
Whole-Transcriptome Sequencing Analyses of Nuclear Antixoxidant-1 in Endothelial Cells: Role in Inflammation and Atherosclerosis
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals
Atherosclerotic Lesion Analysis
2.2. Cell Culture and Reagents
2.2.1. siRNA and Adenovirus Transfection
2.2.2. Immunofluorescence Analysis
2.2.3. Nuclear/Cytosolic Fractionation Assay and Immunoblotting
2.2.4. Monoclonal Atox1 Antibody Production
2.2.5. Chromatin Immunoprecipitation (ChIP) and Sequencing
2.2.6. ChIP Sequencing Analysis
2.2.7. RNA Sequencing
2.2.8. RNA-Seq Analysis
2.2.9. Quantitative Real-Time PCR
2.2.10. Monocyte Adhesion Assay
2.2.11. ROS Measurement
2.3. Statistical Analysis
3. Results
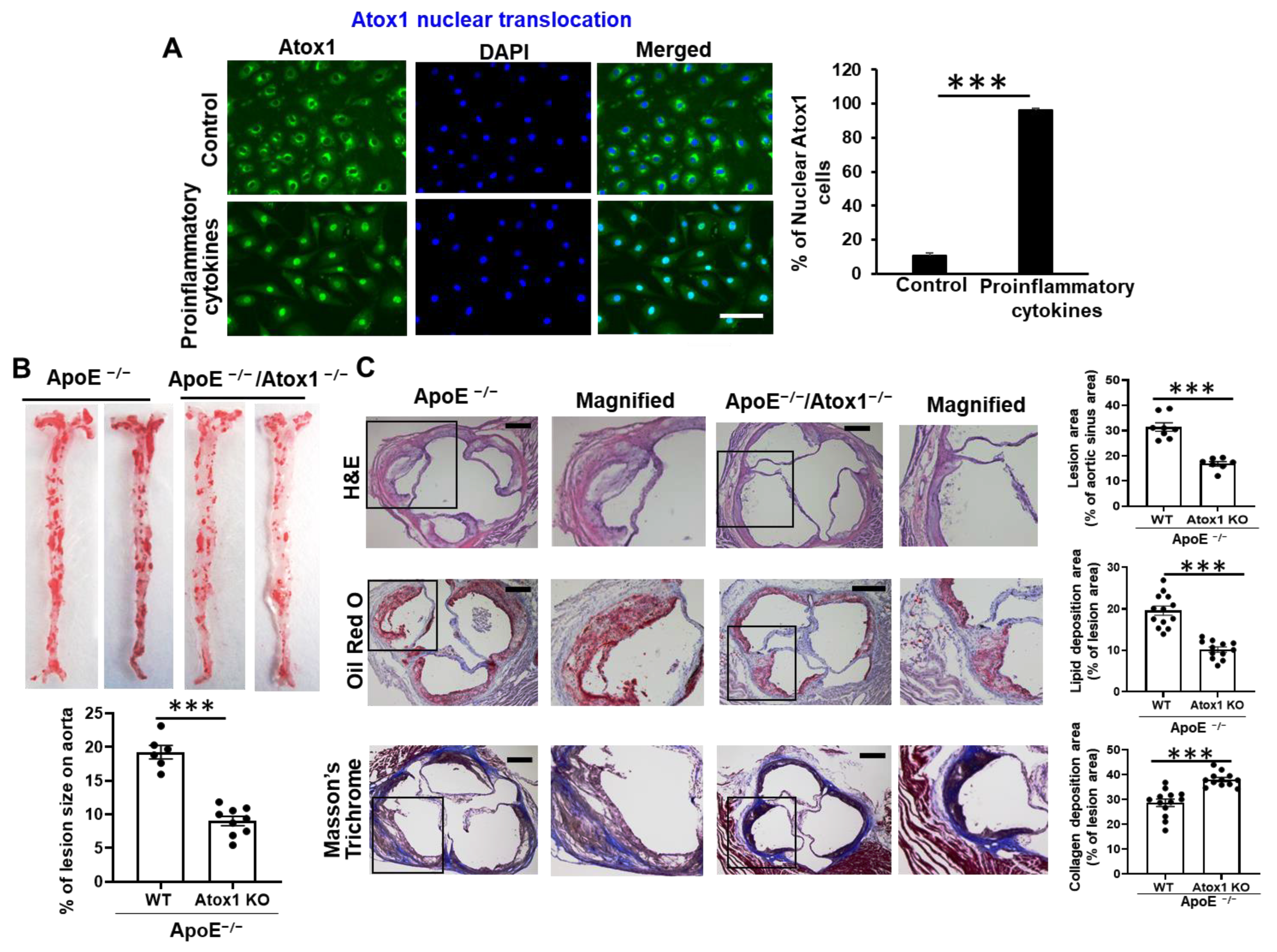
3.1. Role of Atox1 in Inflammatory Disease
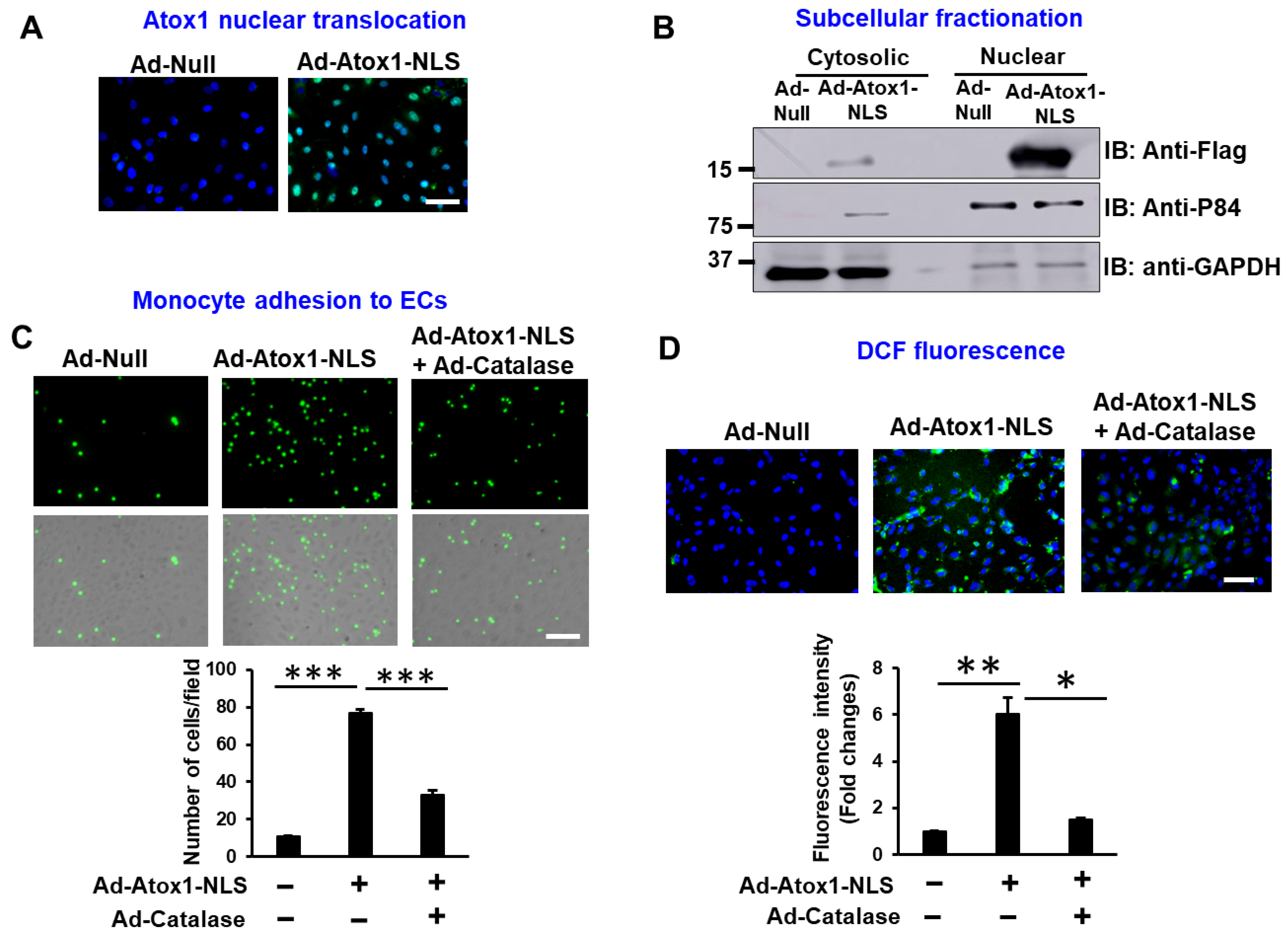
3.2. Nuclear Atox1 Increases Inflammatory Responses and ROS Production in ECs
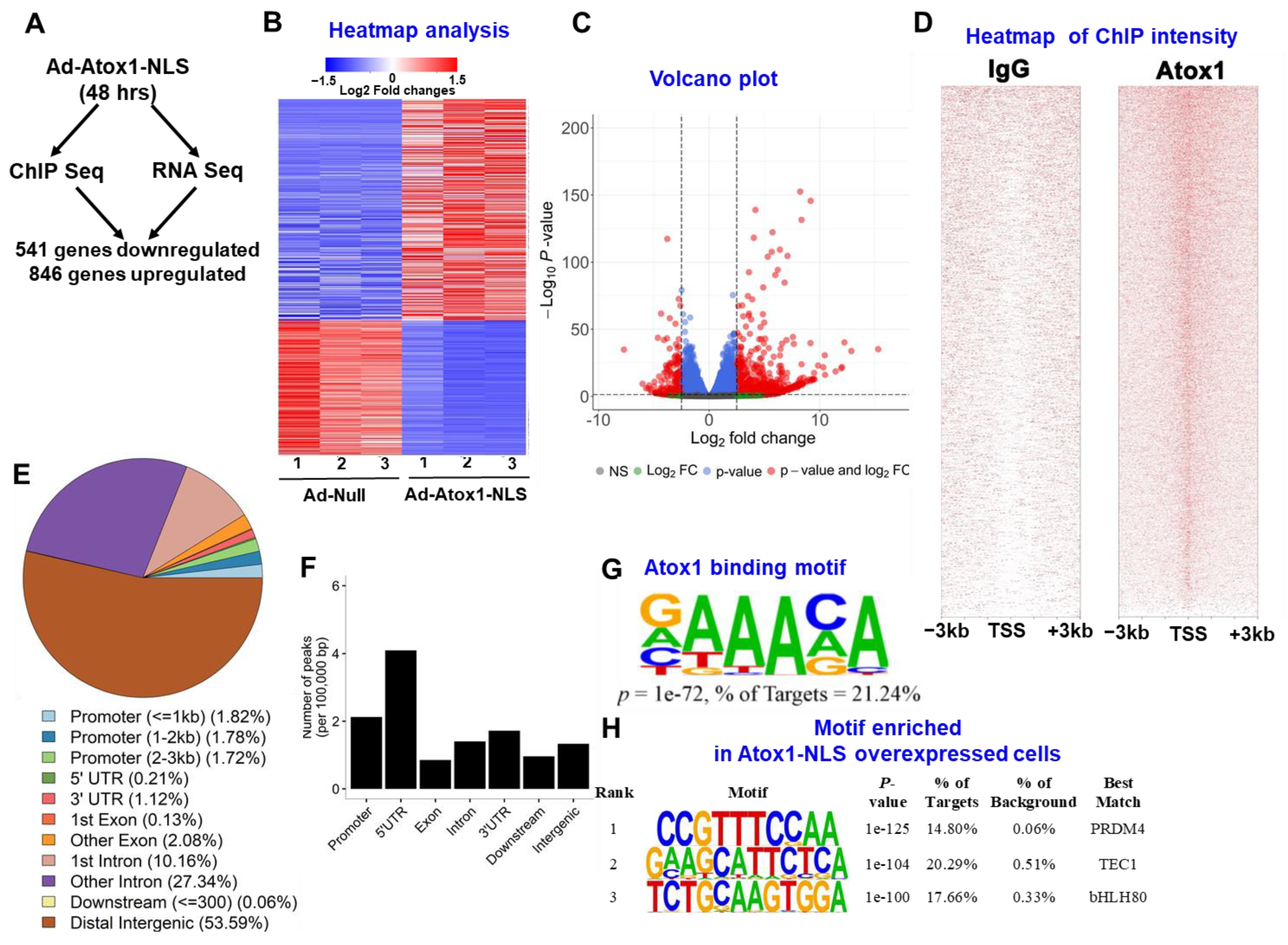
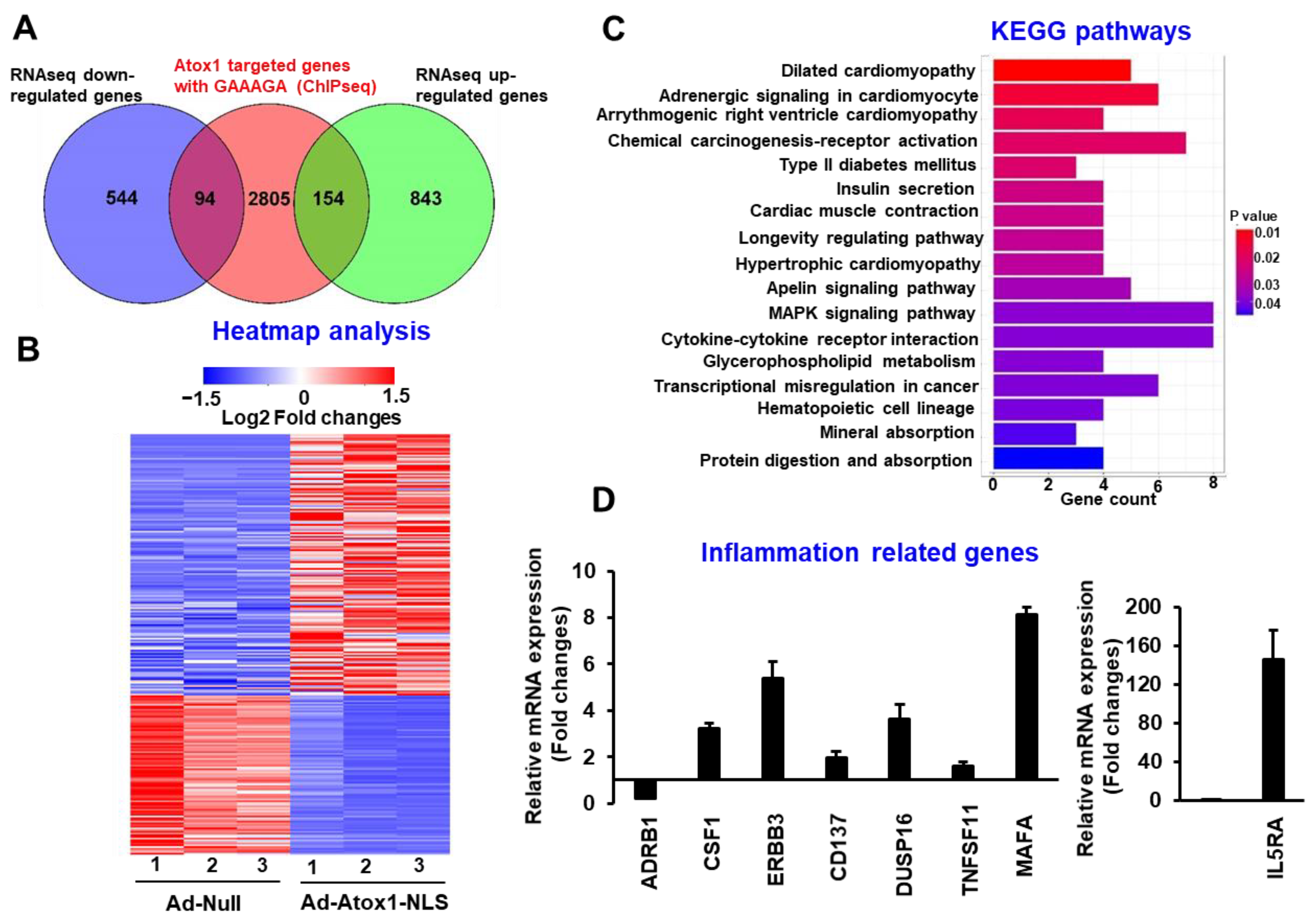
3.3. Atox1 Gene Binding and Gene Expression Signatures
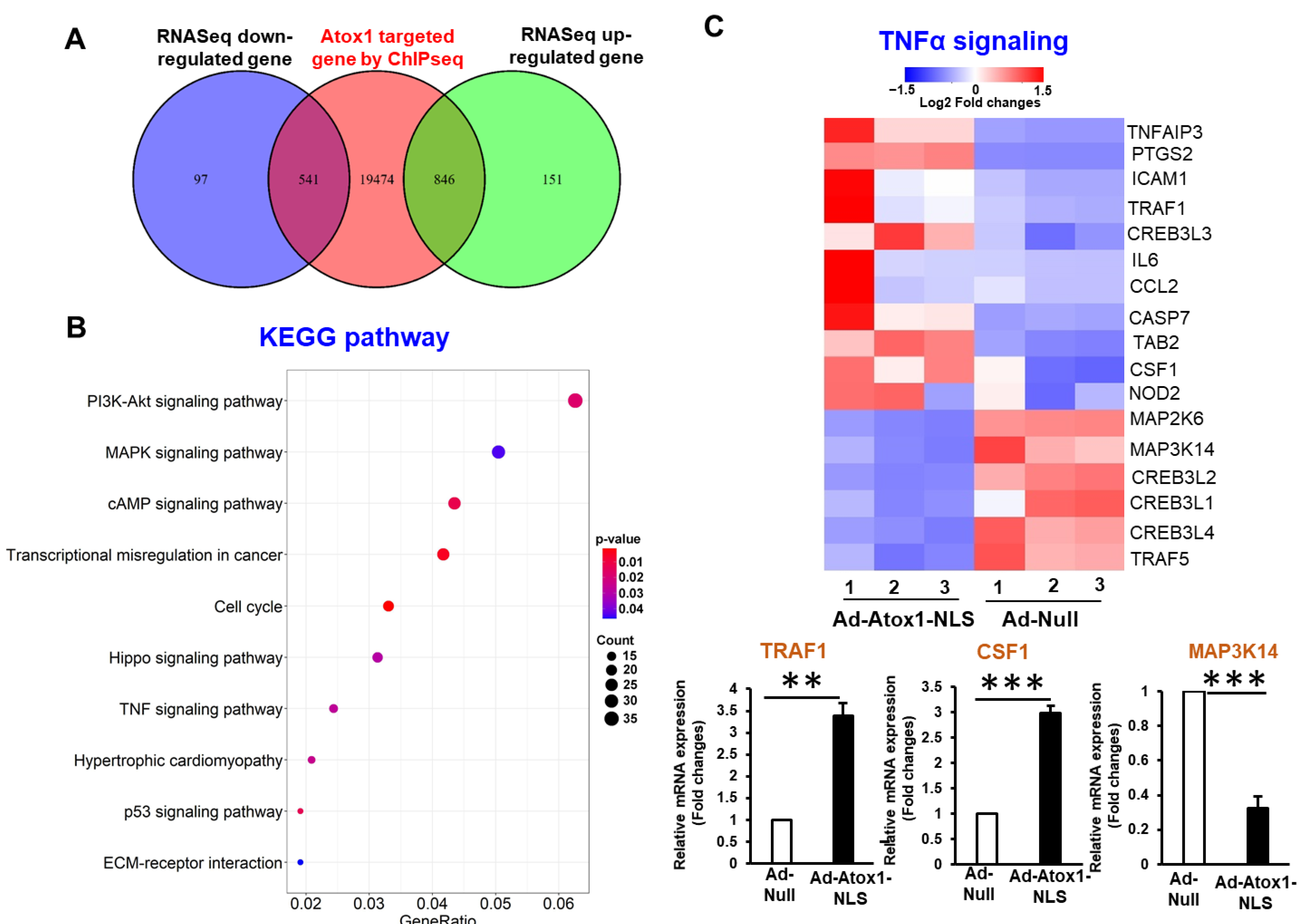
3.4. Analysis of Nuclear Atox1-Regulated Genes
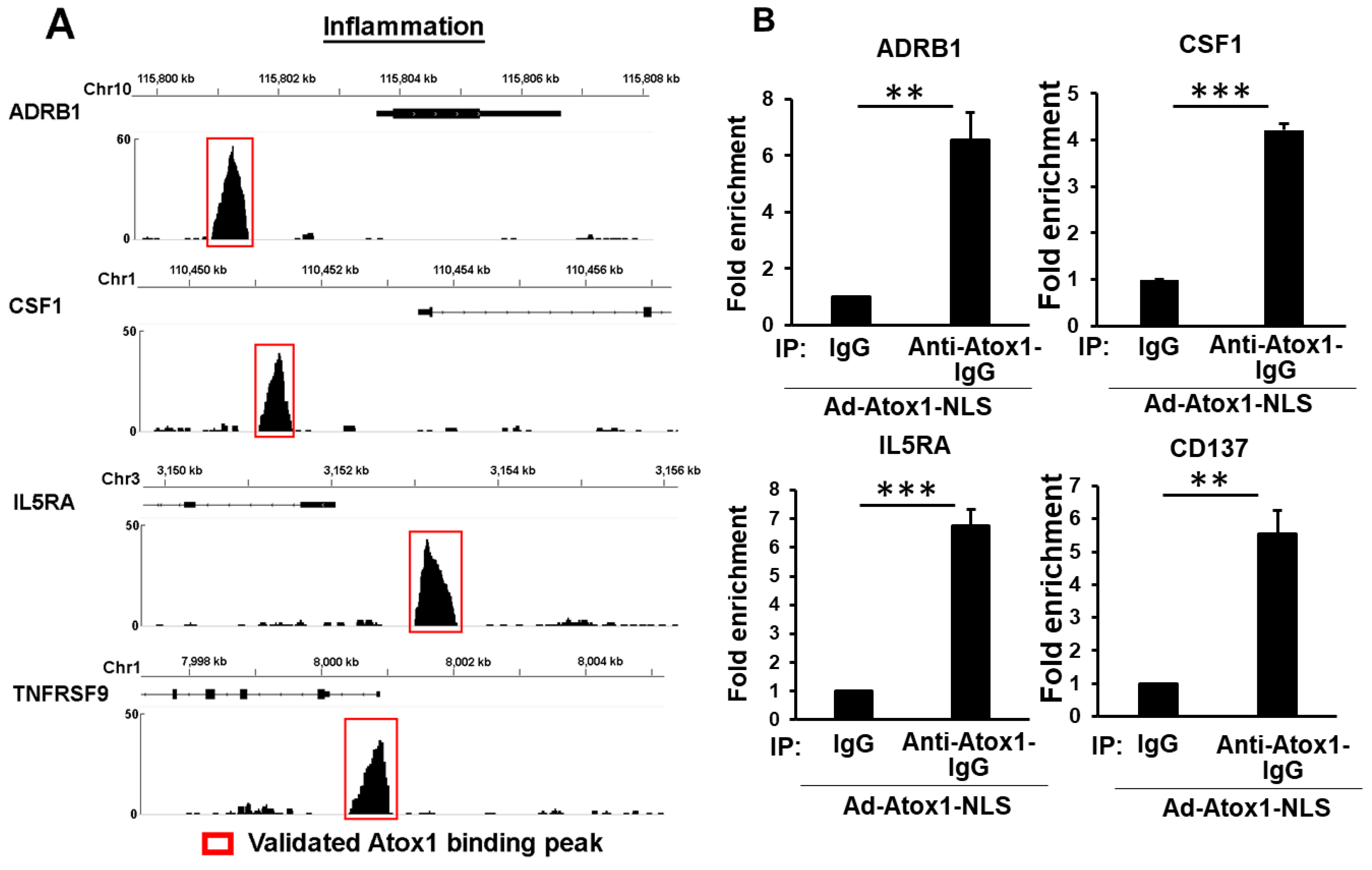
3.5. Genome-Wide Identification of Direct Atox1 Target Genes
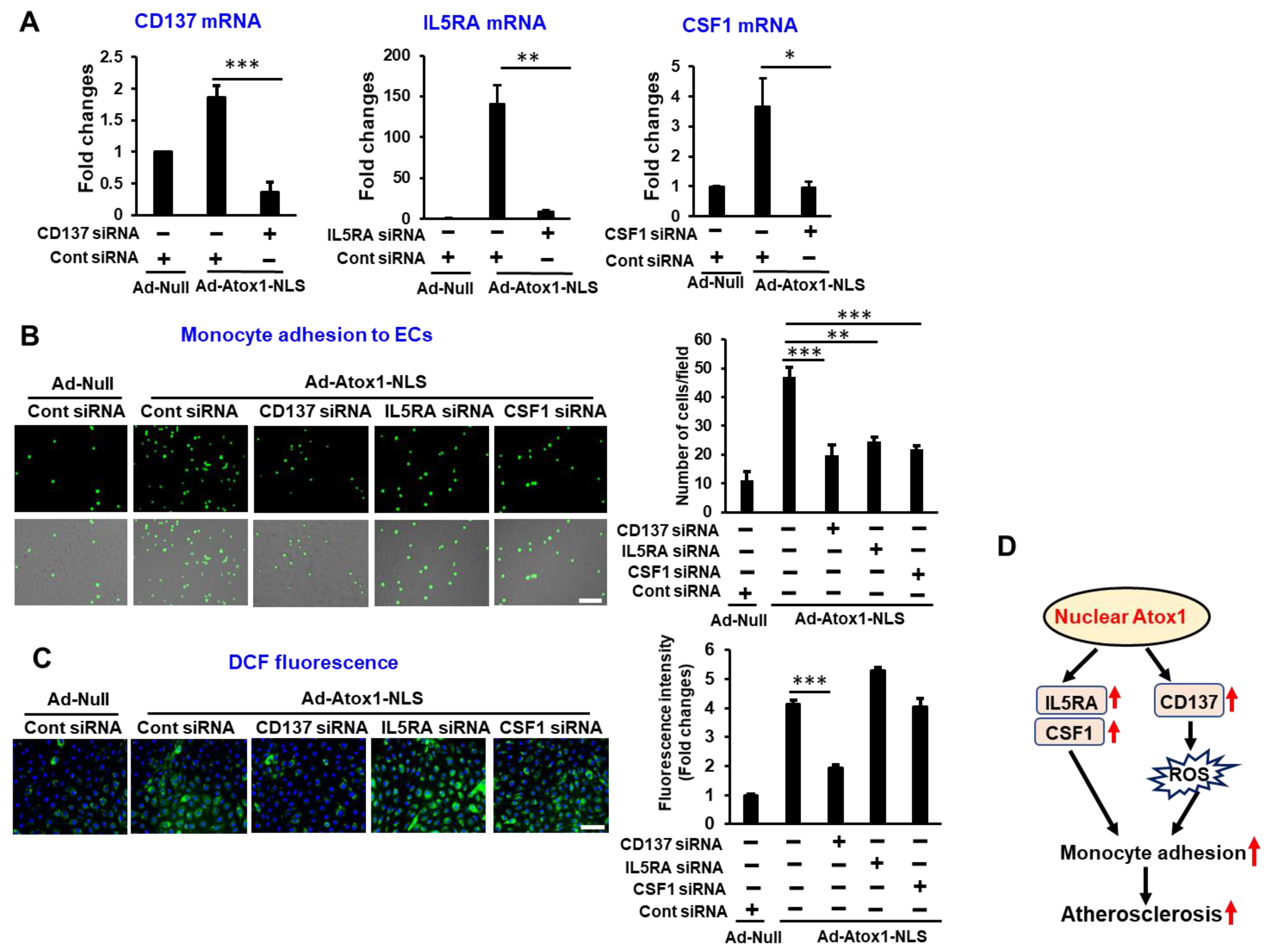
3.6. CD137, IL5RA, and CSF1 as New Atox1 Targets to Regulate Inflammation and ROS Production in ECs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Maatta, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Anticopper therapy against cancer and diseases of inflammation and fibrosis. Drug Discov. Discov. Today 2005, 10, 1103–1109. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M.; Kaplan, J.H. Copper transporters and copper chaperones: Roles in cardiovascular physiology and disease. Am. J. Physiol. Cell Physiol. 2018, 315, C186–C201. [Google Scholar] [CrossRef] [PubMed]
