
C9-ALS-Associated Proline-Arginine Dipeptide Repeat Protein Induces Activation of NLRP3 Inflammasome of HMC3 Microglia Cells by Binding of Complement Component 1 Q Subcomponent-Binding Protein (C1QBP), and Syringin Prevents This Effect

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
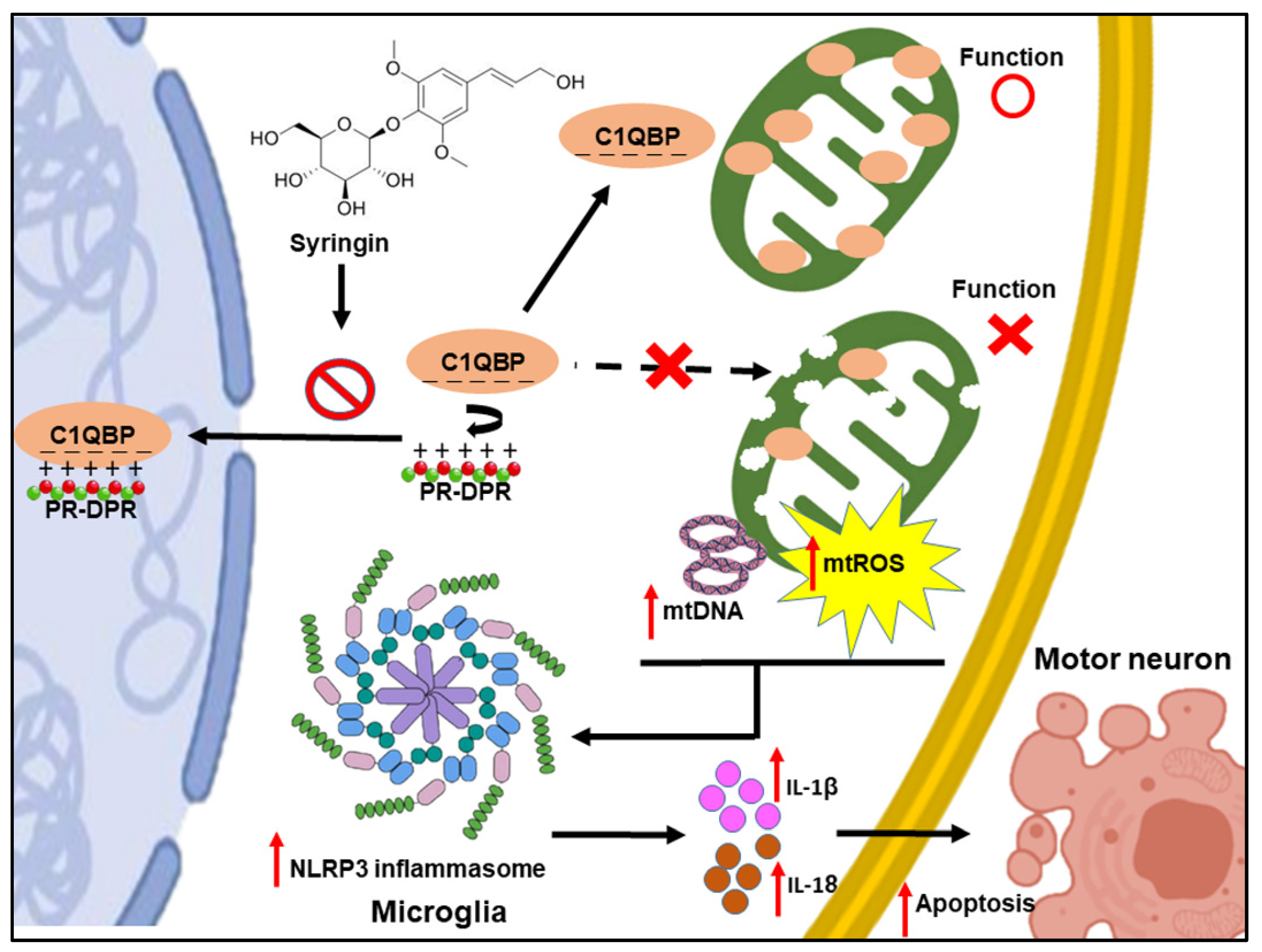
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemicals
2.2. Plasmid Construction and Transfection
2.3. Immunofluorescence Analysis
2.4. Preparation of Nuclear Extract and NF-κB (p65) Transcription Factor Assay
2.5. Western Blotting
2.6. Conditioned Medium Collection and ELISA
2.7. Lactate Dehydrogenase Assay
2.8. In Situ TUNEL Assay
2.9. Measurement of Mitochondrial Membrane Potential (MMP)
2.10. Apoptosis Assay by Flow Cytometry
2.11. Treatment of Inhibitors of NLRP3 Inflammasomes
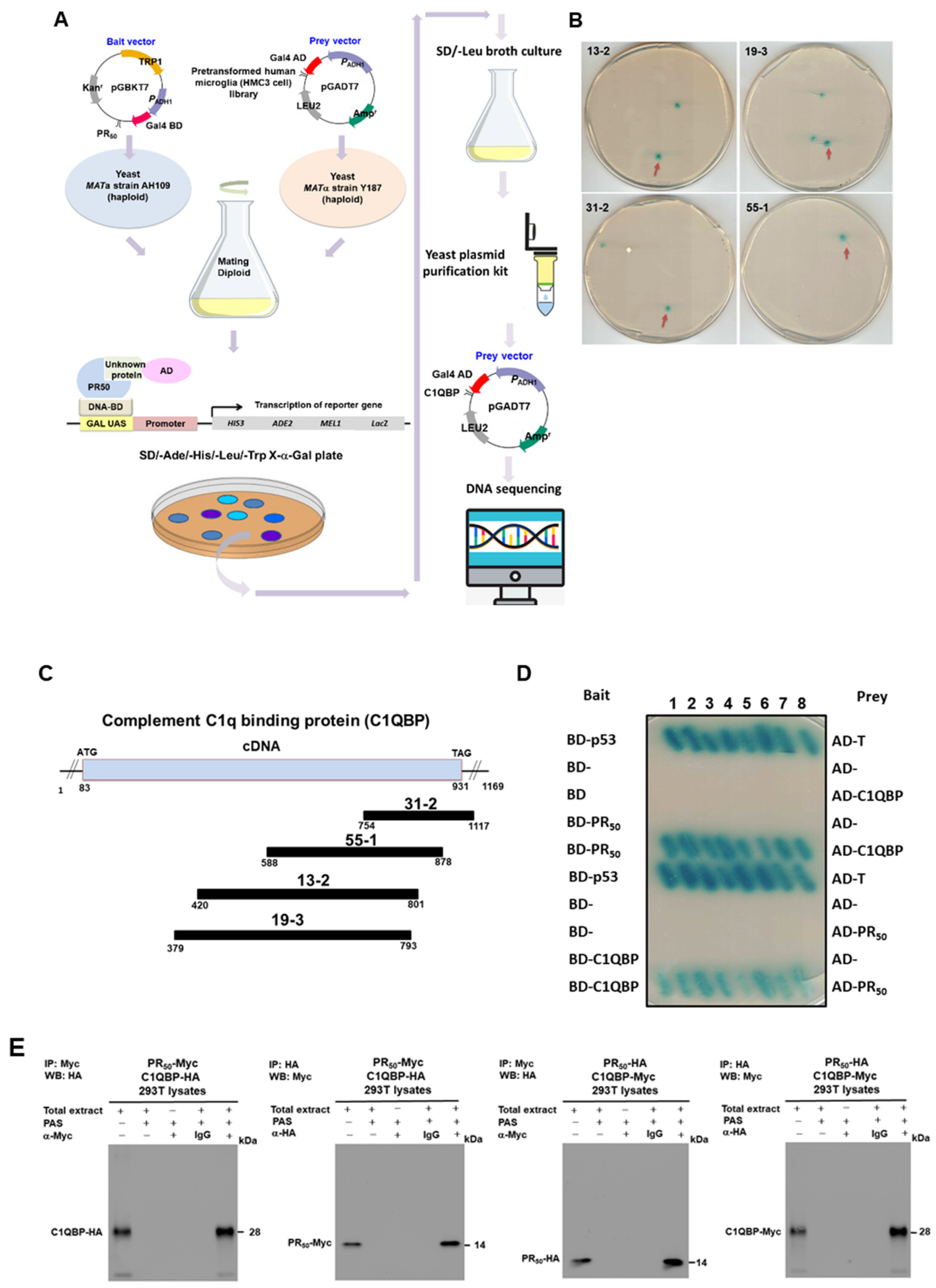
2.12. Yeast Two-Hybrid Library Screening
2.13. Yeast Two-Hybrid Assays
2.14. Co-Immunoprecipitation Analysis
2.15. RNA Interference Technique
2.16. Syringin (SRG) Treatment and Yeast Two-Hybrid-Based Growth Assay
2.17. Cytotoxicity Analysis and Treatment of Syringin (SRG) in HMC3 Cells
2.18. Statistical Analysis
3. Results
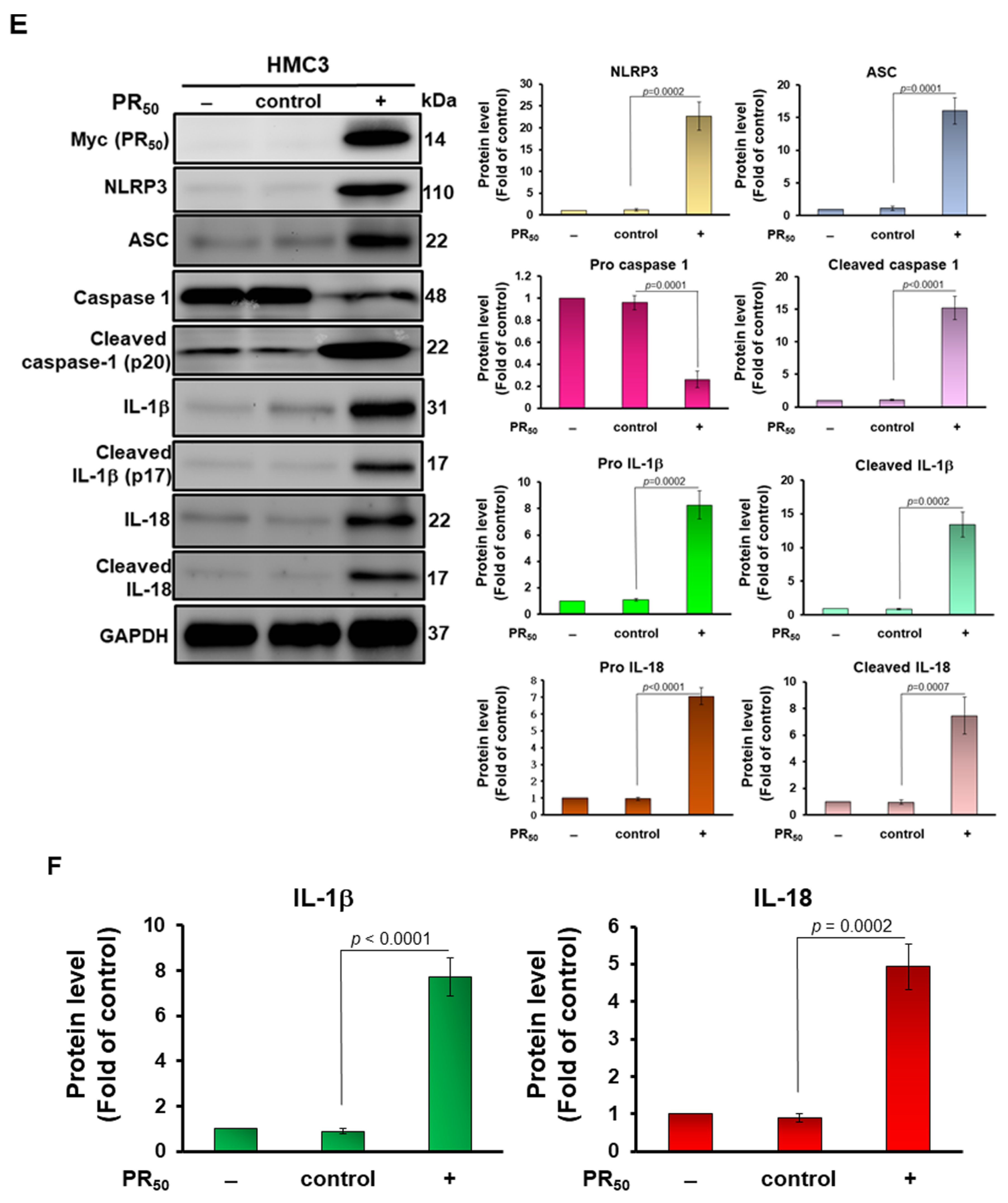
3.1. Expression of Proline-Arginine (PR)-Dipeptide Repeat Protein (PR50) Enhanced Activities of Nuclear Factor kappa B (NF-κB) and NOD-, LRR- and Pyrin Domain-Containing Protein 3 (NLRP3) Inflammasomes in Human HMC3 Microglial Cell
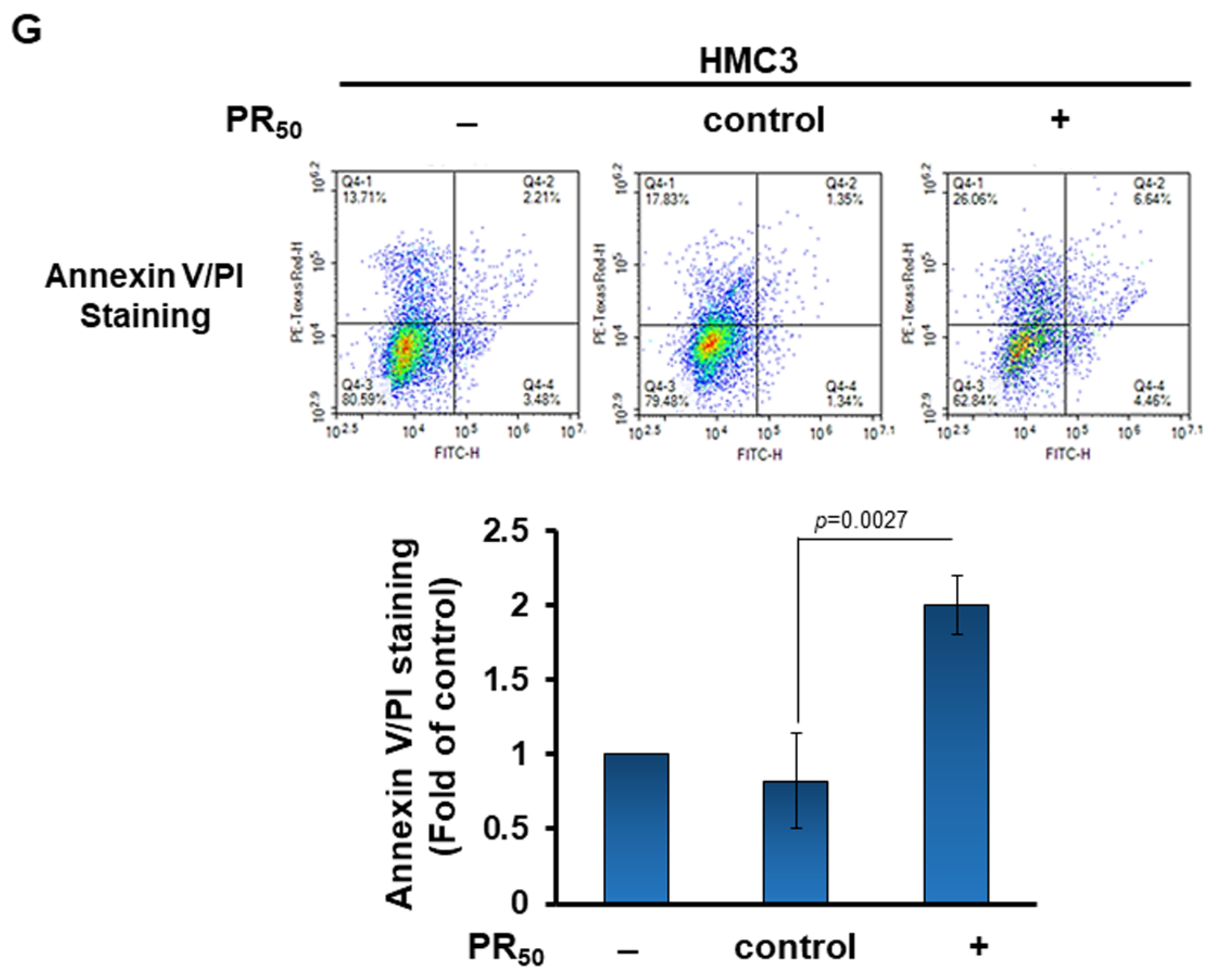
3.2. Conditional Medium of PR-DPR (PR50)-Expressing Human HMC3 Microglial Cells Caused the Damage and Apoptosis of Mouse NSC 34 Motor Neuron Cells
3.3. Treatment of PR50-Expressing HMC3 Cells with the NLRP3 Inflammasome Inhibitor MCC950 Abolished the Capability That Its Conditional Medium Induces Apoptosis for NSC34 Cells
3.4. Complement Component 1 Q Subcomponent-Binding Protein (C1QBP) Is a Candidate Interaction Partner of PR-DPR (PR50) in HMC3 Microglial Cells
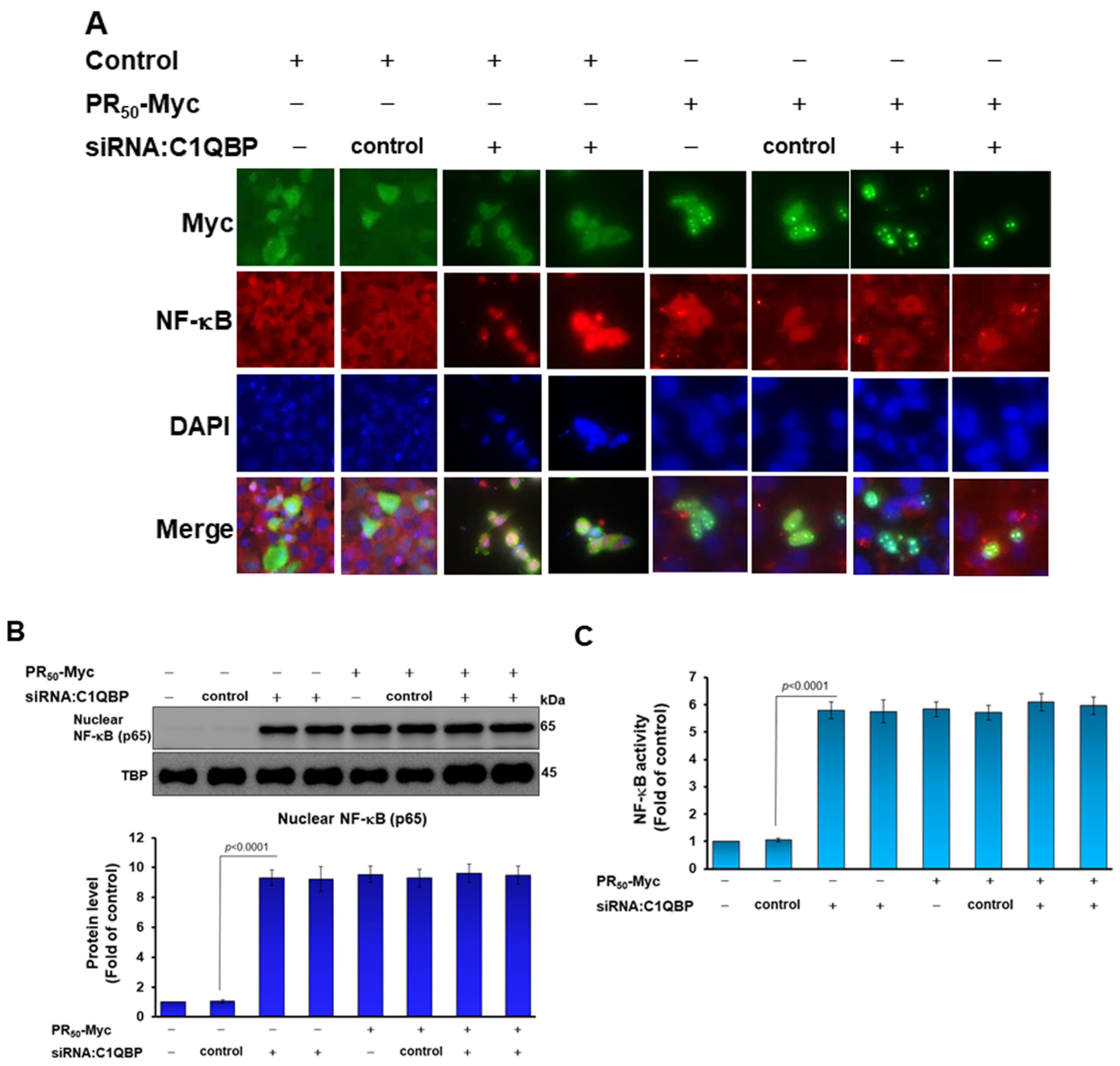
3.5. Down-Regulation of C1QBP Expression Directly Caused NLRP3 Inflammasome Activation in Human HMC3 Microglial Cells and Covered the Effect of PR50 on NLRP3 Inflammasome Activation
3.6. Syringin (SRG) Prevents the Interaction of PR50 with C1QBP in a Yeast Two-Hybrid-Based Growth Assay
3.7. Syringin (SRG) Prevents PR50-Induced NLRP3-Inflammasome Activity in HMC3 Cells and PR-CM-Caused Apoptosis in NSC-34 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chio, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- St Martin, J.L.; Wang, L.; Kaprielian, Z. Toxicity in ALS: TDP-43 modifiers and C9orf72. Neurosci. Lett. 2020, 716, 134621. [Google Scholar] [CrossRef] [PubMed]
