
Treatment of Marmoset Intracerebral Hemorrhage with Humanized Anti-HMGB1 mAb
 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
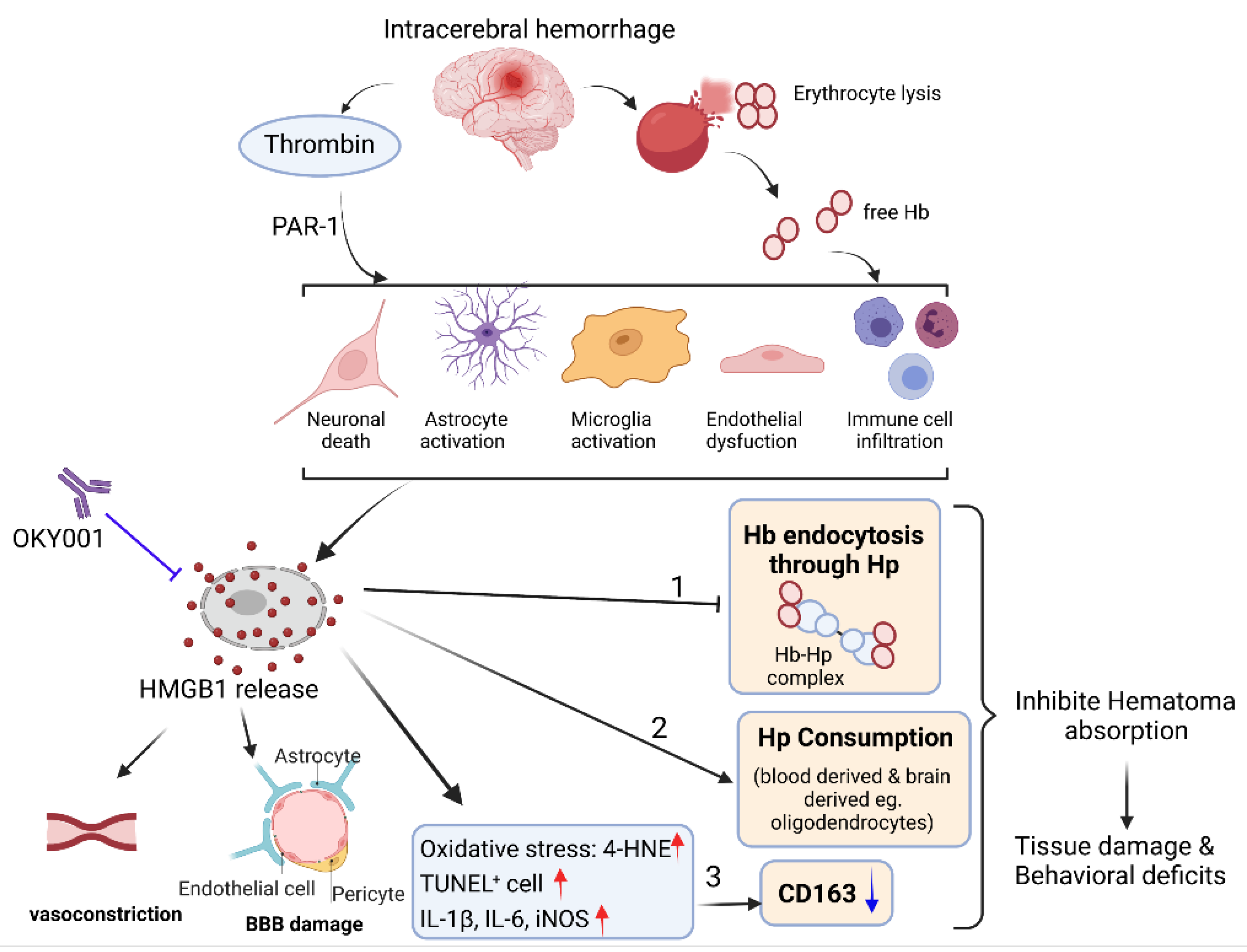
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Production of Humanized Anti-HMGB1 Monoclonal Antibody
2.3. Western Blot Analysis
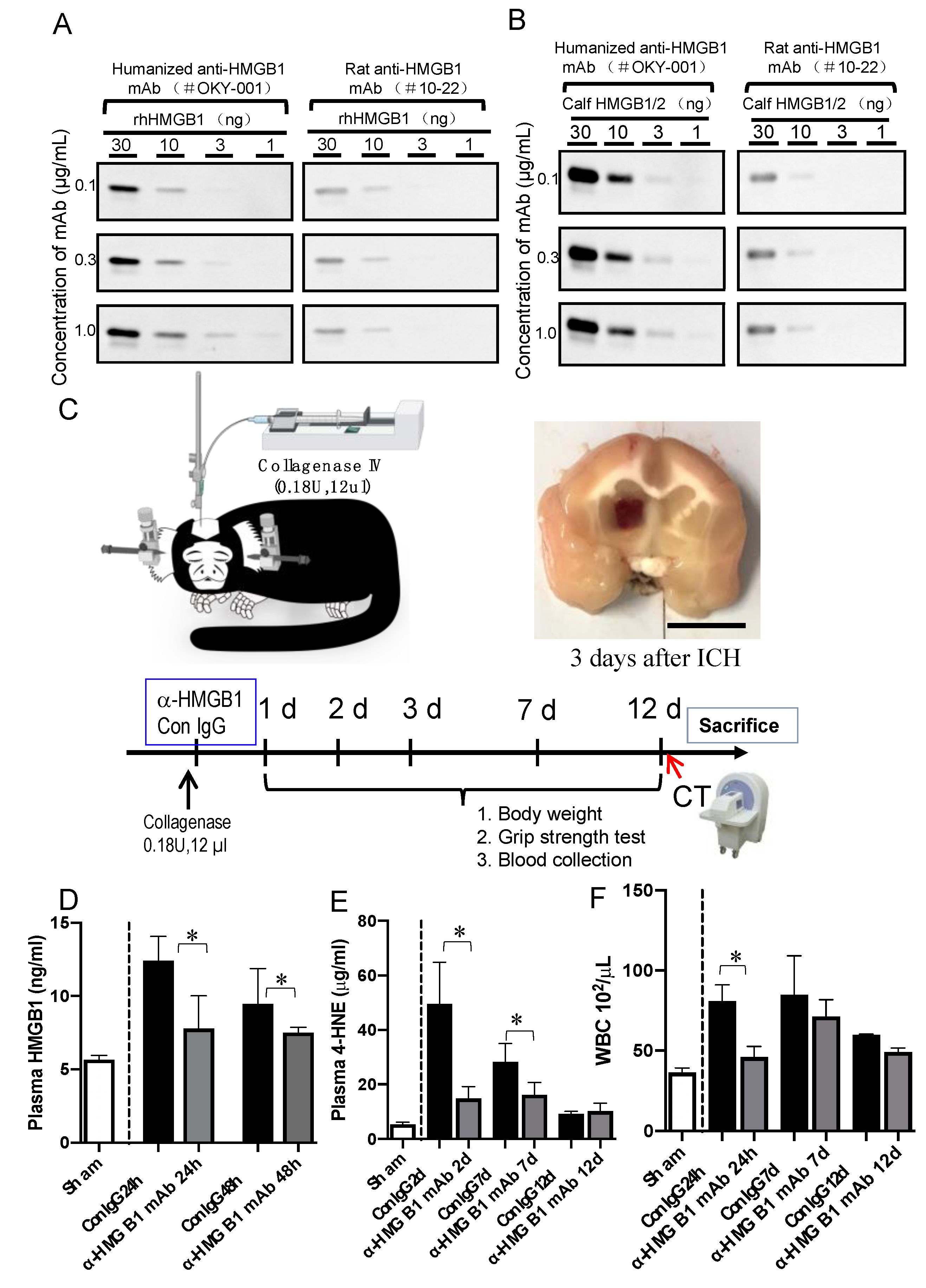
2.4. ICH Induction and the Treatment of Marmosets
2.5. Immunohistochemistry Staining
2.6. Enzyme-Linked Immunosorbent Assay of HMGB1 and 4-HNE Adduct
2.7. TUNEL Staining
2.8. Prussian Blue Staining
2.9. Real-Time PCR
2.10. Computed Tomographic (CT) Examination and CT Sata Analysis
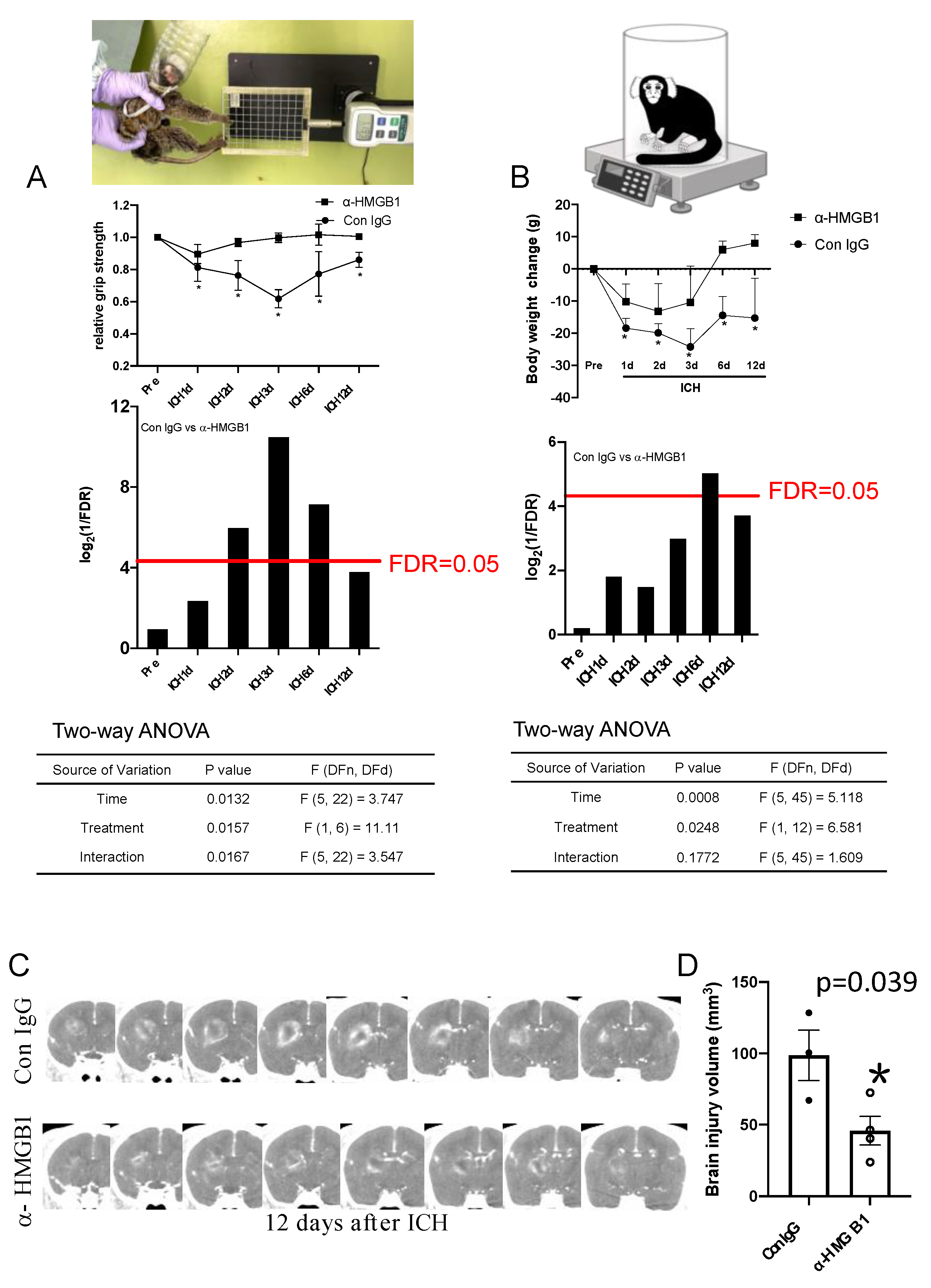
2.11. Evaluation of Neurological Function and Body Weight
2.12. Cell Preparation and Flow Cytometry
2.13. Statistical Analysis
3. Results
3.1. Characterization of Humanized Anti-HMGB1 mAb (OKY-001)
3.2. Effect of Humanized Anti-HMGB1 mAb on HMGB1 Levels in the Plasma after ICH
3.3. Effect of Humanized Anti-HMGB1 mAb on 4-hydroxynonenal (4-HNE) Adduct Levels in the Plasma after ICH
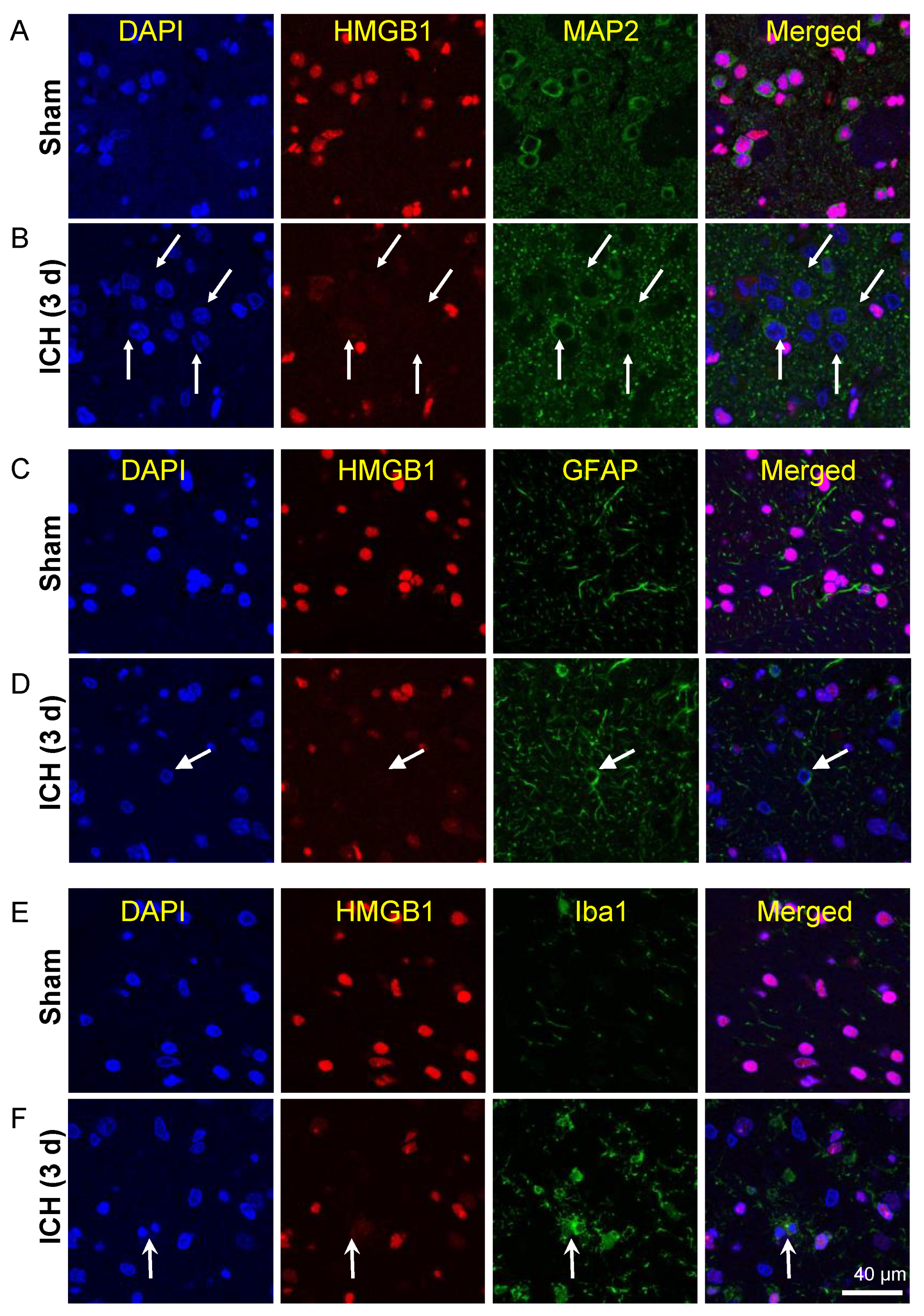
3.4. Release of HMGB1 from Neurons, Astrocytes, and Microglia
3.5. Effects of Humanized Anti-HMGB1 mAb on Brain Injury
3.6. Anti-HMGB1 mAb Improved Neurological Deficits and Ameliorated Body Weight Loss after ICH
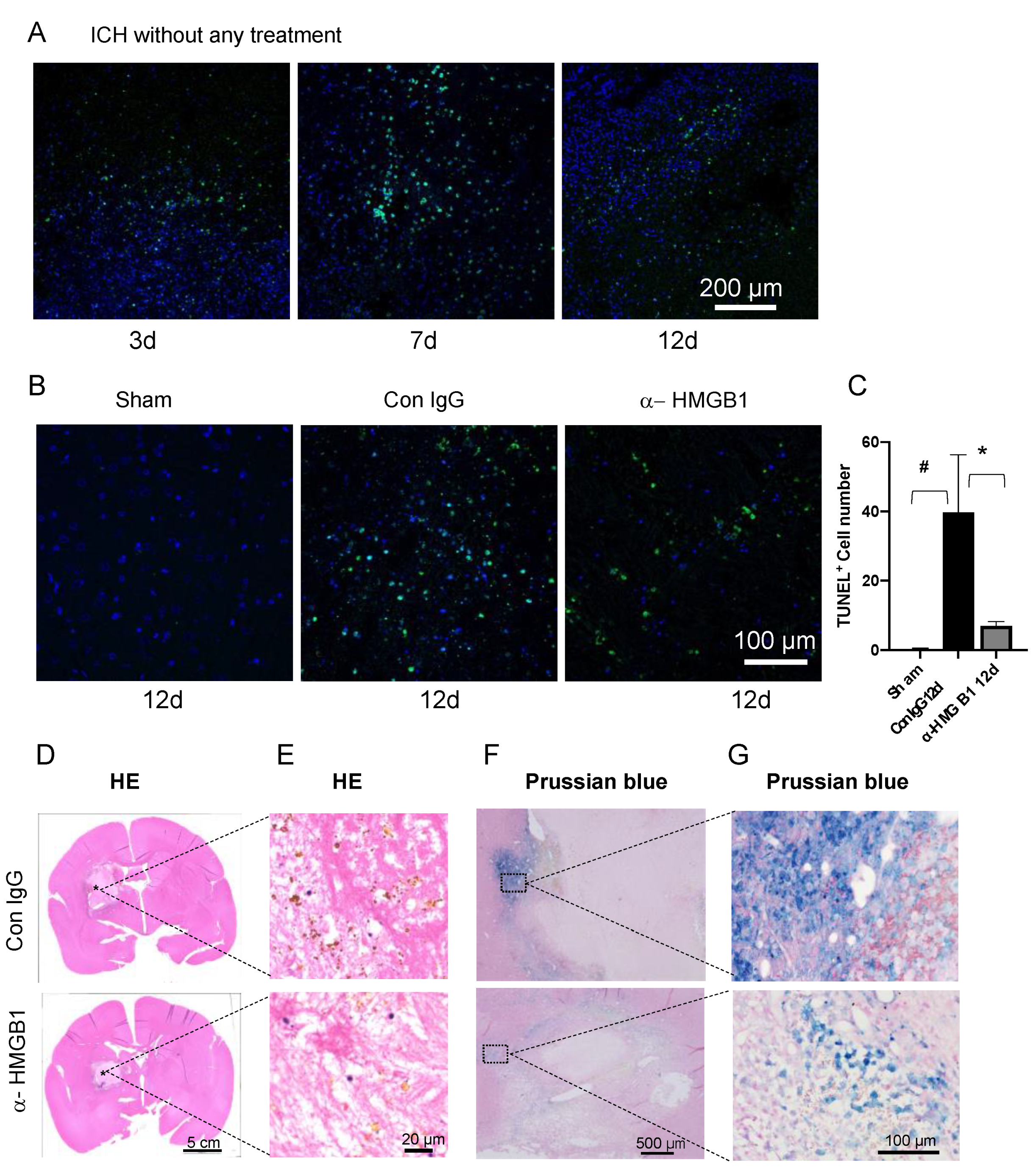
3.7. Anti-HMGB1 mAb Treatment Induced a Decrease in TUNEL-Positive Cells
3.8. Histological Studies on the Effects of Humanized Anti-HMGB1 mAb
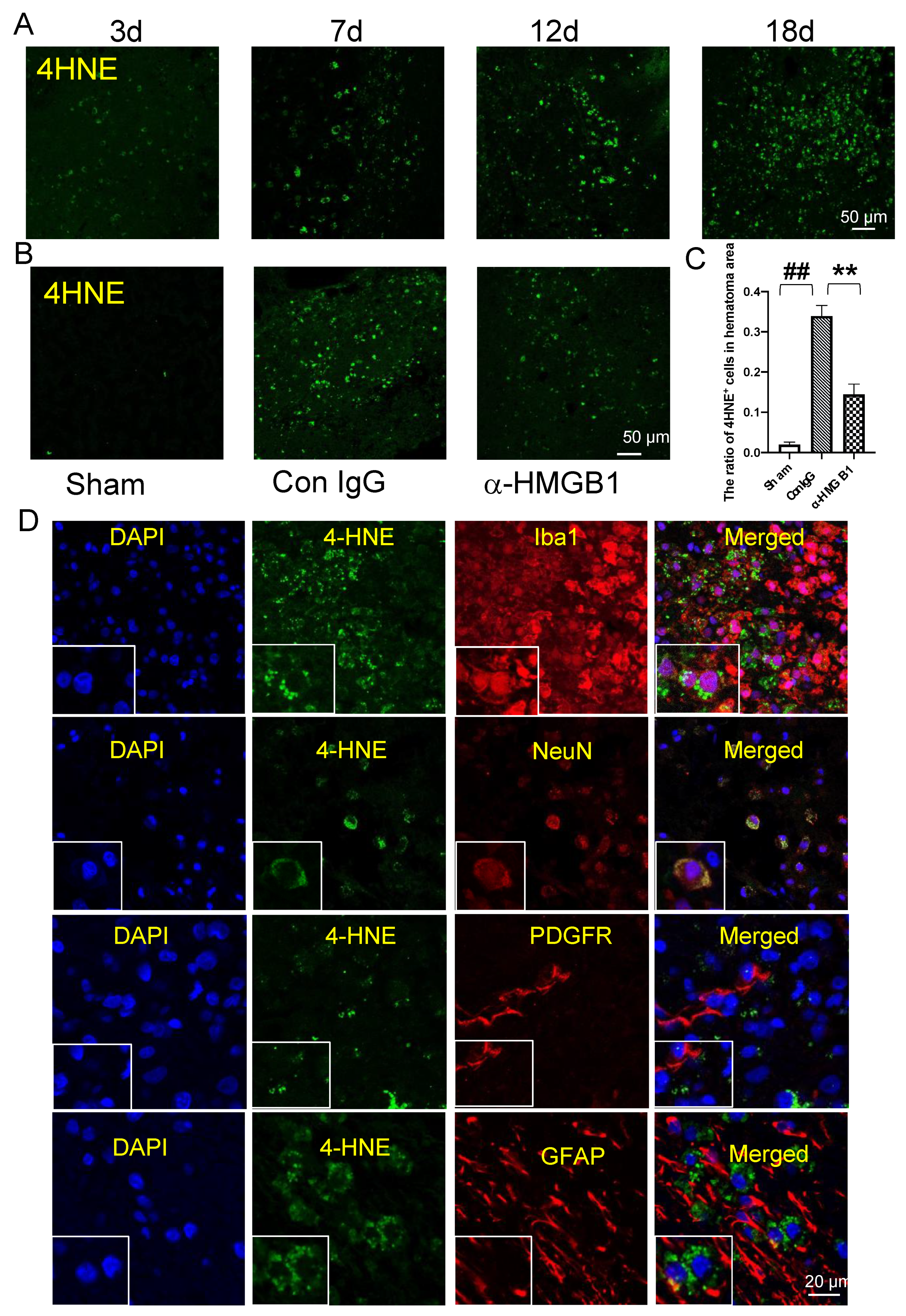
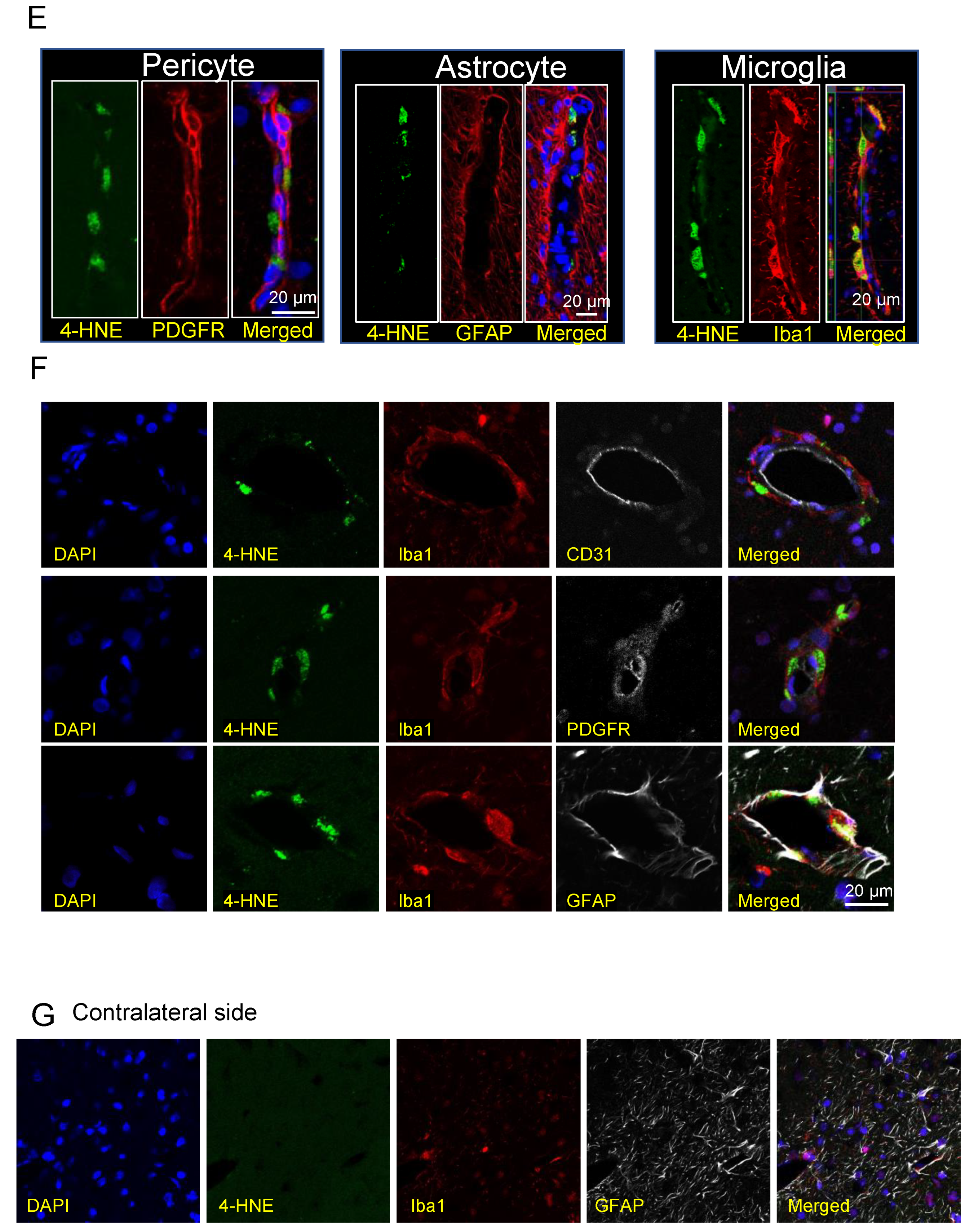
3.9. 4-HNE Induction and Localization in Hematoma and Peri-Hematoma Regions after ICH
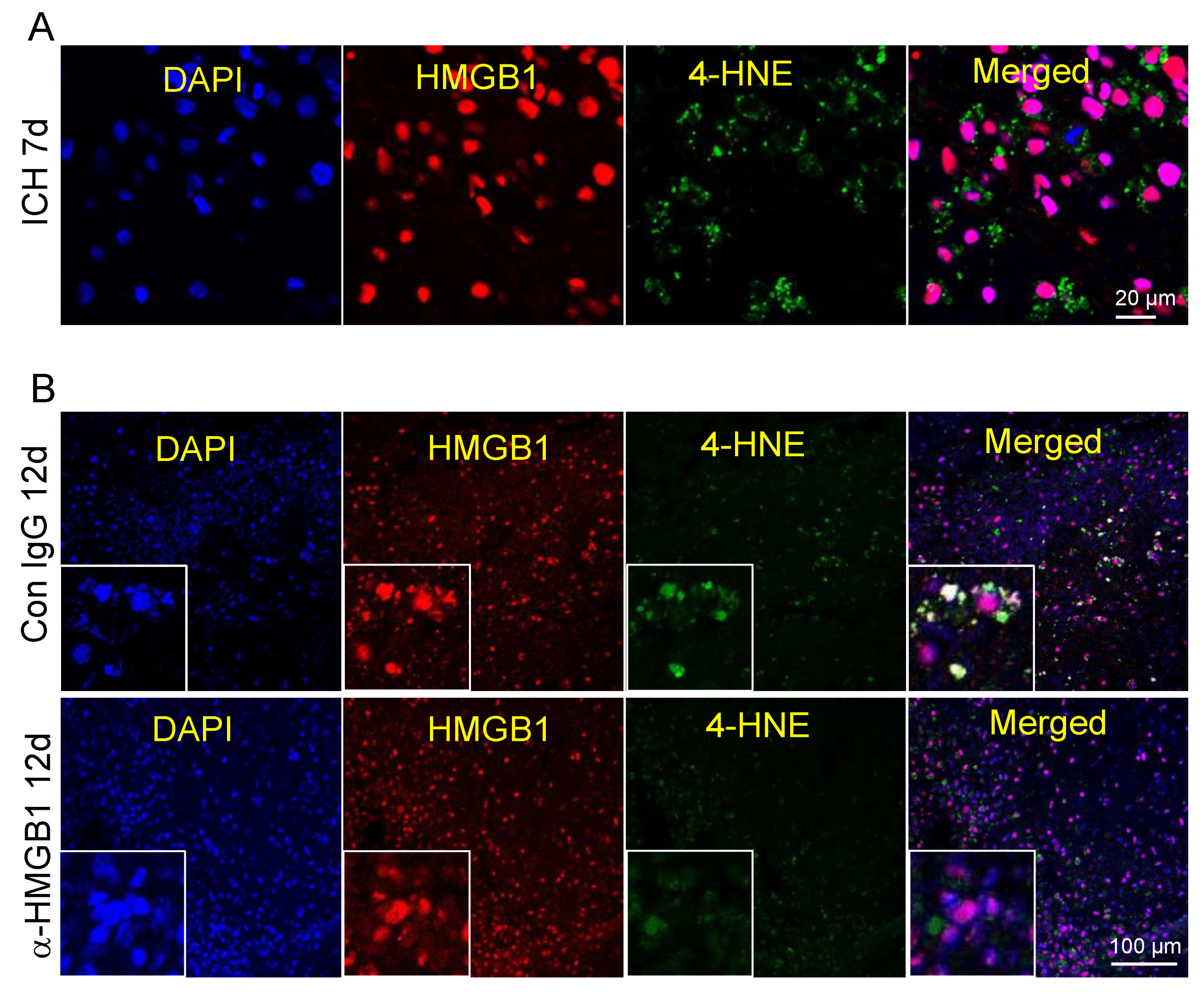
3.10. Co-Localization of HMGB1 and 4-HNE
3.11. Effects of Humanized Anti-HMGB1 on the Expression of Inflammation-Related Molecules
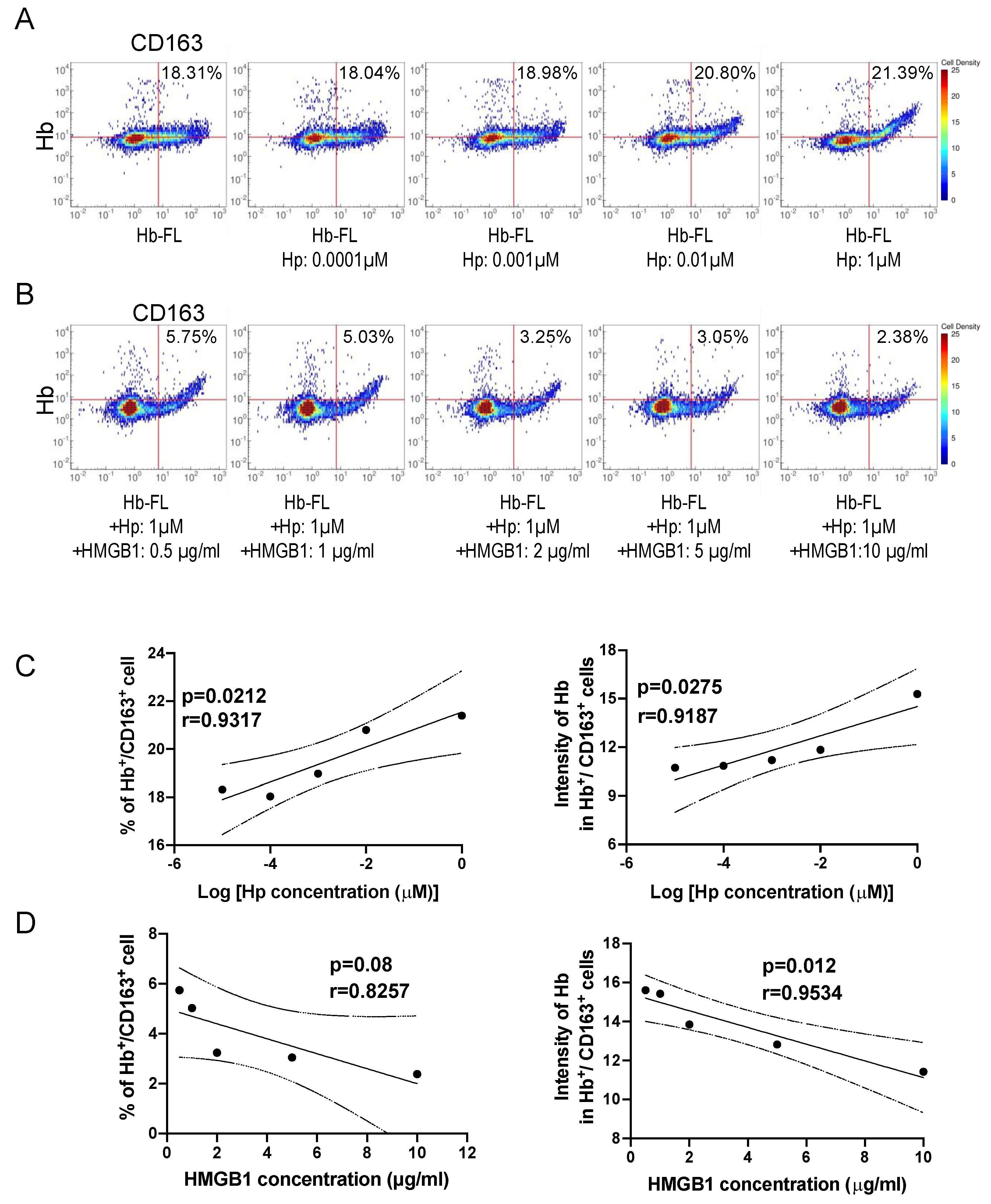
