
KLF9 Aggravates Streptozotocin-Induced Diabetic Cardiomyopathy by Inhibiting PPARγ/NRF2 Signalling

Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Animal Model
2.3. AAV9 Construction and Delivery
2.4. Echocardiography and Pressure-Volume Loop Evaluation
2.5. ELISA Detection of Inflammatory Cytokines
2.6. Immunohistochemical Staining
2.7. Oxidative Stress Assay
2.8. Cardiomyocyte Isolation and Culture
2.9. Western Blotting and qPCR
2.10. Luciferase Assay
2.11. Statistical Analysis
3. Results
3.1. KLF9 Is Upregulated in DCM
3.2. KLF9 Overexpression Suppresses Cardiac Dysfunction in DCM
3.3. KLF9 Exacerbates Inflammation and Oxidative Stress in DCM
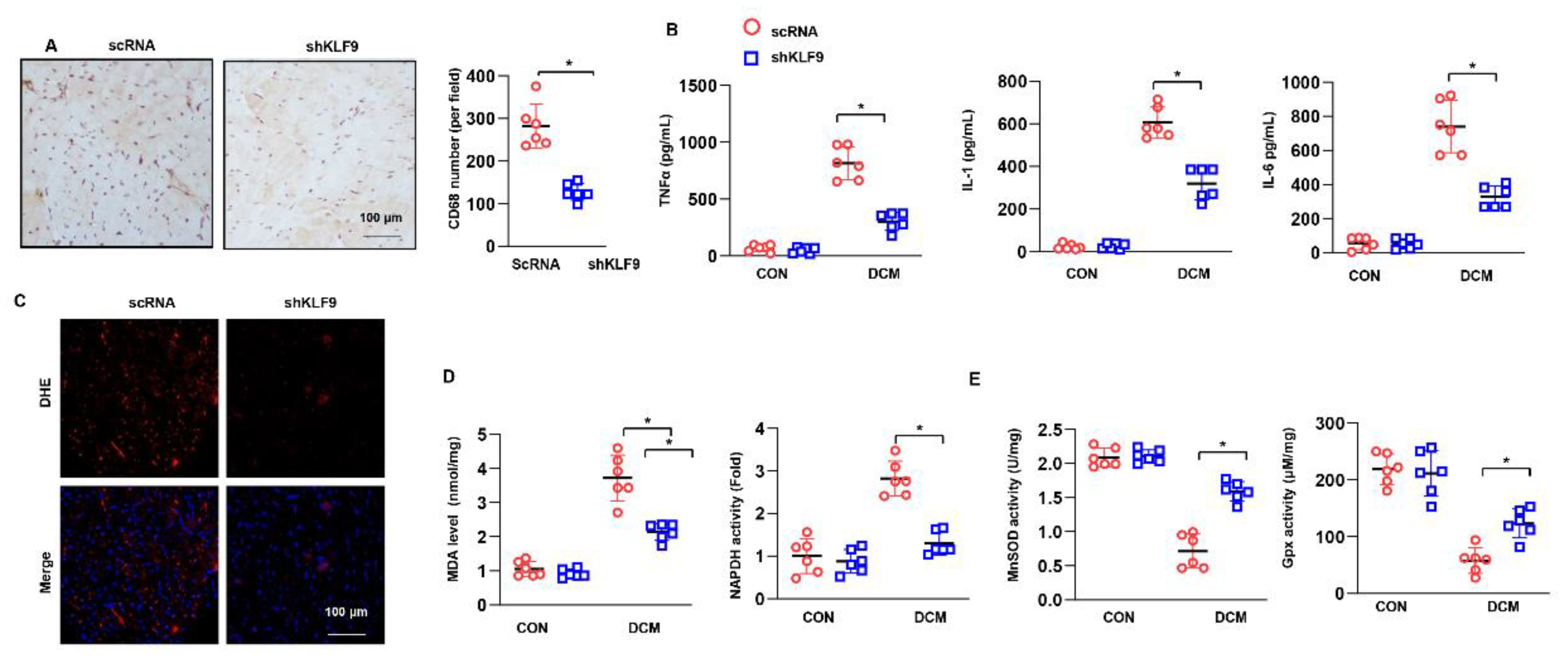
3.4. KLF9 Knockdown Ameliorates Cardiac Dysfunction in DCM
3.5. KLF9 Knockdown Suppressed Inflammation and Oxidative Stress in DCM
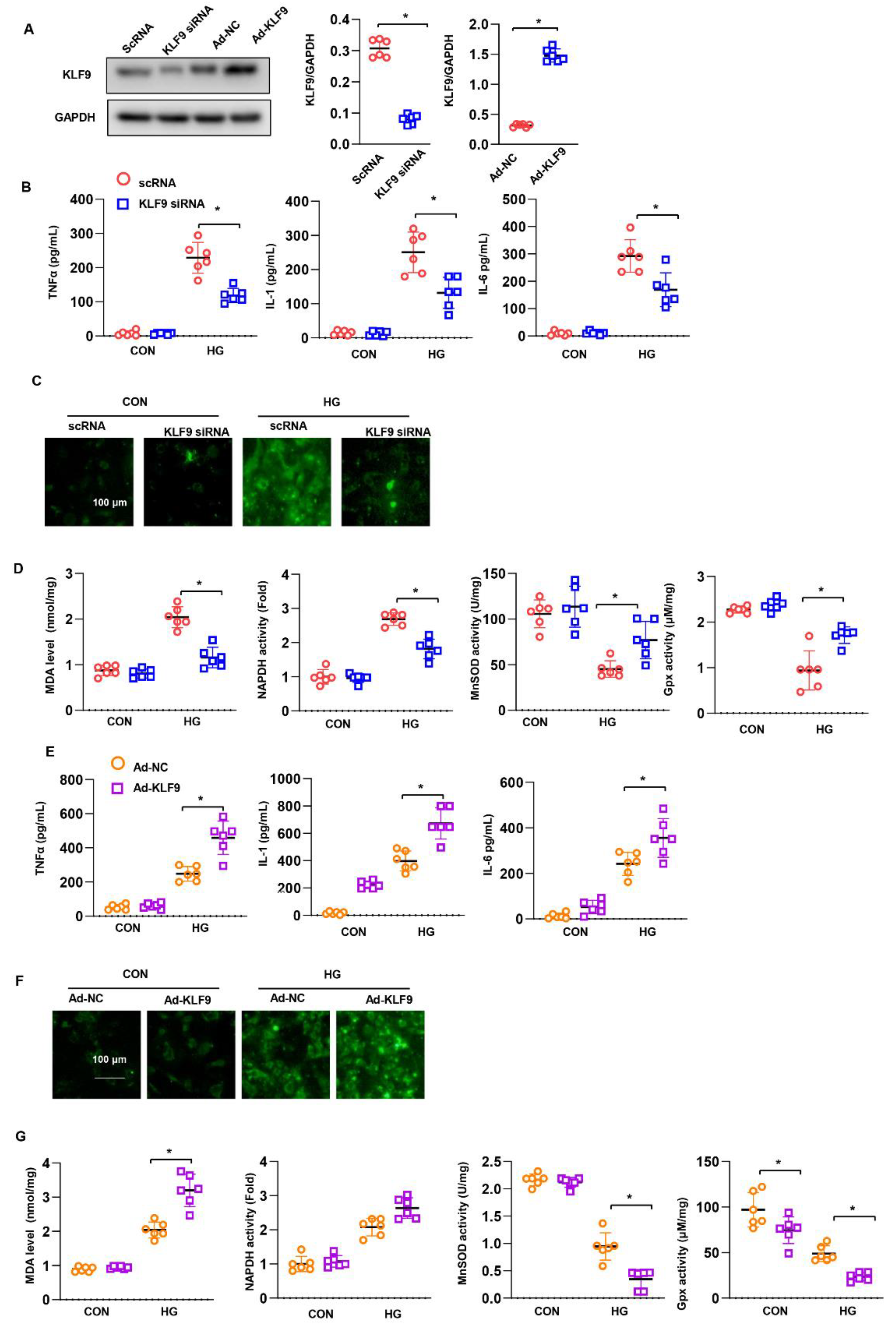
3.6. KLF9 Affects Cardiomyocytes In Vitro
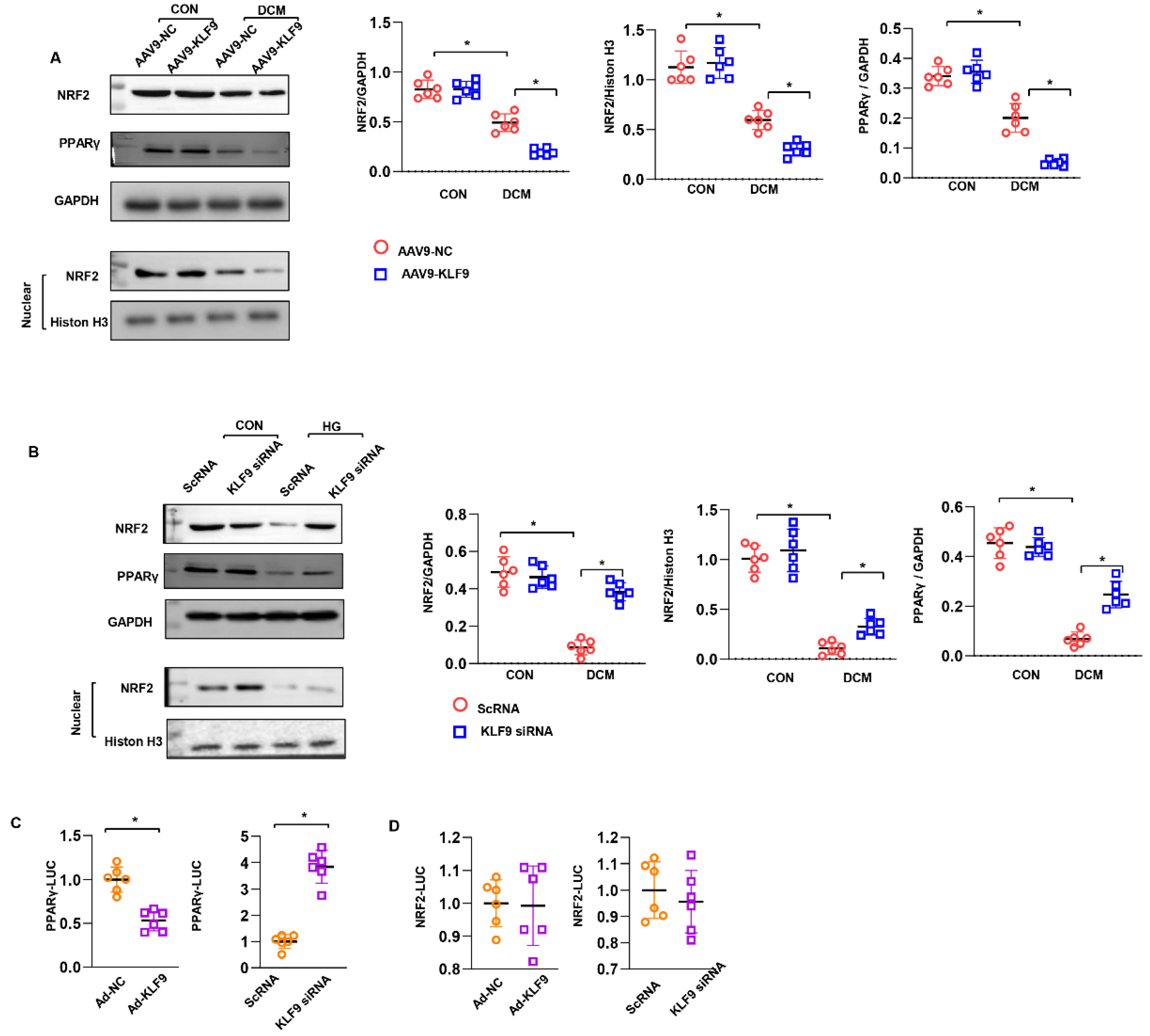
3.7. KLF9 Regulates PPARγ/NRF2 Signalling
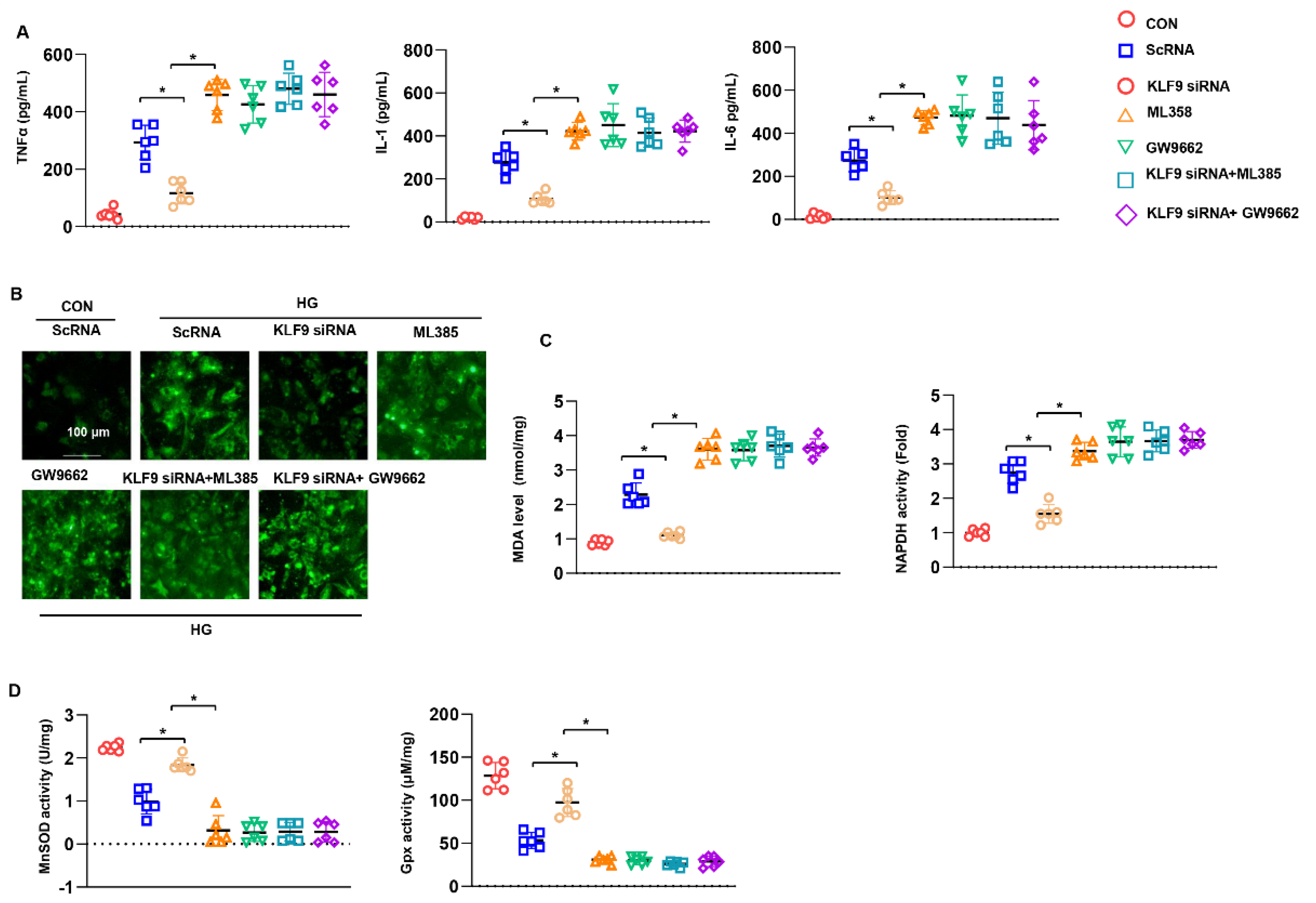
3.8. PPARγ/NRF2 Inhibition Counteracted The Protective Effects of KLF9 Knockdown
3.9. Cardiac Dysfunction Persists in KLF9 Knockdown PPARγ Inhibited Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo, O.; Ramírez, E.; Picatoste, B.; Egido, J.; Tuñón, J. Alteration of Energy Substrates and ROS Production in Diabetic Cardiomyopathy. Mediat. Inflamm. 