- Volker, W.; Dorszewski, A.; Unruh, V.; Robenek, H.; Breithardt, G.; Buddecke, E. Copper-induced inflammatory reactions of rat carotid arteries mimic restenosis/arteriosclerosis-like neointima formation. Atherosclerosis 1997, 130, 29–36. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, W.J.; McMillen, T.S.; Leboeuf, R.C.; Frei, B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis 2012, 223, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandinov, L.; Moodie, K.L.; Mandinova, A.; Zhuang, Z.; Redican, F.; Baklanov, D.; Lindner, V.; Maciag, T.; Simons, M.; de Muinck, E.D. Inhibition of in-stent restenosis by oral copper chelation in porcine coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2692–H2697. [Google Scholar] [CrossRef] [PubMed]
- Mandinov, L.; Mandinova, A.; Kyurkchiev, S.; Kyurkchiev, D.; Kehayov, I.; Kolev, V.; Soldi, R.; Bagala, C.; de Muinck, E.D.; Lindner, V.; et al. Copper chelation represses the vascular response to injury. Proc. Natl. Acad. Sci. USA 2003, 100, 6700–6705. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, V.; Petris, M.J. Copper homeostasis at the host-pathogen interface. J. Biol. Chem. 2012, 287, 13549–13555. [Google Scholar] [CrossRef] [PubMed]
- Percival, S.S. Copper and immunity. Am. J. Clin. Nutr. 1998, 67, 1064S–1068S. [Google Scholar] [CrossRef]
- Schuschke, D.A.; Saari, J.T.; Miller, F.N. Leukocyte-endothelial adhesion is impaired in the cremaster muscle microcirculation of the copper-deficient rat. Immunol. Lett. 2001, 76, 139–144. [Google Scholar] [CrossRef]
- Chen, G.F.; Sudhahar, V.; Youn, S.W.; Das, A.; Cho, J.; Kamiya, T.; Urao, N.; McKinney, R.D.; Surenkhuu, B.; Hamakubo, T.; et al. Copper Transport Protein Antioxidant-1 Promotes Inflammatory Neovascularization via Chaperone and Transcription Factor Function. Sci. Rep. 2015, 5, 14780. [Google Scholar] [CrossRef]
- Das, A.; Sudhahar, V.; Ushio-Fukai, M.; Fukai, T. Novel interaction of antioxidant-1 with TRAF4: Role in inflammatory responses in endothelial cells. Am. J. Physiol. Cell Physiol. 2019, 317, C1161–C1171. [Google Scholar] [CrossRef]
