
Proteomic and Phosphoproteomic Profiling Reveals the Oncogenic Role of Protein Kinase D Family Kinases in Cholangiocarcinoma
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Clinical Samples and Information
2.2. Extraction and Digestion of Protein
2.3. TMT Labelling
2.4. HPLC Fractionation
2.5. Enrichment of Phosphopeptides
2.6. LC-MS/MS Analysis
2.7. Standard Database Search
2.8. Proteomic Analysis
2.9. Phosphoproteomic Analysis
2.10. Public P-Site Resources
2.11. GO Functional Enrichment Analysis
2.12. KEGG Functional Enrichment Analysis
2.13. The CifPK Pipeline
2.14. Cell Culture and Transfection
2.15. Western Blotting
2.16. Cell Proliferation and Drug Cytotoxic Assay In Vitro
2.17. Wound Healing
2.18. Transwell Assay
3. Results
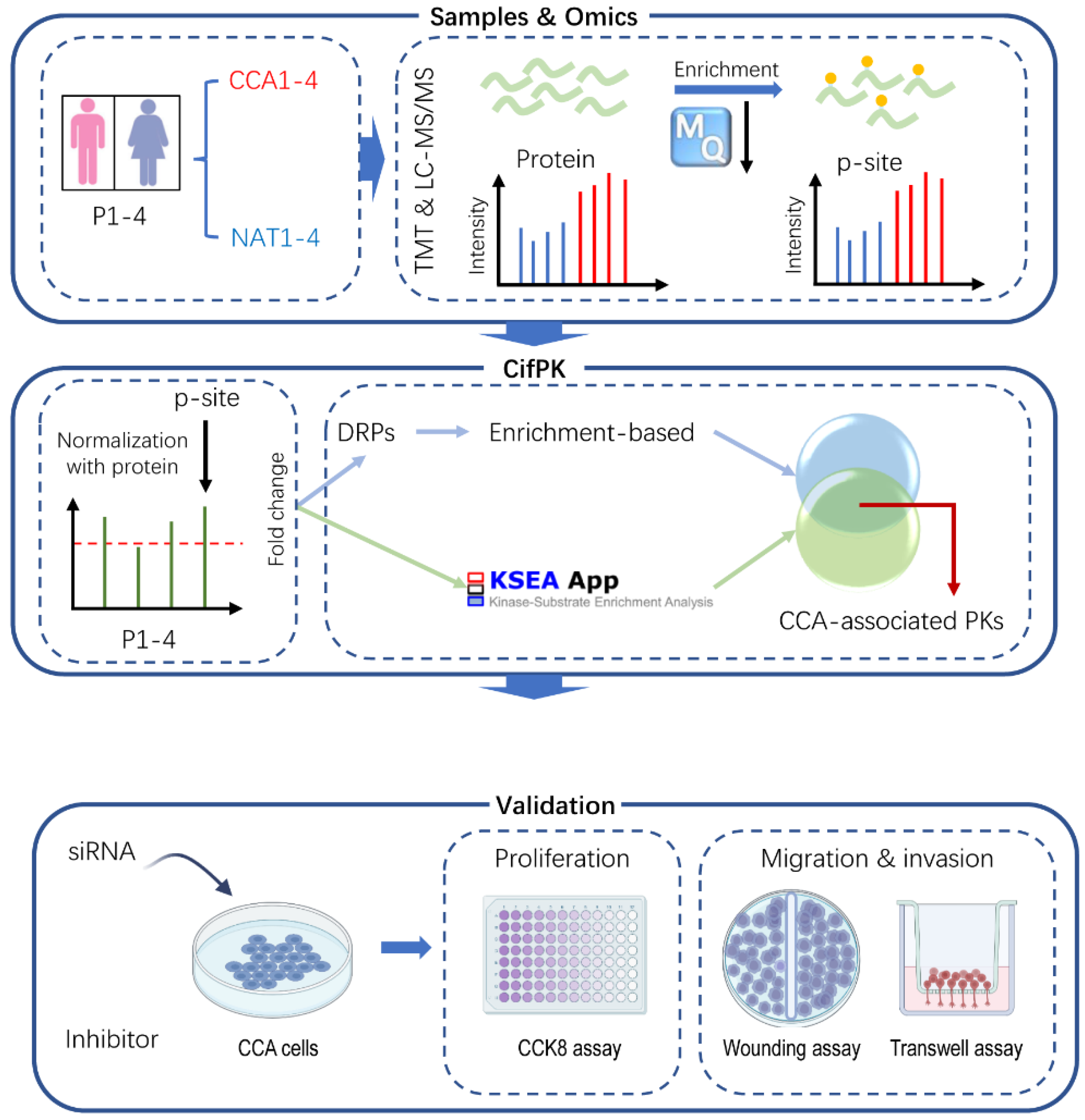
3.1. The Procedure for Analysis of CCA-Associated PKs
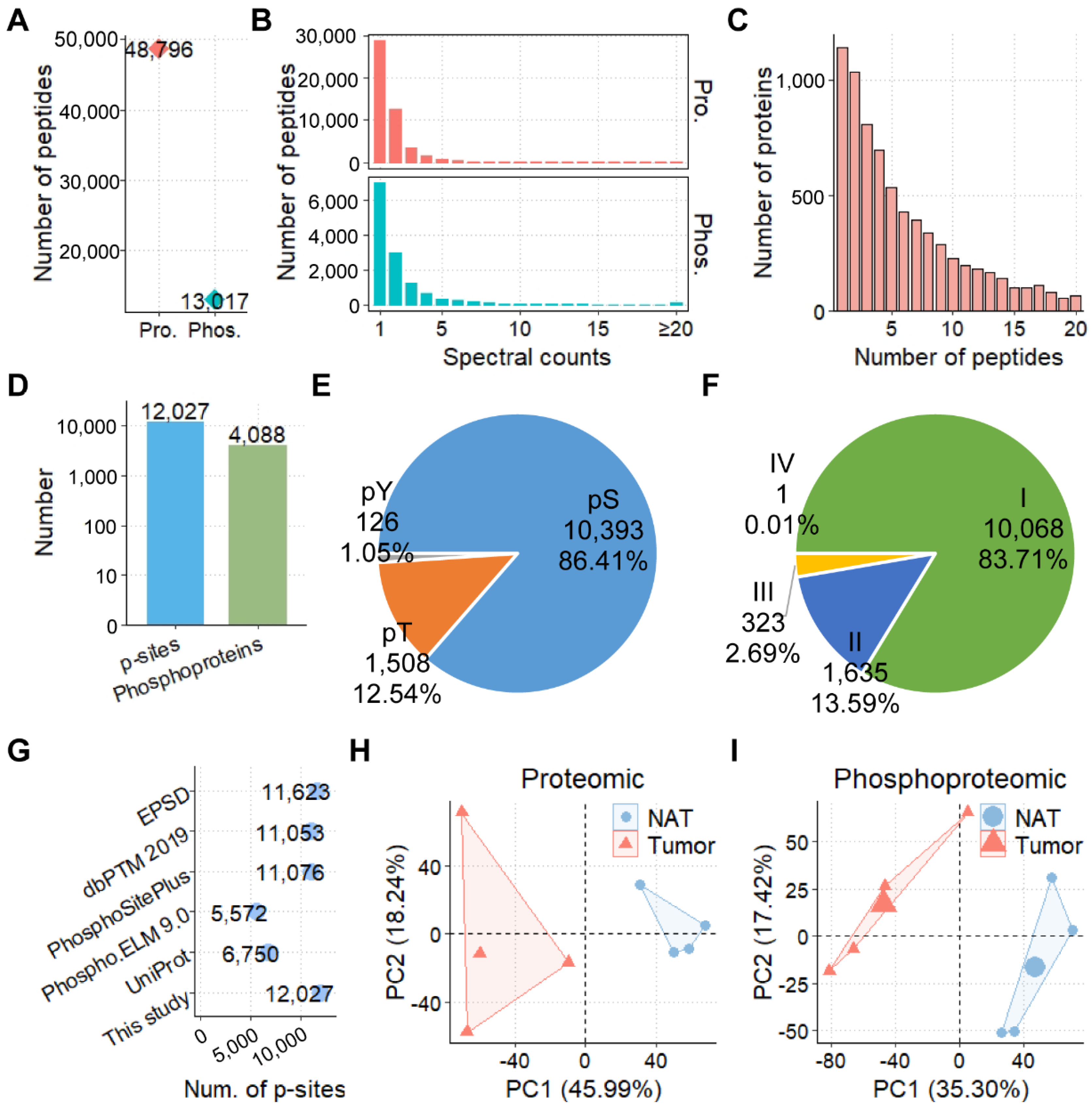
3.2. The Proteomic and Phosphoproteomic Profiling of CCAs
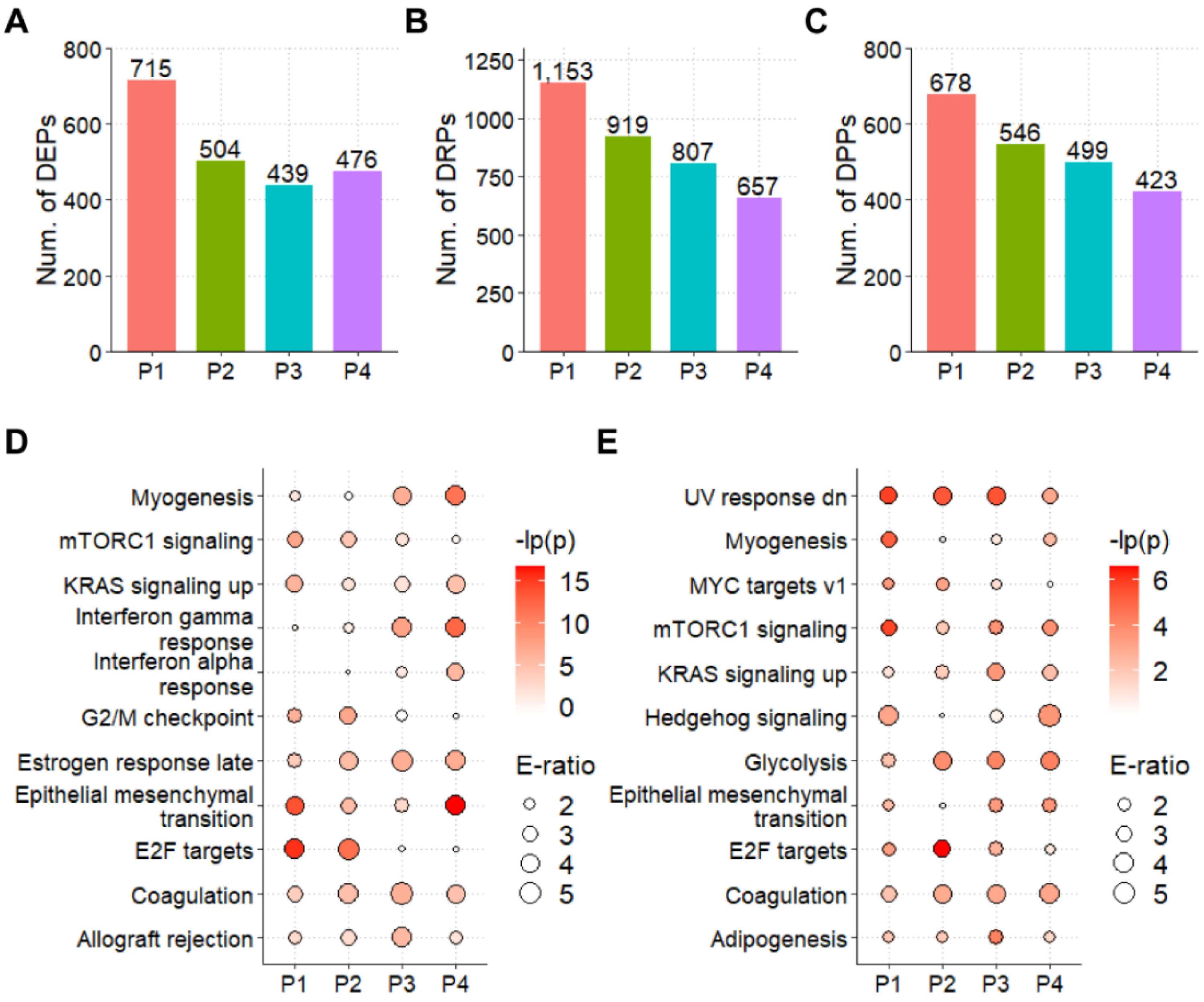
3.3. The Landscapes of Protein Expression and Phosphorylation for CCAs
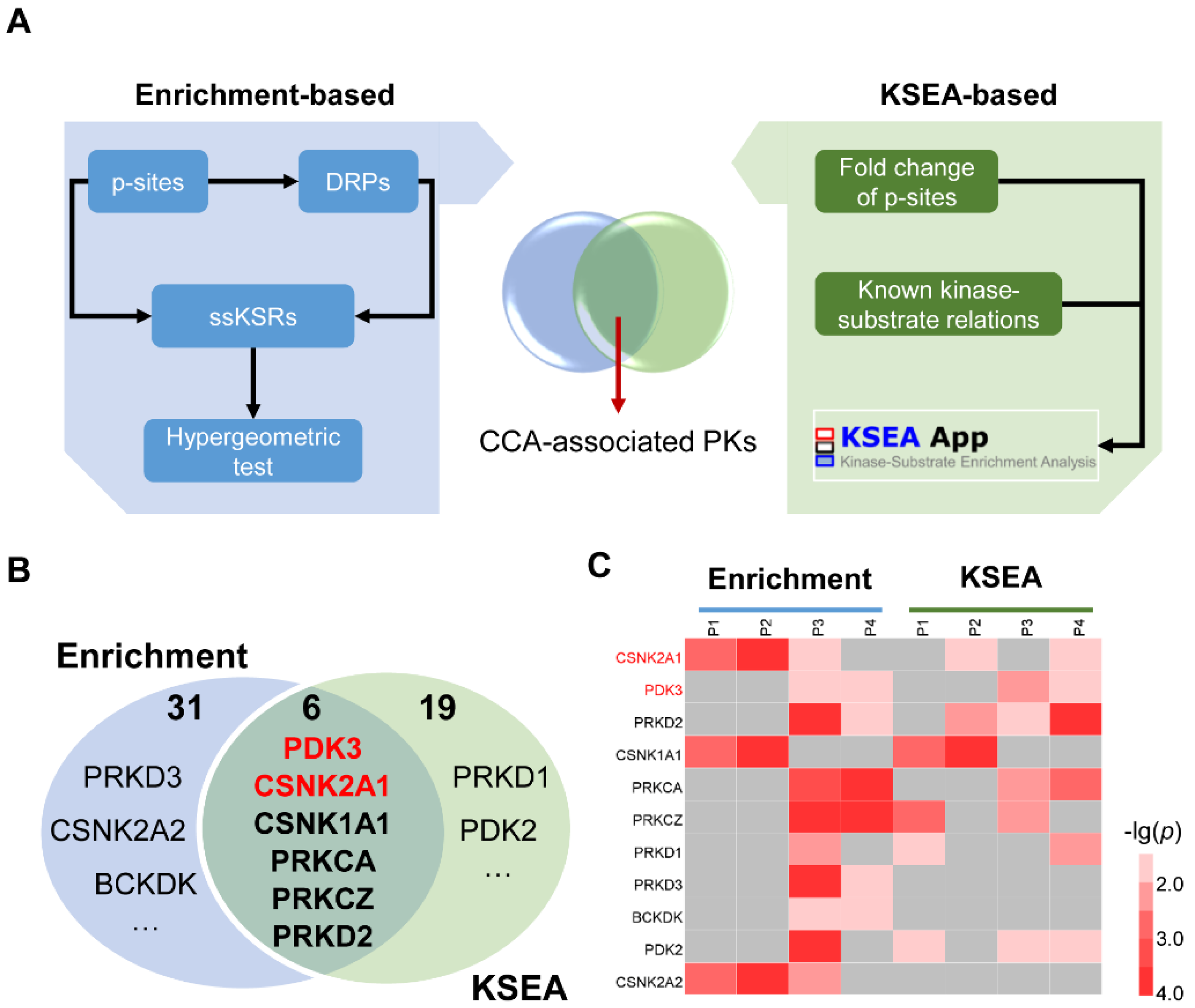
3.4. The Integrated Pipeline for the Prediction of CCA-Associated PKs
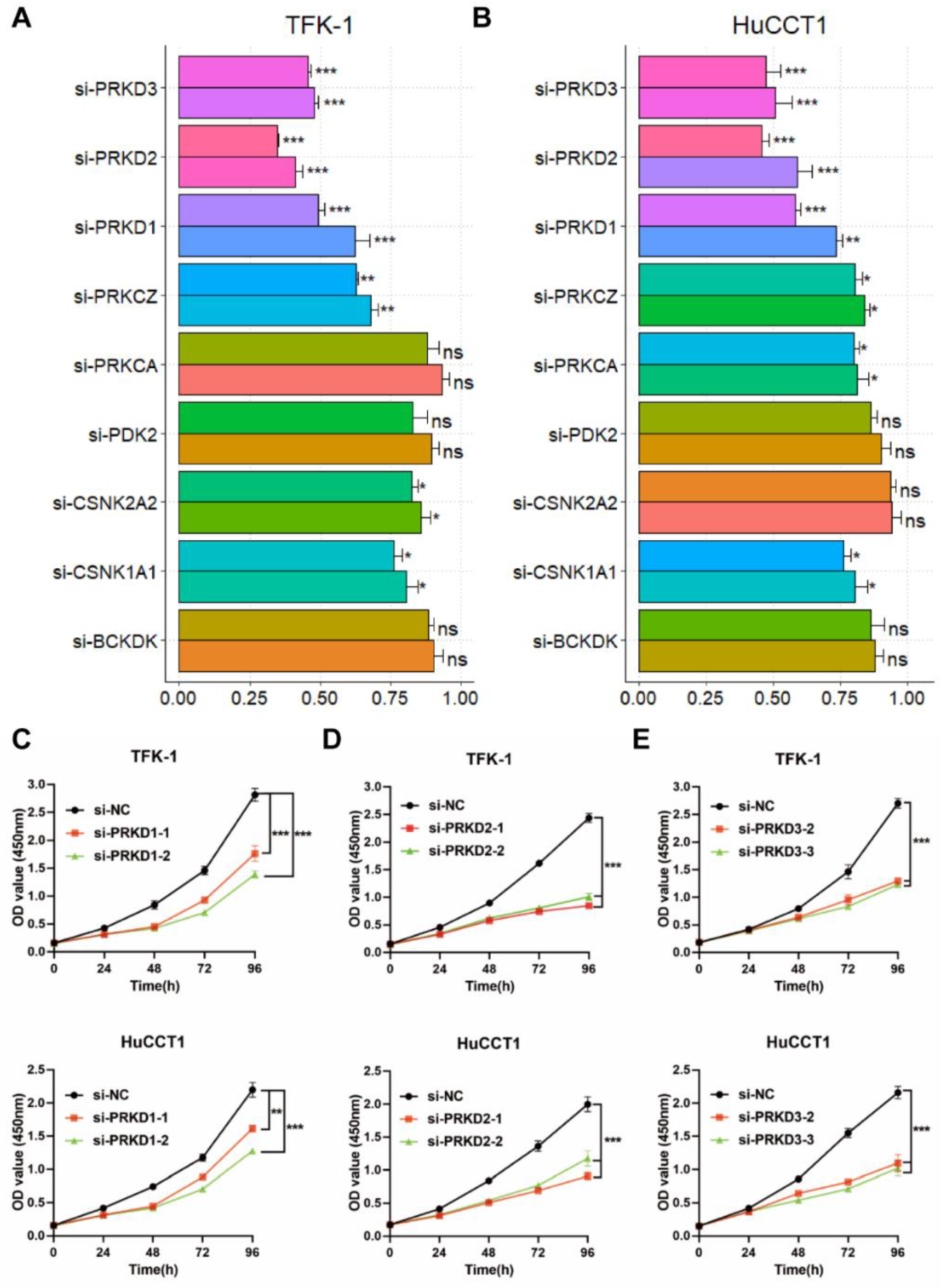
3.5. Screening of PK Candidates
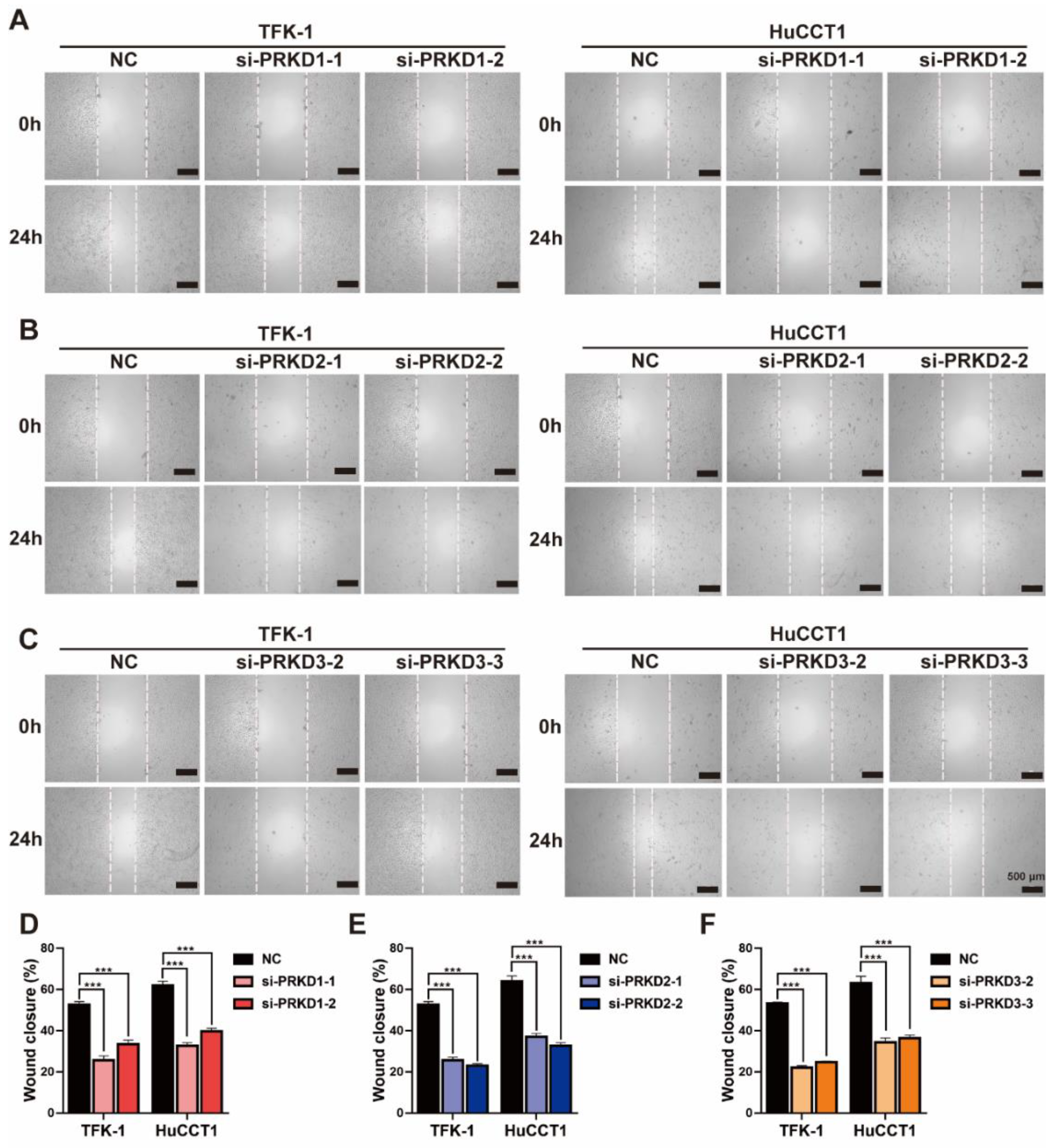
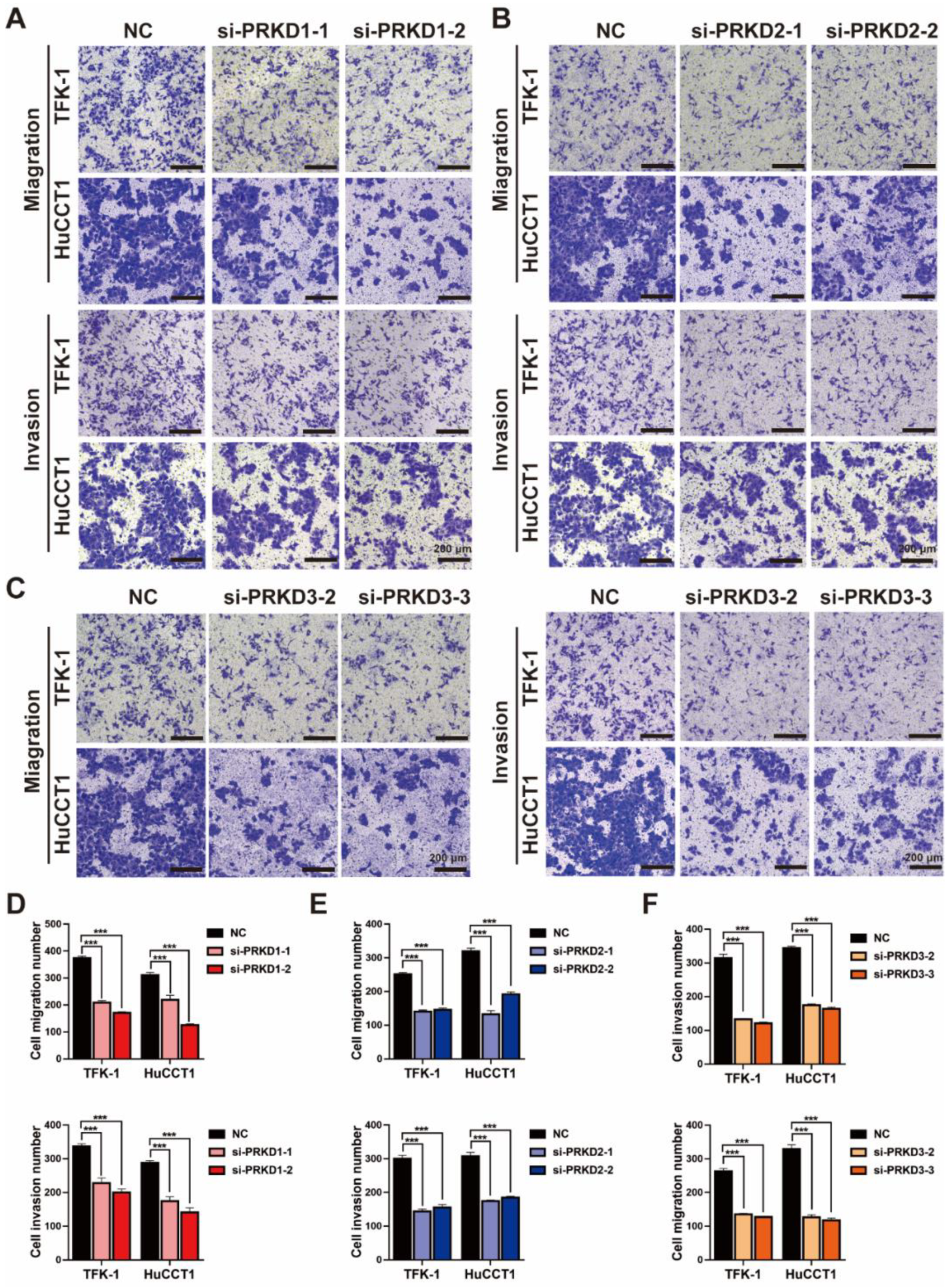
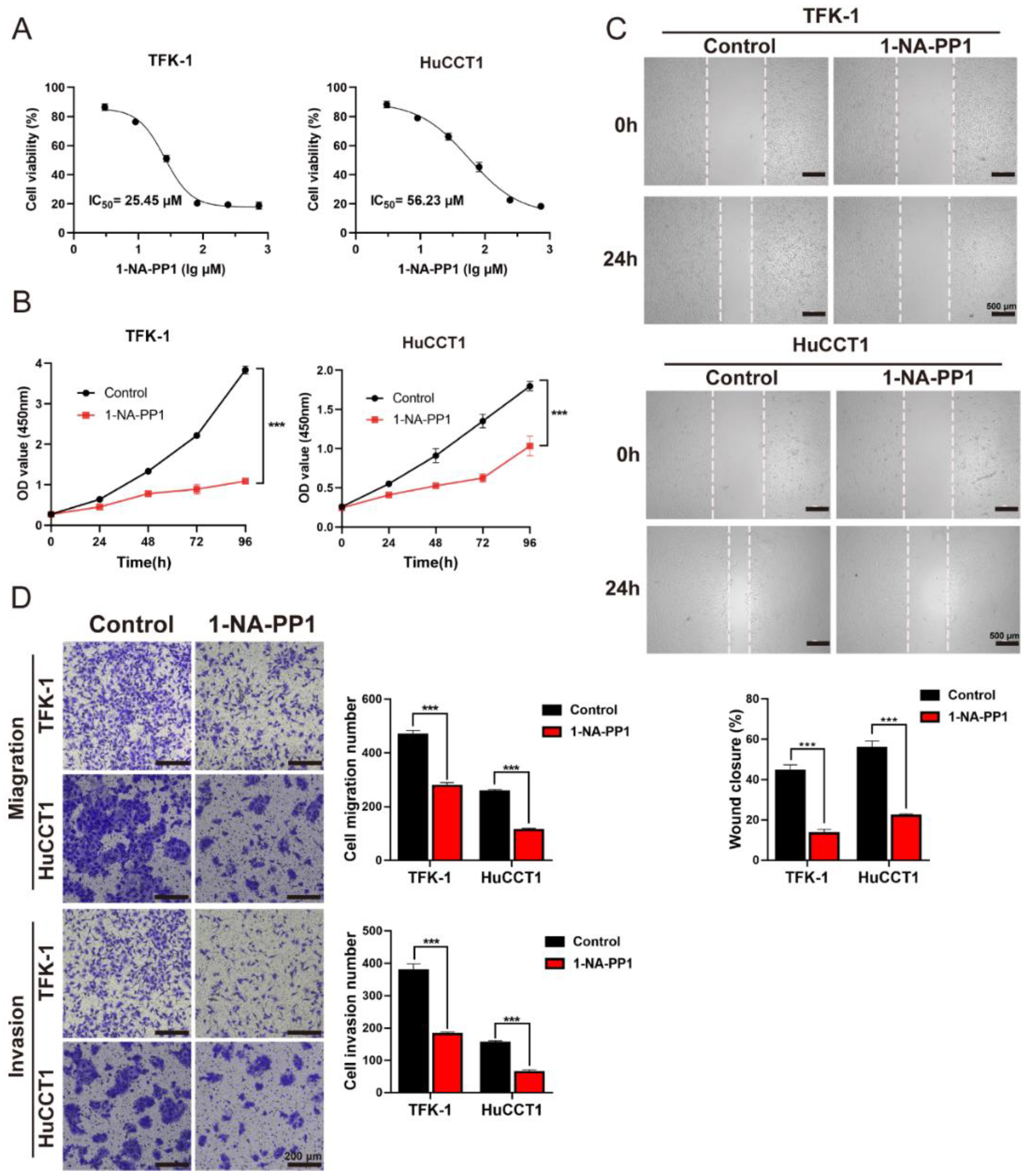
3.6. The Inhibition of PKDs Suppressed the Migration and Invasion of CCA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BRAF | Serine/threonine-protein kinase B-raf |