3.12. Inhibition of Haptoglobin-dependent Uptake of Hb by Recombinant HMGB1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ziai, W.C.; Carhuapoma, J.R. Intracerebral Hemorrhage. Continuum 2018, 24, 1603–1622. [Google Scholar] [CrossRef]
- Hemphill III, J.C.; Greenberg, S.M.; Anderson, C.S.; Becker, K.; Bendok, B.R.; Cushman, M.; Fung, G.L.; Goldstein, J.N.; Macdonald, R.L.; Mitchell, P.H. Guidelines for the management of spontaneous intracerebral hemorrhage: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2015, 46, 2032–2060. [Google Scholar] [CrossRef] [PubMed]
- Thabet, A.M.; Kottapally, M.; Hemphill, J.C., 3rd. Management of intracerebral hemorrhage. Handb. Clin. Neurol. 2017, 140, 177–194. [Google Scholar] [CrossRef]
- Hanley, D.F.; Thompson, R.E.; Rosenblum, M.; Yenokyan, G.; Lane, K.; McBee, N.; Mayo, S.W.; Bistran-Hall, A.J.; Gandhi, D.; Mould, W.A.; et al. Efficacy and safety of minimally invasive surgery with thrombolysis in intracerebral haemorrhage evacuation (MISTIE III): A randomised, controlled, open-label, blinded endpoint phase 3 trial. Lancet 2019, 393, 1021–1032. [Google Scholar] [CrossRef]
- Selim, M.; Foster, L.D.; Moy, C.S.; Xi, G.; Hill, M.D.; Morgenstern, L.B.; Greenberg, S.M.; James, M.L.; Singh, V.; Clark, W.M.; et al. Deferoxamine mesylate in patients with intracerebral haemorrhage (i-DEF): A multicentre, randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2019, 18, 428–438. [Google Scholar] [CrossRef]
- Naidech, A.M.; Maas, M.B.; Levasseur-Franklin, K.E.; Liotta, E.M.; Guth, J.C.; Berman, M.; Rosenow, J.M.; Lindholm, P.F.; Bendok, B.R.; Prabhakaran, S.; et al. Desmopressin improves platelet activity in acute intracerebral hemorrhage. Stroke 2014, 45, 2451–2453. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ward, M.F.; Sama, A.E.; Wang, H. Extracellular HMGB1 as a proinflammatory cytokine. J. Interf. Cytokine Res. 2004, 24, 329–333. [Google Scholar] [CrossRef]