- Hatori, Y.; Lutsenko, S. An expanding range of functions for the copper chaperone/antioxidant protein Atox1. Antioxid. Redox Signal. 2013, 19, 945–957. [Google Scholar] [CrossRef]
- Kim, Y.J.; Bond, G.J.; Tsang, T.; Posimo, J.M.; Busino, L.; Brady, D.C. Copper chaperone ATOX1 is required for MAPK signaling and growth in BRAF mutation-positive melanoma. Metallomics 2019, 11, 1430–1440. [Google Scholar] [CrossRef]
- Hamza, I.; Schaefer, M.; Klomp, L.W.; Gitlin, J.D. Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc. Natl. Acad. Sci. USA 1999, 96, 13363–13368. [Google Scholar] [CrossRef] [Green Version]
- Beaino, W.; Guo, Y.; Chang, A.J.; Anderson, C.J. Roles of Atox1 and p53 in the trafficking of copper-64 to tumor cell nuclei: Implications for cancer therapy. J. Biol. Inorg. Chem. 2014, 19, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Celauro, E.; Hildesjo, C.; Feizi, A.; Stal, O.; Fierro-Gonzalez, J.C.; Wittung-Stafshede, P. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 2017, 9, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Ohrvik, H.; Wittung-Stafshede, P. Identification of New Potential Interaction Partners for Human Cytoplasmic Copper Chaperone Atox1: Roles in Gene Regulation? Int. J. Mol. Sci. 2015, 16, 16728–16739. [Google Scholar] [CrossRef] [PubMed]
- Kahra, D.; Mondol, T.; Niemiec, M.S.; Wittung-Stafshede, P. Human Copper Chaperone Atox1 Translocates to the Nucleus but does not Bind DNA In Vitro. Protein Pept. Lett. 2015, 22, 532–538. [Google Scholar] [CrossRef]
- Itoh, S.; Kim, H.W.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef]
- Itoh, S.; Ozumi, K.; Kim, H.W.; Nakagawa, O.; McKinney, R.D.; Folz, R.J.; Zelko, I.N.; Ushio-Fukai, M.; Fukai, T. Novel mechanism for regulation of extracellular SOD transcription and activity by copper: Role of antioxidant-1. Free Radic Biol. Med. 2009, 46, 95–104. [Google Scholar] [CrossRef]