| CCA | Cholangiocarcinoma |
| CifPK | Comprehensive inference of functional protein kinases |
| CCK-8 | Cell Counting Kit-8 |
| DEPs | Differentially expressed proteins |
| DRPs | Differentially regulated p-sites |
| DPPs | Differentially phosphorylated proteins |
| EMT | Epithelial mesenchymal transition |
| eCCA | Extrahepatic cholangiocarcinoma |
| FGFR2 | Fibroblast growth factor receptor 2 |
| FDA | Food and Drug Administration |
| HCC | Hepatocellular carcinoma |
| IC50 | Half-maximal inhibitory concentration |
| iCCA | Intrahepatic cholangiocarcinoma |
| LC-MS/MS | Liquid chromatography with tandem mass spectrometry |
| mTOR | Mammalian target of rapamycin |
| NATs | Normal adjacent tissues |
| PKs | Protein kinases |
| PKD/PRKD | Protein kinase D |
| PI3K | Phosphoinositide 3-kinase |
| PRKCI | Atypical protein kinase C iota |
| RTK | Receptor tyrosine kinase |
| Sp1 | Specificity protein 1 |
| siRNAs | Small interfering RNAs |
| 1-NA-PP1 | 1-naphthyl PP1 |
References
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The Next Horizon in Mechanisms and Management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.; Olaizola, P.; Paiva, N.A.; Olaizola, I.; Agirre-Lizaso, A.; Landa, A.; Bujanda, L.; Perugorria, M.J.; Banales, J.M. Pathogenesis of Cholangiocarcinoma. Annu. Rev. Pathol. 2021, 16, 433–463. [Google Scholar] [CrossRef] [PubMed]