- Todd, T.W.; McEachin, Z.T.; Chew, J.; Burch, A.R.; Jansen-West, K.; Tong, J.; Yue, M.; Song, Y.; Castanedes-Casey, M.; Kurti, A.; et al. Hexanucleotide Repeat Expansions in c9FTD/ALS and SCA36 Confer Selective Patterns of Neurodegeneration in Vivo. Cell Rep. 2020, 31, 107616. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Mizielinska, S.; Gronke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Liu, L.; Tao, Z.; Wang, R.; Ren, H.; Sun, H.; Lin, Z.; Zhang, Z.; Mu, C.; Zhou, J.; et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat. Commun. 2019, 10, 2906. [Google Scholar] [CrossRef] [Green Version]
- Tao, Z.; Wang, H.; Xia, Q.; Li, K.; Li, K.; Jiang, X.; Xu, G.; Wang, G.; Ying, Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 2015, 24, 2426–2441. [Google Scholar] [CrossRef]
- Suzuki, H.; Shibagaki, Y.; Hattori, S.; Matsuoka, M. The proline-arginine repeat protein linked to C9-ALS/FTD causes neuronal toxicity by inhibiting the DEAD-box RNA helicase-mediated ribosome biogenesis. Cell Death Dis. 2018, 9, 975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.R.; Mitrea, D.M.; Zhang, P.; Stanley, C.B.; Cassidy, D.E.; Nourse, A.; Phillips, A.H.; Tolbert, M.; Taylor, J.P.; Kriwacki, R.W. C9orf72 Poly(PR) Dipeptide Repeats Disturb Biomolecular Phase Separation and Disrupt Nucleolar Function. Mol. Cell 2019, 74, 713–728 e6. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Bao, P.; Jiang, X.; Wang, H.; Qin, M.; Wang, R.; Wang, T.; Yang, Y.; Lorenzini, I.; Liao, L.; et al. Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain 2019, 142, 1349–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Matsuoka, M. Proline-arginine poly-dipeptide encoded by the C9orf72 repeat expansion inhibits adenosine deaminase acting on RNA. J. Neurochem. 2021, 158, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Lopez-Gonzalez, R.; Kunz, R.C.; Gangopadhyay, J.; Borufka, C.; Gygi, S.P.; Gao, F.B.; Reed, R. Evidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-splicing in ALS/FTD Patients. Cell Rep. 2017, 19, 2244–2256. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Chung, C.G.; Park, J.H.; Ko, B.S.; Park, S.S.; Kim, Y.H.; Cha, I.J.; Kim, J.; Ha, C.M.; Kim, H.J.; et al. C9orf72-associated arginine-rich dipeptide repeats induce RNA-dependent nuclear accumulation of Staufen in neurons. Hum. Mol. Genet. 2021, 30, 1084–1100. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Shibagaki, Y.; Hattori, S.; Matsuoka, M. C9-ALS/FTD-linked proline-arginine dipeptide repeat protein associates with paraspeckle components and increases paraspeckle formation. Cell Death Dis. 2019, 10, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maor-Nof, M.; Shipony, Z.; Lopez-Gonzalez, R.; Nakayama, L.; Zhang, Y.J.; Couthouis, J.; Blum, J.A.; Castruita, P.A.; Linares, G.R.; Ruan, K.; et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 2021, 184, 689–708 e20. [Google Scholar] [CrossRef] [PubMed]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Hayes, L.R.; Duan, L.; Bowen, K.; Kalab, P.; Rothstein, J.D. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. Elife 2020, 9, e51685. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–E1117. [Google Scholar] [CrossRef] [Green Version]
- Kanekura, K.; Yagi, T.; Cammack, A.J.; Mahadevan, J.; Kuroda, M.; Harms, M.B.; Miller, T.M.; Urano, F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet. 2016, 25, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- Moens, T.G.; Niccoli, T.; Wilson, K.M.; Atilano, M.L.; Birsa, N.; Gittings, L.M.; Holbling, B.V.; Dyson, M.C.; Thoeng, A.; Neeves, J.; et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 2019, 137, 487–500. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Xu, J. C9orf72 Dipeptide Repeats Cause Selective Neurodegeneration and Cell-Autonomous Excitotoxicity in Drosophila Glutamatergic Neurons. J. Neurosci. 2018, 38, 7741–7752. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.; Lee, J.; Lee, S.Y.; Kwon, I.; Cho, H. Poly-dipeptides produced from C9orf72 hexanucleotide repeats cause selective motor neuron hyperexcitability in ALS. Proc. Natl. Acad. Sci. USA 2022, 119, e2113813119. [Google Scholar] [CrossRef]
- Wang, R.; Xu, X.; Hao, Z.; Zhang, S.; Wu, D.; Sun, H.; Mu, C.; Ren, H.; Wang, G. Poly-PR in C9ORF72-Related Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Causes Neurotoxicity by Clathrin-Dependent Endocytosis. Neurosci. Bull. 2019, 35, 889–900. [Google Scholar] [CrossRef]
- Babu, M.; Favretto, F.; de Opakua, A.I.; Rankovic, M.; Becker, S.; Zweckstetter, M. Proline/arginine dipeptide repeat polymers derail protein folding in amyotrophic lateral sclerosis. Nat. Commun. 2021, 12, 3396. [Google Scholar] [CrossRef]
- Fumagalli, L.; Young, F.L.; Boeynaems, S.; De Decker, M.; Mehta, A.R.; Swijsen, A.; Fazal, R.; Guo, W.; Moisse, M.; Beckers, J.; et al. C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv. 2021, 7, eabg3013. [Google Scholar] [CrossRef]
- Premasiri, A.S.; Gill, A.L.; Vieira, F.G. Type I PRMT Inhibition Protects Against C9ORF72 Arginine-Rich Dipeptide Repeat Toxicity. Front. Pharmacol. 2020, 11, 569661. [Google Scholar] [CrossRef]