- Jana, A.; Das, A.; Krett, N.L.; Guzman, G.; Thomas, A.; Mancinelli, G.; Bauer, J.; Ushio-Fukai, M.; Fukai, T.; Jung, B. Nuclear translocation of Atox1 potentiates activin A-induced cell migration and colony formation in colon cancer. PLoS ONE 2020, 15, e0227916. [Google Scholar] [CrossRef]
- Jeney, V.; Itoh, S.; Wendt, M.; Gradek, Q.; Ushio-Fukai, M.; Harrison, D.G.; Fukai, T. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ. Res. 2005, 96, 723–729. [Google Scholar] [CrossRef]
- Ozumi, K.; Sudhahar, V.; Kim, H.W.; Chen, G.F.; Kohno, T.; Finney, L.; Vogt, S.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Role of copper transport protein antioxidant 1 in angiotensin II-induced hypertension: A key regulator of extracellular superoxide dismutase. Hypertension 2012, 60, 476–486. [Google Scholar] [CrossRef]
- Kamiya, T.; Takeuchi, K.; Fukudome, S.; Hara, H.; Adachi, T. Copper chaperone antioxidant-1, Atox-1, is involved in the induction of SOD3 in THP-1 cells. Biometals 2018, 31, 61–68. [Google Scholar] [CrossRef]
- Das, A.; Sudhahar, V.; Chen, G.F.; Kim, H.W.; Youn, S.W.; Finney, L.; Vogt, S.; Yang, J.; Kweon, J.; Surenkhuu, B.; et al. Endothelial Antioxidant-1: A Key Mediator of Copper-dependent Wound Healing in vivo. Sci. Rep. 2016, 6, 33783. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, N.; Zhang, M.; Sun, M.; Bian, J.; Yang, B.; Li, Z.; Wang, J.; Li, F.; Shi, X.; et al. APEX2-based Proximity Labeling of Atox1 Identifies CRIP2 as a Nuclear Copper-binding Protein that Regulates Autophagy Activation. Angew. Chem. Int. Ed. Engl. 2021, 60, 25346–25355. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Ma, M.; Shi, S.; Wang, J.; Xiao, P.; Yu, H.F.; Zhang, C.; Guo, Q.; Yu, Z.; Lou, Z.; et al. Copper enhances genotoxic drug resistance via ATOX1 activated DNA damage repair. Cancer Lett. 2022, 536, 215651. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, C.; Shan, C.; You, Q.; Lu, J.; Elf, S.; Zhou, Y.; Wen, Y.; Vinkenborg, J.L.; Fan, J.; et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem. 2015, 7, 968–979. [Google Scholar] [CrossRef]
