

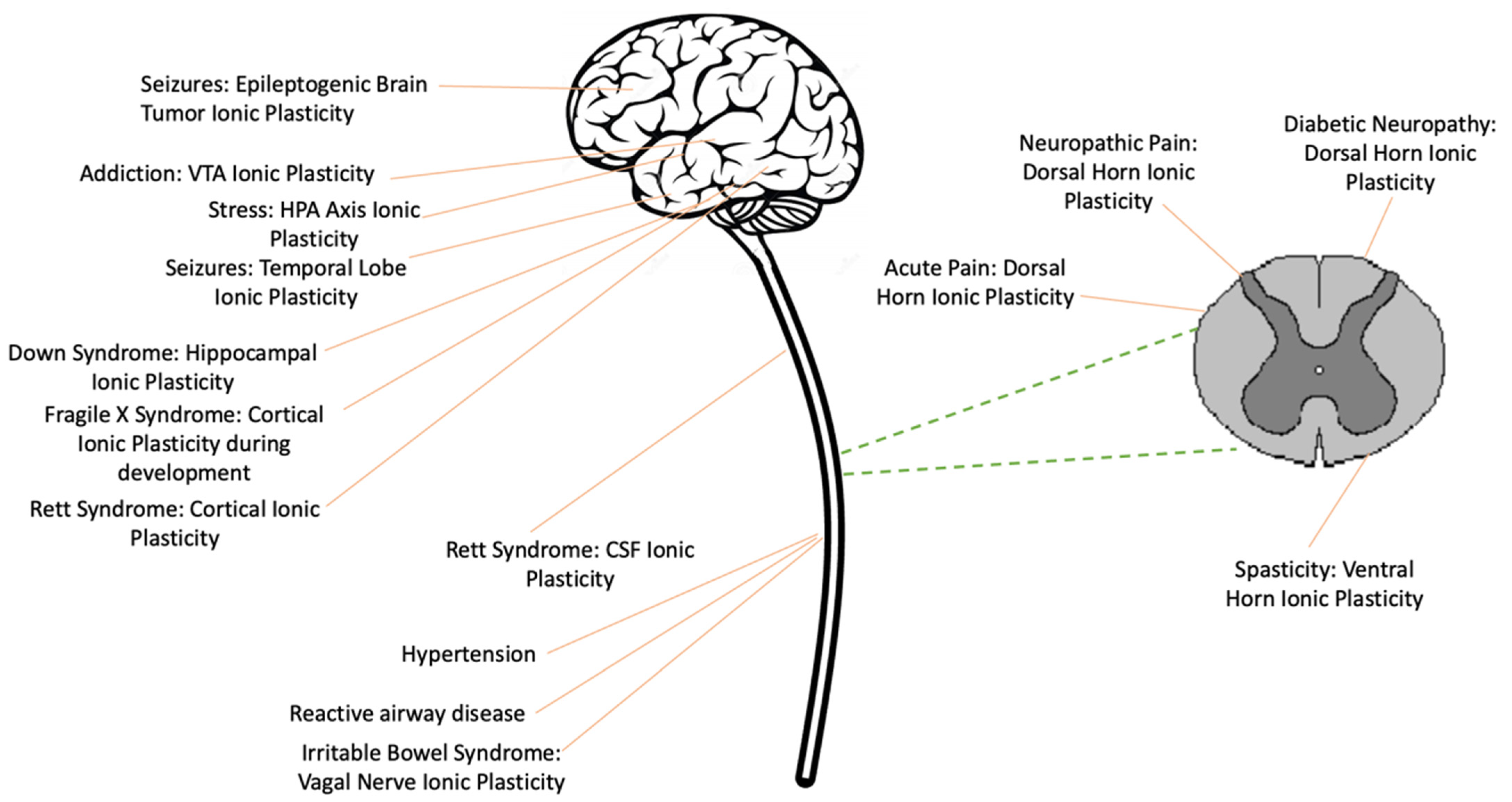
Ionic Plasticity: Common Mechanistic Underpinnings of Pathology in Spinal Cord Injury and the Brain
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Foundations: GABA, Ionic Plasticity, and KCC2
2.1. Regulation of Neural Excitability
2.2. GABA Receptor: Pharmacology
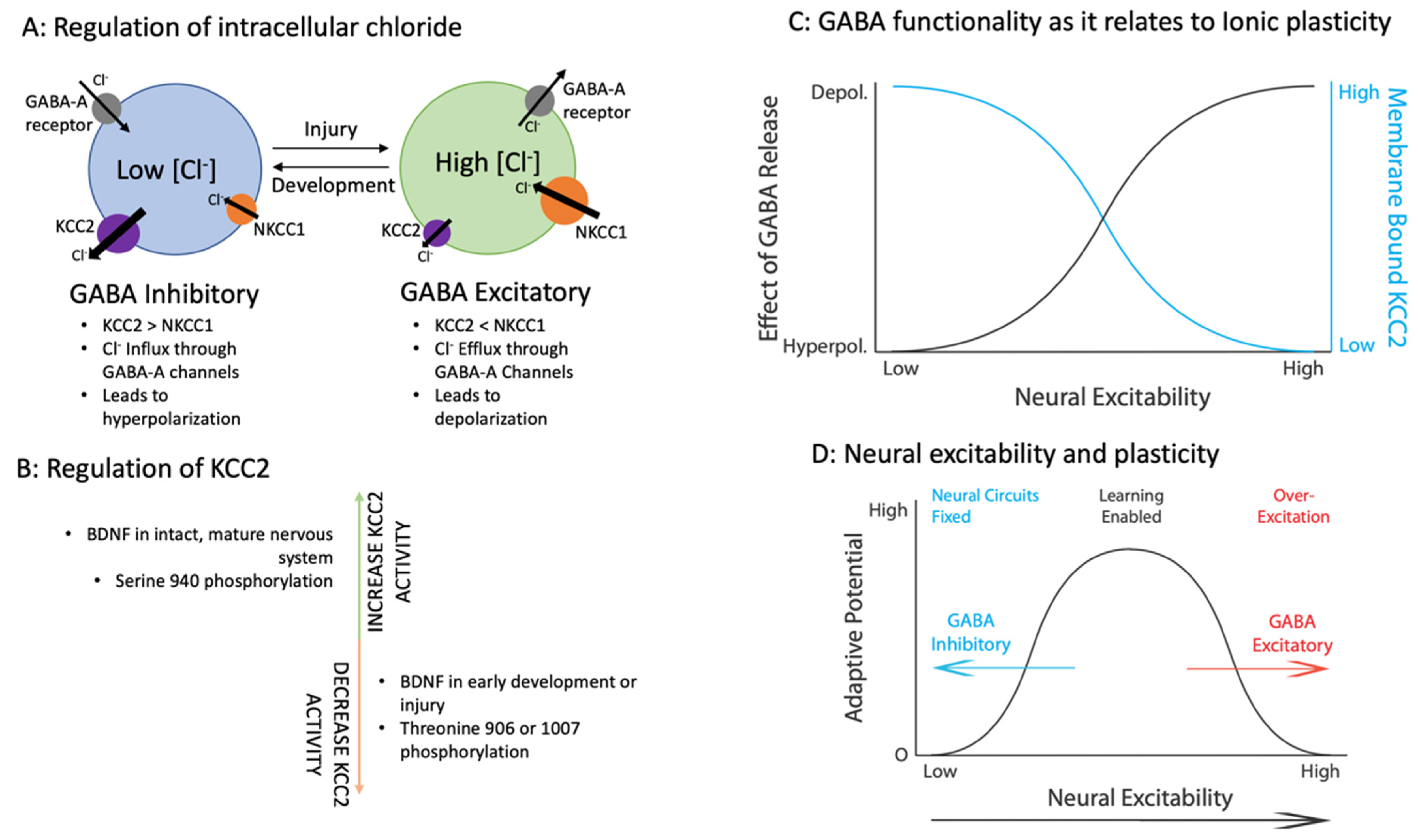
2.3. Regulation of Intracellular Cl− Concentrations
2.3.1. Co-Transporters KCC2 and NKCC1
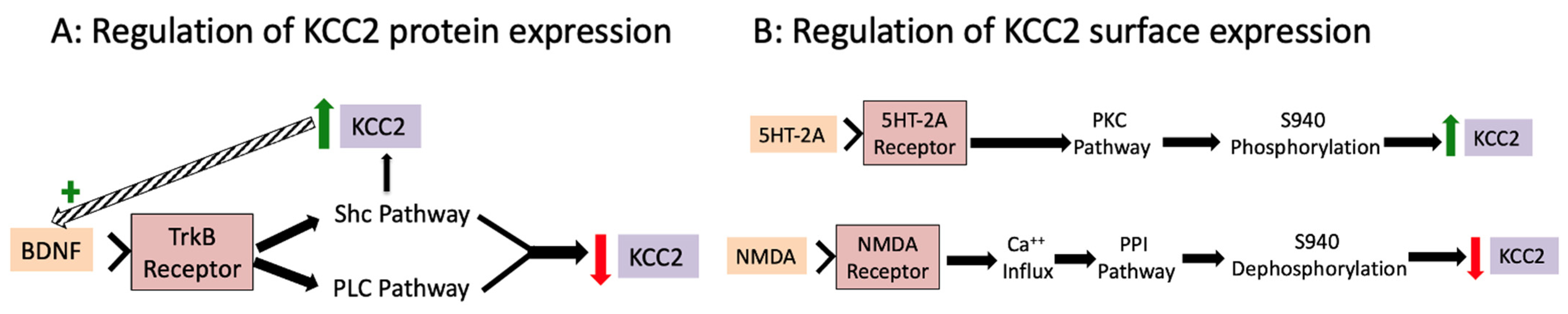
2.3.2. Cellular Processes That Regulate Ionic Plasticity
2.3.3. Pharmacology That Regulates Ionic Plasticity
2.4. Summary
3. Spasticity
3.1. Clinical Symptoms and Indices
3.2. Reduced GABAergic Inhibition Enables Spasticity
3.3. Restoring GABAergic Inhibition Attenuates Spasticity
3.3.1. Pharmacotherapeutics for Spasticity
3.3.2. Exercise a Spasticity Therapeutic
3.4. Ionic Plasticity Disrupts the Recovery of Motor Function
3.5. Summary
4. Pain
4.1. Kinds of Pain
4.2. Pain after Spinal Cord Injury
4.2.1. Noxious Stimulation Sensitizes Nociceptive Circuits in the Spinal Cord
4.2.2. Ionic Plasticity Fosters Nociceptive Sensitization after SCI
4.2.3. Ionic Plasticity Fosters Chronic Pain after SCI
4.2.4. Restoring GABAergic Inhibition Attenuates Pain after SCI
4.3. Ionic Plasticity Can Fuel Pain in the Absence of Spinal Cord Injury
4.3.1. Acute Pain
4.3.2. Neuropathic Pain
4.3.3. Restoring GABAergic Inhibition Attenuates Pain
4.3.4. Role of Sex in Pain
4.3.5. Opiate Hyperalgesia