- Trageser, K.J.; Smith, C.; Herman, F.J.; Ono, K.; Pasinetti, G.M. Mechanisms of Immune Activation by c9orf72-Expansions in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2019, 13, 1298. [Google Scholar] [CrossRef]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef] [Green Version]
- Haukedal, H.; Freude, K. Implications of Microglia in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. J. Mol. Biol. 2019, 431, 1818–1829. [Google Scholar] [CrossRef]
- Masrori, P.; Beckers, J.; Gossye, H.; Van Damme, P. The role of inflammation in neurodegeneration: Novel insights into the role of the immune system in C9orf72 HRE-mediated ALS/FTD. Mol. Neurodegener. 2022, 17, 22. [Google Scholar] [CrossRef]
- Burberry, A.; Wells, M.F.; Limone, F.; Couto, A.; Smith, K.S.; Keaney, J.; Gillet, G.; van Gastel, N.; Wang, J.Y.; Pietilainen, O.; et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 2020, 582, 89–94. [Google Scholar] [CrossRef]
- McCauley, M.E.; O’Rourke, J.G.; Yanez, A.; Markman, J.L.; Ho, R.; Wang, X.; Chen, S.; Lall, D.; Jin, M.; Muhammad, A.; et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 2020, 585, 96–101. [Google Scholar] [CrossRef]
- Rostalski, H.; Leskela, S.; Huber, N.; Katisko, K.; Cajanus, A.; Solje, E.; Marttinen, M.; Natunen, T.; Remes, A.M.; Hiltunen, M.; et al. Astrocytes and Microglia as Potential Contributors to the Pathogenesis of C9orf72 Repeat Expansion-Associated FTLD and ALS. Front. Neurosci. 2019, 13, 486. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting Microglial alpha-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, W.; Zhang, Y.; Yu, Q.; Zeng, M.; Gan, J.; Zhang, W.; Jiang, X.; Li, H. Focus on the role of mitochondria in NLRP3 inflammasome activation: A prospective target for the treatment of ischemic stroke (Review). Int. J. Mol. Med. 2022, 49, 74. [Google Scholar] [CrossRef]
- Haque, M.E.; Akther, M.; Jakaria, M.; Kim, I.S.; Azam, S.; Choi, D.K. Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov. Disord. 2020, 35, 20–33. [Google Scholar] [CrossRef]
- Sagulenko, V.; Vitak, N.; Vajjhala, P.R.; Vince, J.E.; Stacey, K.J. Caspase-1 Is an Apical Caspase Leading to Caspase-3 Cleavage in the AIM2 Inflammasome Response, Independent of Caspase-8. J. Mol. Biol. 2018, 430, 238–247. [Google Scholar] [CrossRef]
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020, 68, 407–421. [Google Scholar] [CrossRef]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef]
- Casarotto, E.; Sproviero, D.; Corridori, E.; Gagliani, M.C.; Cozzi, M.; Chierichetti, M.; Cristofani, R.; Ferrari, V.; Galbiati, M.; Mina, F.; et al. Neurodegenerative Disease-Associated TDP-43 Fragments Are Extracellularly Secreted with CASA Complex Proteins. Cells 2022, 11, 516. [Google Scholar] [CrossRef]