- Nishibori, M.; Mori, S.; Takahashi, H.K. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J. Pharmacol. Sci. 2019, 140, 94–101. [Google Scholar] [CrossRef]
- Nishibori, M.; Wang, D.; Ousaka, D.; Wake, H. High Mobility Group Box-1 and Blood-Brain Barrier Disruption. Cells 2020, 9, 2650. [Google Scholar] [CrossRef]
- Wang, D.; Liu, K.; Fukuyasu, Y.; Teshigawara, K.; Fu, L.; Wake, H.; Ohtsuka, A.; Nishibori, M. HMGB1 Translocation in Neurons after Ischemic Insult: Subcellular Localization in Mitochondria and Peroxisomes. Cells 2020, 9, 643. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, K.; Wake, H.; Teshigawara, K.; Mori, S.; Nishibori, M. Anti-high mobility group box-1 (HMGB1) antibody inhibits hemorrhage-induced brain injury and improved neurological deficits in rats. Sci. Rep. 2017, 7, 46243. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 3904–3916. [Google Scholar] [CrossRef]
- Okuma, Y.; Liu, K.; Wake, H.; Zhang, J.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Otani, N.; Tomura, S.; et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann. Neurol. 2012, 72, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Liu, K.; Agari, T.; Yasuhara, T.; Morimoto, J.; Okazaki, M.; Takeuchi, H.; Toyoshima, A.; Sasada, S.; Shinko, A.; et al. Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson’s disease. Exp. Neurol. 2016, 275 Pt 1, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Morioka, N.; Abe, H.; Zhang, F.F.; Hisaoka-Nakashima, K.; Liu, K.; Nishibori, M.; Nakata, Y. Neuropathic pain in rats with a partial sciatic nerve ligation is alleviated by intravenous injection of monoclonal antibody to high mobility group box-1. PLoS ONE 2013, 8, e73640. [Google Scholar] [CrossRef]
- Fu, L.; Liu, K.; Wake, H.; Teshigawara, K.; Yoshino, T.; Takahashi, H.; Mori, S.; Nishibori, M. Therapeutic effects of anti-HMGB1 monoclonal antibody on pilocarpine-induced status epilepticus in mice. Sci. Rep. 2017, 7, 1179. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.A.; Keep, R.F.; Hua, Y.; Xi, G. Hematoma clearance as a therapeutic target in intracerebral hemorrhage: From macro to micro. J. Cereb. Blood Flow Metab. 2018, 38, 741–745. [Google Scholar] [CrossRef]