- Juvonen, J.; Surcel, H.M.; Satta, J.; Teppo, A.M.; Bloigu, A.; Syrjala, H.; Airaksinen, J.; Leinonen, M.; Saikku, P.; Juvonen, T. Elevated circulating levels of inflammatory cytokines in patients with abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2843–2847. [Google Scholar] [CrossRef] [PubMed]
- Sudhahar, V.; Das, A.; Horimatsu, T.; Ash, D.; Leanhart, S.; Antipova, O.; Vogt, S.; Singla, B.; Csanyi, G.; White, J.; et al. Copper Transporter ATP7A (Copper-Transporting P-Type ATPase/Menkes ATPase) Limits Vascular Inflammation and Aortic Aneurysm Development: Role of MicroRNA-125b. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2320–2337. [Google Scholar] [CrossRef]
- Lin, Z.; Zhou, P.; von Gise, A.; Gu, F.; Ma, Q.; Chen, J.; Guo, H.; van Gorp, P.R.; Wang, D.Z.; Pu, W.T. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ. Res. 2015, 116, 35–45. [Google Scholar] [CrossRef]
- Biddle, J.W.; Nguyen, M.; Gunawardena, J. Negative reciprocity, not ordered assembly, underlies the interaction of Sox2 and Oct4 on DNA. Elife 2019, 8, e41017. [Google Scholar] [CrossRef]
- Suter, D.M. Transcription Factors and DNA Play Hide and Seek. Trends Cell Biol. 2020, 30, 491–500. [Google Scholar] [CrossRef]
- Matson Dzebo, M.; Blockhuys, S.; Valenzuela, S.; Celauro, E.; Esbjorner, E.K.; Wittung-Stafshede, P. Copper Chaperone Atox1 Interacts with Cell Cycle Proteins. Comput. Struct. Biotechnol. J. 2018, 16, 443–449. [Google Scholar] [CrossRef]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.; Koalick, D.; Fabricius, A.; Parplys, A.C.; Borgmann, K.; Pospiech, H.; Grosse, F. Cdc45 is limiting for replication initiation in humans. Cell Cycle 2016, 15, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Xu, X.; Cho, S.; Lee, A.H. Loss of Transcription Factor CREBH Accelerates Diet-Induced Atherosclerosis in Ldlr-/- Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1772–1781. [Google Scholar] [CrossRef]