- Wong, J.H.; Alfatah, M.; Sin, M.F.; Sim, H.M.; Verma, C.S.; Lane, D.P.; Arumugam, P. A yeast two-hybrid system for the screening and characterization of small-molecule inhibitors of protein-protein interactions identifies a novel putative Mdm2-binding site in p53. BMC Biol. 2017, 15, 108. [Google Scholar] [CrossRef]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Invest. 2017, 127, 3250–3258. [Google Scholar] [CrossRef]
- Chung, H.J.; Korm, S.; Lee, S.I.; Phorl, S.; Noh, S.; Han, M.; Naskar, R.; Kim, H.; Lee, J.Y. RAP80 binds p32 to preserve the functional integrity of mitochondria. Biochem. Biophys. Res. Commun. 2017, 492, 441–446. [Google Scholar] [CrossRef]
- Wang, J.; Huang, C.L.; Zhang, Y. Complement C1q Binding Protein (C1QBP): Physiological Functions, Mutation-Associated Mitochondrial Cardiomyopathy and Current Disease Models. Front. Cardiovasc. Med. 2022, 9, 843853. [Google Scholar] [CrossRef]
- Ghebrehiwet, B.; Geisbrecht, B.V.; Xu, X.; Savitt, A.G.; Peerschke, E.I.B. The C1q Receptors: Focus on gC1qR/p33 (C1qBP, p32, HABP-1)1. Semin. Immunol. 2019, 45, 101338. [Google Scholar] [CrossRef] [PubMed]
- Egusquiza-Alvarez, C.A.; Robles-Flores, M. An approach to p32/gC1qR/HABP1: A multifunctional protein with an essential role in cancer. J. Cancer Res. Clin. Oncol. 2022, 148, 1831–1854. [Google Scholar] [CrossRef] [PubMed]
- Pednekar, L.; Valentino, A.; Ji, Y.; Tumma, N.; Valentino, C.; Kadoor, A.; Hosszu, K.K.; Ramadass, M.; Kew, R.R.; Kishore, U.; et al. Identification of the gC1qR sites for the HIV-1 viral envelope protein gp41 and the HCV core protein: Implications in viral-specific pathogenesis and therapy. Mol. Immunol. 2016, 74, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barna, J.; Dimen, D.; Puska, G.; Kovacs, D.; Csikos, V.; Olah, S.; Udvari, E.B.; Pal, G.; Dobolyi, A. Complement component 1q subcomponent binding protein in the brain of the rat. Sci. Rep. 2019, 9, 4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiumi, T.; Kang, D. Mitochondrial nucleic acid binding proteins associated with diseases. Front. Biosci. 2017, 22, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.T.; Xu, X.; Alontaga, A.Y.; Chen, Y.; Liu, Y. Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction. Cell Rep. 2014, 7, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Sun, H.; Liang, X.; Lima, W.F.; Crooke, S.T. Human RNase H1 is associated with protein P32 and is involved in mitochondrial pre-rRNA processing. PLoS ONE 2013, 8, e71006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wan, O.W.; Xie, W.; Chung, K.K. p32 regulates mitochondrial morphology and dynamics through parkin. Neuroscience 2011, 199, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Shifman, O.; Oren, M.; Kimchi, A. The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene 2007, 26, 6677–6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, H.; You, H. p32: A new player in autophagy. Mol. Cell. Oncol. 