2013, 2013, 461967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.J.; Gill, E.K.; Abudalo, R.A.; Edgar, K.S.; Watson, C.J.; Grieve, D.J. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart 2018, 104, 293–299. [Google Scholar] [CrossRef]
- Faria, A.; Persaud, S.J. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol. Ther. 2017, 172, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Chen, X.; Zong, B.; Yuan, H.; Wang, Z.; Wei, Y.; Wang, X.; Liu, G.; Zhang, J.; Li, S.; et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS -mediated NLRP 3 inflammasome activation. J. Cell. Mol. Med. 2018, 22, 4437–4448. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Lu, F.; Yu, X.; Wang, B.; Chen, J.; Lu, F.; Peng, S.; Sun, X.; Yu, M.; Chen, H.; et al. Exogenous H2S Promoted USP8 Sulfhydration to Regulate Mitophagy in the Hearts of db/db Mice. Aging Dis. 2020, 11, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Arow, M.; Waldman, M.; Yadin, D.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Freimark, D.; Kornowski, R.; Aravot, D.; Hochhauser, E.; et al. Sodium–glucose cotransporter 2 inhibitor Dapagliflozin attenuates diabetic cardiomyopathy. Cardiovasc. Diabetol. 2020, 19, 7. [Google Scholar] [CrossRef]