- Keep, R.F.; Zhou, N.; Xiang, J.; Andjelkovic, A.V.; Hua, Y.; Xi, G. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS 2014, 11, 18. [Google Scholar] [CrossRef]
- Mracsko, E.; Veltkamp, R. Neuroinflammation after intracerebral hemorrhage. Front. Cell. Neurosci. 2014, 8, 388. [Google Scholar] [CrossRef]
- Lin, T.; Sammy, F.; Yang, H.; Thundivalappil, S.; Hellman, J.; Tracey, K.J.; Warren, H.S. Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J. Immunol. 2012, 189, 2017–2022. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Kawahara, K.; Nakamura, T.; Yamada, S.; Nakamura, T.; Abeyama, K.; Hashiguchi, T.; Maruyama, I. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J. Thromb. Haemost. 2007, 5, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Katsuki, H.; Fukutomi, C.; Takahashi, M.; Motomura, M.; Fukunaga, M.; Matsuoka, Y.; Isohama, Y.; Izumi, Y.; Kume, T.; et al. HMGB1 inhibitor glycyrrhizin attenuates intracerebral hemorrhage-induced injury in rats. Neuropharmacology 2011, 61, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Lin, S.; Zhang, C.; Tao, W.; Dong, W.; Hao, Z.; Liu, M.; Wu, B. High-mobility group box1 protein promotes neuroinflammation after intracerebral hemorrhage in rats. Neuroscience 2013, 228, 190–199. [Google Scholar] [CrossRef]
- Su, X.; Wang, H.; Zhao, J.; Pan, H.; Mao, L. Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expression and TLR4/NF-kappaB pathway after traumatic brain injury in the rat. Mediat. Inflamm. 2011, 2011, 807142. [Google Scholar] [CrossRef]
- Katsuki, H. Exploring neuroprotective drug therapies for intracerebral hemorrhage. J. Pharmacol. Sci. 2010, 114, 366–378. [Google Scholar] [CrossRef]