2016, 3, e1061097. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.; Phorl, S.; Naskar, R.; Oeum, K.; Seo, Y.; Kim, E.; Kweon, H.S.; Lee, J.Y. p32/C1QBP regulates OMA1-dependent proteolytic processing of OPA1 to maintain mitochondrial connectivity related to mitochondrial dysfunction and apoptosis. Sci. Rep. 2020, 10, 10618. [Google Scholar] [CrossRef]
- Hu, M.; Crawford, S.A.; Henstridge, D.C.; Ng, I.H.; Boey, E.J.; Xu, Y.; Febbraio, M.A.; Jans, D.A.; Bogoyevitch, M.A. p32 protein levels are integral to mitochondrial and endoplasmic reticulum morphology, cell metabolism and survival. Biochem. J. 2013, 453, 381–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayarajan, J.; Angandoor, S.; Vedulla, S.H.; Sritharan, S.; Ganesan, K.; War, A.R.; Sivalingam, N. Curcumin induces chemosensitization to doxorubicin in Duke’s type B coloadenocarcinoma cell line. Mol. Biol. Rep. 2020, 47, 7883–7892. [Google Scholar] [CrossRef] [PubMed]
- McGee, A.M.; Baines, C.P. Complement 1q-binding protein inhibits the mitochondrial permeability transition pore and protects against oxidative stress-induced death. Biochem. J. 2011, 433, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.J.; Gu, P.Q.; Fan, W.M.; Liu, Z.; Qiu, F.; Peng, Y.Z.; Guo, X.R. The role of gC1qR in regulating survival of human papillomavirus 16 oncogene-transfected cervical cancer cells. Int. J. Oncol. 2011, 39, 1265–1272. [Google Scholar] [PubMed]
- Choi, K.; Koo, B.H.; Yoon, B.J.; Jung, M.; Yun, H.Y.; Jeon, B.H.; Won, M.H.; Kim, Y.M.; Mun, J.Y.; Lim, H.K.; et al. Overexpressed p32 localized in the endoplasmic reticulum and mitochondria negatively regulates calciumdependent endothelial nitric oxide synthase activit. Mol. Med. Rep. 2020, 22, 2395–2403. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell. Biol. 2021, 136, 106013. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10, 2898. [Google Scholar] [CrossRef]
- Zhang, W.; Li, G.; Luo, R.; Lei, J.; Song, Y.; Wang, B.; Ma, L.; Liao, Z.; Ke, W.; Liu, H.; et al. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp. Mol. Med. 2022, 54, 129–142. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, J.H.; Zhang, H.; Canfran-Duque, A.; Singh, A.K.; Perry, R.J.; Shulman, G.; Fernandez-Hernando, C.; Min, W. Brown adipose TRX2 deficiency activates mtDNA-NLRP3 to impair thermogenesis and protect against diet-induced insulin resistance. J. Clin. Invest. 2022, 132, e148852. [Google Scholar] [CrossRef]