- Gao, L.; Liu, Y.; Guo, S.; Xiao, L.; Wu, L.; Wang, Z.; Zhang, Y. LAZ3 protects cardiac remodeling in diabetic cardiomyopathy via regulating miR-21/PPARa signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3322–3338. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Shi, W.; Zhang, H. The role of KLF14 in multiple disease processes. BioFactors 2020, 46, 276–282. [Google Scholar] [CrossRef]
- Rani, N.; Arya, D.S. Chrysin rescues rat myocardium from ischemia-reperfusion injury via PPAR-gamma/Nrf2 activation. Eur. J. Pharm. 2020, 883, 173389. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 Amplifies Oxidative Stress via Induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parga, J.A.; Rodriguez-Perez, A.I.; Garcia-Garrote, M.; Rodriguez-Pallares, J.; Labandeira-Garcia, J.L. Angiotensin II induces oxidative stress and upregulates neuroprotective signaling from the NRF2 and KLF9 pathway in dopaminergic cells. Free Radic. Biol. Med. 2018, 129, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Lv, Z.; Zhang, H.; Liu, B.; Jiang, H.; Tan, X.; Lu, J.; Baiyun, R.; Zhang, Z. Activation of the Nrf2 Signaling Pathway Involving KLF9 Plays a Critical Role in Allicin Resisting Against Arsenic Trioxide-Induced Hepatotoxicity in Rats. Biol. Trace Elem. Res. 2016, 176, 192–200. [Google Scholar] [CrossRef]
- Zong, J.; Li, F.-F.; Liang, K.; Dai, R.; Zhang, H.; Yan, L.; Liu, J.-L.; Xu, L.-H.; Qian, W.-H. Nuclear Localization Leucine-Rich-Repeat Protein 1 Deficiency Protects Against Cardiac Hypertrophy by Pressure Overload. Cell. Physiol. Biochem. 2018, 48, 75–86. [Google Scholar] [CrossRef]