4.3.6. Diabetic Neuropathy
4.4. Summary
5. Contributions of Ionic Plasticity to Brain-Mediated Pathophysiology
5.1. Seizures and Epilepsy
5.1.1. Role of Ionic Plasticity in Seizures and Epilepsy
5.1.2. Restoring GABAergic Inhibition Attenuates Seizure activity
5.1.3. Neurodevelopmental Diseases
5.2. Addiction
5.2.1. Neural Mechanisms of Addiction
5.2.2. Ionic Plasticity in Addiction
5.3. Summary
6. Other Disease States
6.1. Hypertension
6.2. Asthma
6.3. Irritable Bowel Syndrome
6.4. Summary
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Golan, D.E. Principles of Pharmacology; Armstrong: Lancaster, PA, USA, 2017. [Google Scholar]
- Perez-Sanchez, T.; De Koninck, Y. Regulation of Chloride Gradients and Neural Plasticity. In Oxford Research Encyclopedia: Neuroscience; Oxford University Press: Oxford, UK, 2019. [Google Scholar]
- Boulenguez, P.; Liabeuf, S.; Bos, R.; Bras, H.; Jean-Xavier, C.; Brocard, C.; Stil, A.; Darbon, P.; Cattaert, D.; Delpire, E.; et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 2010, 16, 302–307. [Google Scholar] [CrossRef]
- Cramer, S.W.; Baggott, C.; Cain, J.; Tilghman, J.; Allcock, B.; Miranpuri, G.; Rajpal, S.; Sun, D.; Resnick, D. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol. Pain 2008, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Voipio, J.; Kaila, K. Two developmental switches in GABAergic signalling: The K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. J. Physiol. 2005, 562, 27–36. [Google Scholar] [CrossRef]
