2022. "Aberrant B Cell Signaling in Autoimmune Diseases" Cells 11, no. 21: 3391. https://doi.org/10.3390/cells11213391