- Norton, A.J.; Jordan, S.; Yeomans, P. Brief, high-temperature heat denaturation (pressure cooking): A simple and effective method of antigen retrieval for routinely processed tissues. J. Pathol. 1994, 173, 371–379. [Google Scholar] [CrossRef]
- Sun, G.-R.; Zhang, M.; Sun, J.-W.; Li, F.; Ma, X.-F.; Li, W.-T.; Han, R.-L.; Li, Z.-J.; Jiang, R.-R.; Li, G.-X.; et al. Krüppel-like factor KLF9 inhibits chicken intramuscular preadipocyte differentiation. Br. Poult. Sci. 2019, 60, 790–797. [Google Scholar] [CrossRef]
- Pei, H.; Yao, Y.; Yang, Y.; Liao, K.; Wu, J.R. Kruppel-like factor KLF9 regulates PPARgamma transactivation at the middle stage of adipogenesis. Cell Death Differ. 2011, 18, 315–327. [Google Scholar] [CrossRef]
- Xiong, Z.; Li, Y.; Zhao, Z.; Zhang, Y.; Man, W.; Lin, J.; Dong, Y.; Liu, L.; Wang, B.; Wang, H.; et al. Mst1 knockdown alleviates cardiac lipotoxicity and inhibits the development of diabetic cardiomyopathy in db/db mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165806. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Shah, A.K.; Tappia, P.S. Role of Oxidative Stress in Metabolic and Subcellular Abnormalities in Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 2413. [Google Scholar] [CrossRef]