- Brindley, P.J.; Bachini, M.; Ilyas, S.I.; Khan, S.A.; Loukas, A.; Sirica, A.E.; Teh, B.T.; Wongkham, S.; Gores, G.J. Cholangiocarcinoma. Nat. Rev. Dis. Prim. 2021, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome Sequencing of Liver Fluke-Associated Cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef]
- Chan-On, W.; Nairismägi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome Sequencing Identifies Distinct Mutational Patterns in Liver Fluke-Related and Non-Infection-Related Bile Duct Cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic Spectra of Biliary Tract Cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Shi, L.; Liu, L.Y.; de la Cruz, F.F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. Tas-120 Overcomes Resistance to Atp-Competitive Fgfr Inhibitors in Patients with Fgfr2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for Previously Treated, Locally Advanced or Metastatic Cholangiocarcinoma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Silverman, I.M.; Hollebecque, A.; Friboulet, L.; Owens, S.; Newton, R.C.; Zhen, H.; Féliz, L.; Zecchetto, C.; Melisi, D.; Burn, T.C. Clinicogenomic Analysis of Fgfr2-Rearranged Cholangiocarcinoma Identifies Correlates of Response and Mechanisms of Resistance to Pemigatinib. Cancer Discov. 2021, 11, 326–339. [Google Scholar] [CrossRef]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative Molecular Analysis of Intrahepatic Cholangiocarcinoma Reveals 2 Classes That Have Different Outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Kam, A.E.; Masood, A.; Shroff, R.T. Current and Emerging Therapies for Advanced Biliary Tract Cancers. Lancet Gastroenterol. Hepatol. 2021, 6, 956–969. [Google Scholar] [CrossRef]