- Zhu, S.; Noviello, C.M.; Teng, J.; Walsh, R.M., Jr.; Kim, J.J.; Hibbs, R.E. Structure of a human synaptic GABA(A) receptor. Nature 2018, 559, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Petroff, O.A. GABA and glutamate in the human brain. Neuroscientist 2002, 8, 562–573. [Google Scholar] [CrossRef]
- Li, C.; Lei, Y.; Tian, Y.; Xu, S.; Shen, X.; Wu, H.; Bao, S.; Wang, F. The etiological contribution of GABAergic plasticity to the pathogenesis of neuropathic pain. Mol. Pain 2019, 15, 1744806919847366. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W.; DeLorey, T.M. GABA Receptor Physiology and Pharmacology. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J.A.B., Albers, R.W., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999. [Google Scholar]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Rowley, N.M.; Madsen, K.K.; Schousboe, A.; Steve White, H. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control. Neurochem. Int. 2012, 61, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Ngo, D.H.; Vo, T.S. An Updated Review on Pharmaceutical Properties of Gamma-Aminobutyric Acid. Molecules 2019, 24, 2678. [Google Scholar] [CrossRef] [PubMed]
- Prévot, T.; Sibille, E. Altered GABA-mediated information processing and cognitive dysfunctions in depression and other brain disorders. Mol. Psychiatry 2021, 26, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Maust, D.T.; Lin, L.A.; Blow, F.C. Benzodiazepine Use and