- Yan, Q.; He, B.; Hao, G.; Liu, Z.; Tang, J.; Fu, Q.; Jiang, C. KLF9 aggravates ischemic injury in cardiomyocytes through augmenting oxidative stress. Life Sci. 2019, 233, 116641. [Google Scholar] [CrossRef] [PubMed]
- Bagati, A.; Moparthy, S.; Fink, E.E.; Bianchi-Smiraglia, A.; Yun, D.H.; Kolesnikova, M.; Udartseva, O.O.; Wolff, D.W.; Roll, M.V.; Lipchick, B.C.; et al. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene 2019, 38, 3585–3597. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.R.; Alhallak, I.; Simmen, R.C.M.; Melnyk, S.B.; Heard-Lipsmeyer, M.E.; Montales, M.T.E.; Habenicht, D.; Van, T.T.; Simmen, F.A. Krüppel-like Factor 9 (KLF9) Suppresses Hepatocellular Carcinoma (HCC)-Promoting Oxidative Stress and Inflammation in Mice Fed High-Fat Diet. Cancers 2022, 14, 1737. [Google Scholar] [CrossRef] [PubMed]
- Qu, R.; Liu, J.; Feng, L.; Li, L.; Liu, J.; Sun, F.; Sun, L. Down-regulation of KLF9 ameliorates LPS-caused acute lung injury and inflammation in mice via reducing GSDMD expression. Autoimmunity 2022, 55, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Fu, L.; Xu, Y.; Hu, X.; Yang, H.; Zhang, Y.; Luo, H.; Gan, S.; Tao, L.; Liang, G.; et al. Cyclovirobuxine D protects against diabetic cardiomyopathy by activating Nrf2-mediated antioxidant responses. Sci. Rep. 2020, 10, 6427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Forward Primer | Reverse Primer |
|---|---|---|