- Corti, F.; Nichetti, F.; Raimondi, A.; Niger, M.; Prinzi, N.; Torchio, M.; Tamborini, E.; Perrone, F.; Pruneri, G.; di Bartolomeo, M.; et al. Targeting the Pi3k/Akt/Mtor Pathway in Biliary Tract Cancers: A Review of Current Evidences and Future Perspectives. Cancer Treat. Rev. 2019, 72, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mahipal, A.; Kim, R. Targeted Therapy for Advanced or Metastatic Cholangiocarcinoma: Focus on the Clinical Potential of Infigratinib. Onco Targets Ther. 2021, 14, 5145–5160. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.L.; Deng, X.; Huang, F.; Tucker, M.; Crosby, K.; Rimkunas, V.; Wang, Y.; Deng, G.; Zhu, L.; Tan, Z.; et al. Survey of Tyrosine Kinase Signaling Reveals Ros Kinase Fusions in Human Cholangiocarcinoma. PLoS ONE 2011, 6, e15640. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with Braf V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Khorsandi, S.E.; Dokal, A.D.; Rajeeve, V.; Britton, D.J.; Illingworth, M.; Heaton, N.; Cutillas, P.R. Computational Analysis of Cholangiocarcinoma Phosphoproteomes Identifies Patient-Specific Drug Targets. Cancer Res. 2021, 81, 5765–5776. [Google Scholar] [CrossRef]
- Deng, M.; Ran, P.; Chen, L.; Wang, Y.; Yu, Z.; Cai, K.; Feng, J.; Qin, Z.; Yin, Y.; Tan, S.; et al. Proteogenomic Characterization of Cholangiocarcinoma. Hepatology 2022. [Google Scholar] [CrossRef]