- So, T.; Choi, H.; Croft, M. OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J. Immunol. 2011, 186, 3547–3555. [Google Scholar] [CrossRef] [PubMed]
- Shaposhnik, Z.; Wang, X.; Lusis, A.J. Arterial colony stimulating factor-1 influences atherosclerotic lesions by regulating monocyte migration and apoptosis. J. Lipid Res. 2010, 51, 1962–1970. [Google Scholar] [CrossRef]
- Lin, X.; Lin, Q. MiRNA-495-3p Attenuates TNF-alpha Induced Apoptosis and Inflammation in Human Nucleus Pulposus Cells by Targeting IL5RA. Inflammation 2020, 43, 1797–1805. [Google Scholar] [CrossRef]
- Evans, A.K.; Ardestani, P.M.; Yi, B.; Park, H.H.; Lam, R.K.; Shamloo, M. Beta-adrenergic receptor antagonism is proinflammatory and exacerbates neuroinflammation in a mouse model of Alzheimer’s Disease. Neurobiol. Dis. 2020, 146, 105089. [Google Scholar] [CrossRef]
- Qu, M.; Yu, J.; Liu, H.; Ren, Y.; Ma, C.; Bu, X.; Lan, Q. The Candidate Tumor Suppressor Gene SLC8A2 Inhibits Invasion, Angiogenesis and Growth of Glioblastoma. Mol. Cells 2017, 40, 761–772. [Google Scholar] [CrossRef]
- Matrone, G.; Meng, S.; Gu, Q.; Lv, J.; Fang, L.; Chen, K.; Cooke, J.P. Lmo2 (LIM-Domain-Only 2) Modulates Sphk1 (Sphingosine Kinase) and Promotes Endothelial Cell Migration. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1860–1868. [Google Scholar] [CrossRef]
- Chen, M.; Yi, B.; Zhu, N.; Wei, X.; Zhang, G.X.; Huang, S.; Sun, J. Pim1 kinase promotes angiogenesis through phosphorylation of endothelial nitric oxide synthase at Ser-633. Cardiovasc. Res. 2016, 109, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Xu, C.; Li, B.; Xia, H.; Pan, Y.; Zhong, W.; Xu, L.; Chen, R.; Wang, B. Contributions of Costimulatory Molecule CD137 in Endothelial Cells. J. Am. Heart Assoc. 2021, 10, e020721. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, P.S.; Soderstrom, L.A.; Wagsater, D.; Sheikine, Y.; Ocaya, P.; Lang, F.; Rabu, C.; Chen, L.; Rudling, M.; Aukrust, P.; et al. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation 2008, 117, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.J.; Choi, J.H.; Jung, I.H.; Park, J.G.; Lee, M.R.; Lee, M.N.; Kim, B.; Yoo, J.Y.; Jeong, S.J.; Kim, D.Y.; et al. CD137 (4-1BB) deficiency reduces atherosclerosis in hyperlipidemic mice. Circulation 2010, 121, 1124–1133. [Google Scholar] [CrossRef]
- de Villiers, W.J.; Smith, J.D.; Miyata, M.; Dansky, H.M.; Darley, E.; Gordon, S. Macrophage phenotype in mice deficient in both macrophage-colony-stimulating factor (op) and apolipoprotein E. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 631–640. [Google Scholar] [CrossRef]
- Qiao, J.H.; Tripathi, J.; Mishra, N.K.; Cai, Y.; Tripathi, S.; Wang, X.P.; Imes, S.; Fishbein, M.C.; Clinton, S.K.; Libby, P.; et al. Role of macrophage colony-stimulating factor in atherosclerosis: Studies of osteopetrotic mice. Am. J. Pathol. 1997, 150, 1687–1699. [Google Scholar] [PubMed]
- Rajavashisth, T.; Qiao, J.H.; Tripathi, S.; Tripathi, J.; Mishra, N.; Hua, M.; Wang, X.P.; Loussararian, A.; Clinton, S.; Libby, P.; et al. Heterozygous osteopetrotic (op) mutation reduces atherosclerosis in LDL receptor- deficient mice. J. Clin. Invest. 1998, 101, 2702–2710. [Google Scholar] [CrossRef]
- Rajavashisth, T.B.; Andalibi, A.; Territo, M.C.; Berliner, J.A.; Navab, M.; Fogelman, A.M.; Lusis, A.J. Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature 1990, 344, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Sinha, S.K.; Miikeda, A.; Fouladian, Z.; Mehrabian, M.; Edillor, C.; Shih, D.; Zhou, Z.; Paul, M.K.; Charugundla, S.; Davis, R.C.; et al. Local M-CSF (Macrophage Colony-Stimulating Factor) Expression Regulates Macrophage Proliferation and Apoptosis in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D.; Trogan, E.; Ginsberg, M.; Grigaux, C.; Tian, J.; Miyata, M. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc. Natl. Acad. Sci. USA 1995, 92, 8264–8268. [Google Scholar] [CrossRef] [Green Version]