- Lee, W.-C.; Wong, H.-Y.; Chai, Y.-Y.; Shi, C.-W.; Amino, N.; Kikuchi, S.; Huang, S.-H. Lipid peroxidation dysregulation in ischemic stroke: Plasma 4-HNE as a potential biomarker? Biochem. Biophys. Res. Commun. 2012, 425, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Arbab, A.S.; Yocum, G.T.; Kalish, H.; Jordan, E.K.; Anderson, S.A.; Khakoo, A.Y.; Read, E.J.; Frank, J.A. Efficient magnetic cell labeling with protamine sulfate complexed to ferumoxides for cellular MRI. Blood 2004, 104, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Sultana, R.; Cenini, G.; Di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteom. Clin. Appl. 2009, 3, 682–693. [Google Scholar] [CrossRef]
- Liu, Q.; Raina, A.K.; Smith, M.A.; Sayre, L.M.; Perry, G. Hydroxynonenal, toxic carbonyls, and Alzheimer disease. Mol. Asp. Med. 2003, 24, 305–313. [Google Scholar] [CrossRef]
- Schaer, D.J.; Schaer, C.A.; Buehler, P.W.; Boykins, R.A.; Schoedon, G.; Alayash, A.I.; Schaffner, A. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood 2006, 107, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.W.; Ridley, R.M. Assessment of functional impairment following permanent middle cerebral artery occlusion in a non-human primate species. Neurodegeneration 1996, 5, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wang, H.; Zhu, L.; Zhao, J.; Pan, H.; Ji, X. Ethyl pyruvate ameliorates intracerebral hemorrhage-induced brain injury through anti-cell death and anti-inflammatory mechanisms. Neuroscience 2013, 245, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. HMGB1-Mediated Neuroinflammatory Responses in Brain Injuries: Potential Mechanisms and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 4609. [Google Scholar] [CrossRef]