- Horn, H.; Schoof, E.M.; Kim, J.; Robin, X.; Miller, M.L.; Diella, F.; Palma, A.; Cesareni, G.; Jensen, L.J.; Linding, R. Kinomexplorer: An Integrated Platform for Kinome Biology Studies. Nat. Methods 2014, 11, 603–604. [Google Scholar] [CrossRef]
- Song, C.; Ye, M.; Liu, Z.; Cheng, H.; Jiang, X.; Han, G.; Songyang, Z.; Tan, Y.; Wang, H.; Ren, J.; et al. Systematic Analysis of Protein Phosphorylation Networks from Phosphoproteomic Data. Mol. Cell. Proteom. 2012, 11, 1070–1083. [Google Scholar] [CrossRef]
- Wiredja, D.D.; Koyutürk, M.; Chance, M.R. The Ksea App: A Web-Based Tool for Kinase Activity Inference from Quantitative Phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Cox, J. The Maxquant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Uniprot: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef] [PubMed]
- Lin, S.; Wang, C.; Zhou, J.; Shi, Y.; Ruan, C.; Tu, Y.; Yao, L.; Peng, D.; Xue, Y. Epsd: A Well-Annotated Data Resource of Protein Phosphorylation Sites in Eukaryotes. Brief. Bioinform. 2021, 22, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.Y.; Lee, T.Y.; Kao, H.J.; Ma, C.T.; Lee, C.C.; Lin, T.H.; Chang, W.C.; Huang, H.D. Dbptm in 2019: Exploring Disease Association and Cross-Talk of Post-Translational Modifications. Nucleic Acids Res. 2019, 47, D298–D308. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. Phosphositeplus, 2014: Mutations, Ptms and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.Elm: A Database of Phosphorylation Sites—Update 2011. Nucleic Acids Res. 2011, 39, D261–D267. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (Msigdb) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-Substrate Enrichment Analysis Provides Insights into the Heterogeneity of Signaling Pathway Activation in Leukemia Cells. Sci. Signal. 2013, 6, rs6. [Google Scholar] [CrossRef]
- Deng, Z.; Li, X.; Shi, Y.; Lu, Y.; Yao, W.; Wang, J. A Novel Autophagy-Related Incrnas Signature for Prognostic Prediction and Clinical Value in Patients with Pancreatic Cancer. Front. Cell Dev. Biol. 2020, 8, 606817. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stöckli, J.; Yang, J.Y.; James, D.E. Dynamic Adipocyte Phosphoproteome Reveals That Akt Directly Regulates Mtorc2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, J.; Guedj, N.; Clapéron, A.; Ho-Bouldoires, T.H.N.; Paradis, V.; Fouassier, L. Epithelial-Mesenchymal Transition in Cholangiocarcinoma: From Clinical Evidence to Regulatory Networks. J. Hepatol. 2017, 66, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Lertsuwan, J.; Lertsuwan, K.; Sawasdichai, A.; Tasnawijitwong, N.; Lee, K.Y.; Kitchen, P.; Afford, S.; Gaston, K.; Jayaraman, P.S.; Satayavivad, J. Cx-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a Ck2-Independent Mechanism. Cancers 2018, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Sanmai, S.; Proungvitaya, T.; Limpaiboon, T.; Chua-On, D.; Seubwai, W.; Roytrakul, S.; Wongkham, S.; Wongkham, C.; Somintara, O.; Sangkhamanon, S.; et al. Serum Pyruvate Dehydrogenase Kinase as a Prognostic Marker for Cholangiocarcinoma. Oncol. Lett. 2019, 17, 5275–5282. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Peng, D.; Zhou, J.; Lin, S.; Wang, C.; Ning, W.; Xu, H.; Deng, W.; Xue, Y. Iekpd 2.0: An Update with Rich Annotations for Eukaryotic Protein Kinases, Protein Phosphatases and Proteins Containing Phosphoprotein-Binding Domains. Nucleic Acids Res. 2019, 47, D344–D350. [Google Scholar] [CrossRef]
- Tandon, M.; Johnson, J.; Li, Z.; Xu, S.; Wipf, P.; Wang, Q.J. New Pyrazolopyrimidine Inhibitors of Protein Kinase D as Potent Anticancer Agents for Prostate Cancer Cells. PLoS ONE 2013, 8, e75601. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Qian, Y.; Yao, W.; Yang, T.; Yang, Y.; Liu, Y.; Shen, Q.; Zhang, J.; Qi, W.; Wang, J. Apkc-Ι/P-Sp1/Snail Signaling Induces Epithelial-Mesenchymal Transition and Immunosuppression in Cholangiocarcinoma. Hepatology 2017, 66, 1165–1182. [Google Scholar] [CrossRef]
- Dong, L.; Lu, D.; Chen, R.; Lin, Y.; Zhu, H.; Zhang, Z.; Cai, S.; Cui, P.; Song, G.; Rao, D.; et al. Proteogenomic Characterization Identifies Clinically Relevant Subgroups of Intrahepatic Cholangiocarcinoma. Cancer Cell 2022, 40, 70–87 e15. [Google Scholar] [CrossRef]
- Fu, Y.; Rubin, C.S. Protein Kinase D: Coupling Extracellular Stimuli to the Regulation of Cell Physiology. EMBO Rep. 2011, 12, 785–796. [Google Scholar] [CrossRef]