- Zhao, S.; Li, X.; Wang, J.; Wang, H. The Role of the Effects of Autophagy on NLRP3 Inflammasome in Inflammatory Nervous System Diseases. Front Cell Dev. Biol. 2021, 9, 657478. [Google Scholar] [CrossRef] [PubMed]
- Watthanasurorot, A.; Jiravanichpaisal, P.; Soderhall, K.; Soderhall, I. A calreticulin/gC1qR complex prevents cells from dying: A conserved mechanism from arthropods to humans. J. Mol. Cell. Biol. 2013, 5, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Wu, Y.; Fu, B.; Wang, L.; Hao, W.; Hua, F.; Sun, Y.; Dorf, M.E.; Li, S. Leaked Mitochondrial C1QBP Inhibits Activation of the DNA Sensor cGAS. J. Immunol. 2021, 207, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fort, B.P.; Dubyak, G.R.; Greenfield, E.M. Lysosomal disruption by orthopedic wear particles induces activation of the NLRP3 inflammasome and macrophage cell death by distinct mechanisms. J. Orthop. Res. 2021, 39, 493–505. [Google Scholar] [CrossRef]
- Lv, S.; Li, X.; Wang, H. The Role of the Effects of Endoplasmic Reticulum Stress on NLRP3 Inflammasome in Diabetes. Front Cell Dev. Biol. 2021, 9, 663528. [Google Scholar] [CrossRef] [PubMed]
- Burstein, S.R.; Valsecchi, F.; Kawamata, H.; Bourens, M.; Zeng, R.; Zuberi, A.; Milner, T.A.; Cloonan, S.M.; Lutz, C.; Barrientos, A.; et al. In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet. 2018, 27, 160–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Wu, M.; Wang, Y.; Du, W.; He, Y.; Shi, Z. Total Syntheses and Anti-inflammatory Activities of Syringin and Its Natural Analogues. J. Nat. Prod. 2021, 84, 2866–2874. [Google Scholar] [CrossRef]
- Jin, L.; Schmiech, M.; El Gaafary, M.; Zhang, X.; Syrovets, T.; Simmet, T. A comparative study on root and bark extracts of Eleutherococcus senticosus and their effects on human macrophages. Phytomedicine 2020, 68, 153181. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Niu, M.; Wang, N.; Wang, Y. Syringin alleviates ovalbumin-induced lung inflammation in BALB/c mice asthma model via NF-kappaB signaling pathway. Environ. Toxicol. 2021, 36, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, H.; Han, Z.; Sheng, H.; Wu, Y.; Wang, Y.; Guo, X.; Zhu, Y.; Li, X.; Wang, Y. Guhong injection promotes post-stroke functional recovery via attenuating cortical inflammation and apoptosis in subacute stage of ischemic stroke. Phytomedicine 2022, 99, 154034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhao, L.; Su, Y.; Liu, X.; Zhang, F.; Gao, Y. Syringin Prevents Abeta25-35-Induced Neurotoxicity in SK-N-SH and SK-N-BE Cells by Modulating