- Okuma, Y.; Liu, K.; Wake, H.; Liu, R.; Nishimura, Y.; Hui, Z.; Teshigawara, K.; Haruma, J.; Yamamoto, Y.; Yamamoto, H.; et al. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology 2014, 85, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Kao, K.K.; Fink, M.P. The biochemical basis for the anti-inflammatory and cytoprotective actions of ethyl pyruvate and related compounds. Biochem. Pharmacol. 2010, 80, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Park, E.J.; Kim, J.H.; Park, S.W.; Kim, H.J.; Chang, K.C. Ethyl pyruvate inhibits the acetylation and release of HMGB1 via effects on SIRT1/STAT signaling in LPS-activated RAW264.7 cells and peritoneal macrophages. Int. Immunopharmacol. 2016, 41, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, H.; Ohta, T.; Yoshikawa, Y.; Funatsu, N.; Yamamoto, M.; Someda, K. Cerebral vasospasm and hemoglobins--clinical and experimental studies. Neurol. Med.-Chir. 1979, 19, 61–71. [Google Scholar] [CrossRef]
- Wu, H.; Wu, T.; Li, M.; Wang, J. Efficacy of the lipid-soluble iron chelator 2,2’-dipyridyl against hemorrhagic brain injury. Neurobiol. Dis. 2012, 45, 388–394. [Google Scholar] [CrossRef]
- Gaasch, J.A.; Lockman, P.R.; Geldenhuys, W.J.; Allen, D.D.; Van der Schyf, C.J. Brain iron toxicity: Differential responses of astrocytes, neurons, and endothelial cells. Neurochem. Res. 2007, 32, 1196–1208. [Google Scholar] [CrossRef]
- Wu, H.; Wu, T.; Xu, X.; Wang, J.; Wang, J. Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2011, 31, 1243–1250. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R. Quantitative analysis of cell death and ferritin expression in response to cortical iron: Implications for hypoxia-ischemia and stroke. Brain Res. 2001, 907, 175–187. [Google Scholar] [CrossRef]
- Veltkamp, R.; Purrucker, J. Management of Spontaneous Intracerebral Hemorrhage. Curr. Neurol. Neurosci. Rep. 2017, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Xu, L.; Zhang, H.; Liu, Y.; Lu, X.; Chen, G.; Tang, H.; Wu, J. A Review of Hematoma Components Clearance Mechanism after Subarachnoid Hemorrhage. Front. Neurosci. 2020, 14, 685. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Song, W.; Gonzales, N.; Grotta, J.C.; Aronowski, J. Hematoma resolution as a target for intracerebral hemorrhage treatment: Role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann. Neurol. 2007, 61, 352–362. [Google Scholar] [CrossRef]