| KLF9-Mouse | GCACAAGTGCCCCTACAGT | TGTATGCACTCTGTAATGGGCTTT |
| KLF9-Rat | GTTTGCCCCTGTAAGTAGTAAGTG | GGTTCAGGCCATTGTGTAGAC |
| β-MHC-Mouse | CCGAGTCCCAGGTCAACAA | CTTCACGGGCACCCTTGGA |
| α-MHC-Mouse | GTCCAAGTTCCGCAAGGT | AGGGTCTGCTGGAGAGGTTA |
| Collagen I-Mouse | AGGCTTCAGTGGTTTGGATG | CACCAACAGCACCATCGTTA |
| Collagen III-Mouse | CCCAACCCAGAGATCCCATT | GAAGCACAGGAGCAGGTGTAGA |
| GAPDH-Mouse | ACTTGAAGGGTGGAGCCAAA | GACTGTGGTCATGAGCCCTT |
| GAPDH-Rat | GACATGCCGCCTGGAGAAAC | AGCCCAGGATGCCCTTTAGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Peng, J.; Feng, H.; Yang, Y.; Gao, J.; Liu, C.; Xu, J.; Zhao, Y.; Pan, S.; Wang, Y.; et al. KLF9 Aggravates Streptozotocin-Induced Diabetic Cardiomyopathy by Inhibiting PPARγ/NRF2 Signalling. Cells 2022, 11, 3393. https://doi.org/10.3390/cells11213393
Li F, Peng J, Feng H, Yang Y, Gao J, Liu C, Xu J, Zhao Y, Pan S, Wang Y, et al. KLF9 Aggravates Streptozotocin-Induced Diabetic Cardiomyopathy by Inhibiting PPARγ/NRF2 Signalling. Cells. 2022; 11(21):3393. https://doi.org/10.3390/cells11213393
Chicago/Turabian StyleLi, Fangfang, Jingfeng Peng, Hui Feng, Yiming Yang, Jianbo Gao, Chunrui Liu, Jie Xu, Yanru Zhao, Siyu Pan, Yixiao Wang, and et al. 2022. "KLF9 Aggravates Streptozotocin-Induced Diabetic Cardiomyopathy by Inhibiting PPARγ/NRF2 Signalling" Cells 11, no. 21: 3393. https://doi.org/10.3390/cells11213393