- Bernhart, E.; Damm, S.; Wintersperger, A.; DeVaney, T.; Zimmer, A.; Raynham, T.; Ireson, C.; Sattler, W. Protein Kinase D2 Regulates Migration and Invasion of U87mg Glioblastoma Cells in vitro. Exp. Cell Res. 2013, 319, 2037–2048. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Y.; Wang, J.; Zhang, S.; Zhao, Y.; Ren, H.; Chu, Y.; Feng, L.; Wang, C. Protein Kinase D3 Promotes Gastric Cancer Development through P65/6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 Activation of Glycolysis. Exp. Cell Res. 2019, 380, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, H.; Zhou, Y.; Ma, X.; Xu, J.; Yu, Z.; Chen, L. The Oncogenic Role of Protein Kinase D3 in Cancer. J. Cancer 2021, 12, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The Pride Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Li, X.; Zhao, K.; Shi, Y.; Deng, Z.; Yao, W.; Wang, J. Proteomic and Phosphoproteomic Profiling Reveals the Oncogenic Role of Protein Kinase D Family Kinases in Cholangiocarcinoma. Cells 2022, 11, 3088. https://doi.org/10.3390/cells11193088
Lu Y, Li X, Zhao K, Shi Y, Deng Z, Yao W, Wang J. Proteomic and Phosphoproteomic Profiling Reveals the Oncogenic Role of Protein Kinase D Family Kinases in Cholangiocarcinoma. Cells. 2022; 11(19):3088. https://doi.org/10.3390/cells11193088
Chicago/Turabian StyleLu, Yun, Xiangyu Li, Kai Zhao, Yuanxin Shi, Zhengdong Deng, Wei Yao, and Jianming Wang. 2022. "Proteomic and Phosphoproteomic Profiling Reveals the Oncogenic Role of Protein Kinase D Family Kinases in Cholangiocarcinoma" Cells 11, no. 19: 3088. https://doi.org/10.3390/cells11193088