- Zhao, X.; Song, S.; Sun, G.; Strong, R.; Zhang, J.; Grotta, J.C.; Aronowski, J. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J. Neurosci. 2009, 29, 15819–15827. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Levine, Y.A.; Gunasekaran, M.K.; Wang, Y.; Addorisio, M.; Zhu, S.; Li, W.; Li, J.; de Kleijn, D.P.; et al. Identification of CD163 as an antiinflammatory receptor for HMGB1-haptoglobin complexes. JCI Insight 2016, 1, e85375. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, M.; Jing, C.; Keep, R.F.; Hua, Y.; Xi, G. Multinucleated Giant Cells in Experimental Intracerebral Hemorrhage. Transl. Stroke Res. 2020, 11, 1095–1102. [Google Scholar] [CrossRef]
- Duan, X.; Wen, Z.; Shen, H.; Shen, M.; Chen, G. Intracerebral Hemorrhage, Oxidative Stress, and Antioxidant Therapy. Oxidative Med. Cell. Longev. 2016, 2016, 1203285. [Google Scholar] [CrossRef] [PubMed]
- Zarkovic, N. Roles and Functions of ROS and RNS in Cellular Physiology and Pathology. Cells 2020, 9, 767. [Google Scholar] [CrossRef]
- Žarković, N.; Žarković, K.; Schaur, R.J.; Štolc, S.; Schlag, G.; Redl, H.; Waeg, G.; Borović, S.; Lončarić, I.; Jurić, G.; et al. 4-Hydroxynonenal as a second messenger of free radicals and growth modifying factor. Life Sci. 1999, 65, 1901–1904. [Google Scholar] [CrossRef]
- Allegra, M.; Restivo, I.; Fucarino, A.; Pitruzzella, A.; Vasto, S.; Livrea, M.A.; Tesoriere, L.; Attanzio, A. Proeryptotic Activity of 4-Hydroxynonenal: A New Potential Physiopathological Role for Lipid Peroxidation Products. Biomolecules 2020, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- De Herdt, V.; Dumont, F.; Henon, H.; Derambure, P.; Vonck, K.; Leys, D.; Cordonnier, C. Early seizures