- Gregory, B.; Kirchem, A.; Phipps, S.; Gevaert, P.; Pridgeon, C.; Rankin, S.M.; Robinson, D.S. Differential regulation of human eosinophil IL-3, IL-5, and GM-CSF receptor alpha-chain expression by cytokines: IL-3, IL-5, and GM-CSF down-regulate IL-5 receptor alpha expression with loss of IL-5 responsiveness, but up-regulate IL-3 receptor alpha expression. J. Immunol. 2003, 170, 5359–5366. [Google Scholar] [CrossRef] [PubMed]
- Hellman, C.; Hallden, G.; Hylander, B.; Lundahl, J. Regulation of the interleukin-5 receptor alpha-subunit on peripheral blood eosinophils from healthy subjects. Clin. Exp. Immunol. 2003, 131, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Tavernier, J.; Van der Heyden, J.; Verhee, A.; Brusselle, G.; Van Ostade, X.; Vandekerckhove, J.; North, J.; Rankin, S.M.; Kay, A.B.; Robinson, D.S. Interleukin 5 regulates the isoform expression of its own receptor alpha-subunit. Blood 2000, 95, 1600–1607. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudhahar, V.; Shi, Y.; Kaplan, J.H.; Ushio-Fukai, M.; Fukai, T. Whole-Transcriptome Sequencing Analyses of Nuclear Antixoxidant-1 in Endothelial Cells: Role in Inflammation and Atherosclerosis. Cells 2022, 11, 2919. https://doi.org/10.3390/cells11182919
Sudhahar V, Shi Y, Kaplan JH, Ushio-Fukai M, Fukai T. Whole-Transcriptome Sequencing Analyses of Nuclear Antixoxidant-1 in Endothelial Cells: Role in Inflammation and Atherosclerosis. Cells. 2022; 11(18):2919. https://doi.org/10.3390/cells11182919
Chicago/Turabian StyleSudhahar, Varadarajan, Yang Shi, Jack H. Kaplan, Masuko Ushio-Fukai, and Tohru Fukai. 2022. "Whole-Transcriptome Sequencing Analyses of Nuclear Antixoxidant-1 in Endothelial Cells: Role in Inflammation and Atherosclerosis" Cells 11, no. 18: 2919. https://doi.org/10.3390/cells11182919