miR-124-3p/BID Pathway. Neurochem. Res. 2021, 46, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Zhang, Q.; Xun, Z.; Yuan, L.; Li, R.; Li, X.; Tian, S.Y.; Xin, N.; Xu, Y. Increases of iASPP-Keap1 interaction mediated by syringin enhance synaptic plasticity and rescue cognitive impairments via stabilizing Nrf2 in Alzheimer’s models. Redox. Biol. 2020, 36, 101672. [Google Scholar] [CrossRef]
- Ding, H.G.; Deng, Y.Y.; Yang, R.Q.; Wang, Q.S.; Jiang, W.Q.; Han, Y.L.; Huang, L.Q.; Wen, M.Y.; Zhong, W.H.; Li, X.S.; et al. Hypercapnia induces IL-1beta overproduction via activation of NLRP3 inflammasome: Implication in cognitive impairment in hypoxemic adult rats. J. Neuroinflammation 2018, 15, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chen, Y.; Wang, H.; Wei, Y.; Yuan, Y.; Zhou, Q.; Fang, F.; Shi, S.; Jiang, X.; Dong, Y.; et al. Microglia-derived IL-1beta promoted neuronal apoptosis through ER stress-mediated signaling pathway PERK/eIF2alpha/ATF4/CHOP upon arsenic exposure. J. Hazard Mater. 2021, 417, 125997. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Kong, K.M.; Qi, W.L.; Ye, W.L.; Song, P.S. Interleukin-1 beta induction of neuron apoptosis depends on p38 mitogen-activated protein kinase activity after spinal cord injury. Acta Pharmacol. Sin. 2005, 26, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed]





















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, R.-H.; Tsai, C.-W.; Chiu, S.-C.; Liu, S.-P.; Chiang, Y.-T.; Kuo, Y.-H.; Shyu, W.-C.; Lin, S.-Z. C9-ALS-Associated Proline-Arginine Dipeptide Repeat Protein Induces Activation of NLRP3 Inflammasome of HMC3 Microglia Cells by Binding of Complement Component 1 Q Subcomponent-Binding Protein (C1QBP), and Syringin Prevents This Effect. Cells 2022, 11, 3128. https://doi.org/10.3390/cells11193128
Fu R-H, Tsai C-W, Chiu S-C, Liu S-P, Chiang Y-T, Kuo Y-H, Shyu W-C, Lin S-Z. C9-ALS-Associated Proline-Arginine Dipeptide Repeat Protein Induces Activation of NLRP3 Inflammasome of HMC3 Microglia Cells by Binding of Complement Component 1 Q Subcomponent-Binding Protein (C1QBP), and Syringin Prevents This Effect. Cells. 2022; 11(19):3128. https://doi.org/10.3390/cells11193128
Chicago/Turabian StyleFu, Ru-Huei, Chia-Wen Tsai, Shao-Chih Chiu, Shih-Ping Liu, Yu-Ting Chiang, Yun-Hua Kuo, Woei-Cherng Shyu, and Shinn-Zong Lin. 2022. "C9-ALS-Associated Proline-Arginine Dipeptide Repeat Protein Induces Activation of NLRP3 Inflammasome of HMC3 Microglia Cells by Binding of Complement Component 1 Q Subcomponent-Binding Protein (C1QBP), and Syringin Prevents This Effect" Cells 11, no. 19: 3128. https://doi.org/10.3390/cells11193128