in intracerebral hemorrhage: Incidence, associated factors, and outcome. Neurology 2011, 77, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Rattani, A.; Anderson, C.D.; Ayres, A.M.; Gurol, E.M.; Greenberg, S.M.; Rosand, J.; Viswanathan, A. Delayed seizures after intracerebral haemorrhage. Brain 2016, 139, 2694–2705. [Google Scholar] [CrossRef] [PubMed]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Nishibori, M. The Role of High Mobility Group Box-1 in Epileptogenesis. Acta Med. Okayama 2019, 73, 383–386. [Google Scholar] [CrossRef]
- Henderson, S.J.; Weitz, J.I.; Kim, P.Y. Fibrinolysis: Strategies to enhance the treatment of acute ischemic stroke. J. Thromb. Haemost. 2018, 16, 1932–1940. [Google Scholar] [CrossRef]
- Kim, J.S. tPA Helpers in the Treatment of Acute Ischemic Stroke: Are They Ready for Clinical Use? J. Stroke 2019, 21, 160–174. [Google Scholar] [CrossRef]
- Zhang, J.; Takahashi, H.K.; Liu, K.; Wake, H.; Liu, R.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Mori, S.; et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 2011, 42, 1420–1428. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Ousaka, D.; Qiao, H.; Wang, Z.; Zhao, K.; Gao, S.; Liu, K.; Teshigawara, K.; Takada, K.; Nishibori, M. Treatment of Marmoset Intracerebral Hemorrhage with Humanized Anti-HMGB1 mAb. Cells 2022, 11, 2970. https://doi.org/10.3390/cells11192970
Wang D, Ousaka D, Qiao H, Wang Z, Zhao K, Gao S, Liu K, Teshigawara K, Takada K, Nishibori M. Treatment of Marmoset Intracerebral Hemorrhage with Humanized Anti-HMGB1 mAb. Cells. 2022; 11(19):2970. https://doi.org/10.3390/cells11192970
Chicago/Turabian StyleWang, Dengli, Daiki Ousaka, Handong Qiao, Ziyi Wang, Kun Zhao, Shangze Gao, Keyue Liu, Kiyoshi Teshigawara, Kenzo Takada, and Masahiro Nishibori. 2022. "Treatment of Marmoset Intracerebral Hemorrhage with Humanized Anti-HMGB1 mAb" Cells 11, no. 19: 2970. https://doi.org/10.3390/cells11192970