Misuse Among Adults in the United States. Psychiatr. Serv. 2019, 70, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABA(A) receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Ertzgaard, P.; Campo, C.; Calabrese, A. Efficacy and safety of oral baclofen in the management of spasticity: A rationale for intrathecal baclofen. J. Rehabil. Med. 2017, 49, 193–203. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Godwin, J. Flumazenil in benzodiazepine overdose. CMAJ 2016, 188, E537. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. The GABA excitatory/inhibitory developmental sequence: A personal journey. Neuroscience 2014, 279, 187–219. [Google Scholar] [CrossRef]
- Grau, J.W.; Huang, Y.J. Metaplasticity within the spinal cord: Evidence brain-derived neurotrophic factor (BDNF), tumor necrosis factor (TNF), and alterations in GABA function (ionic plasticity) modulate pain and the capacity to learn. Neurobiol. Learn. Mem. 2018, 154, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Fukuda, A. Development and regulation of chloride homeostasis in the central nervous system. Front. Cell Neurosci. 2015, 9, 371. [Google Scholar] [CrossRef]
- Payne, J.A.; Rivera, C.; Voipio, J.; Kaila, K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003, 26, 199–206. [Google Scholar] [CrossRef]
- Dzhala, V.I.; Staley, K.J. KCC2 chloride transport contributes to the termination of ictal epileptiform activity. eNeuro 2020, 8, ENEURO.0208-20.2020. [Google Scholar] [CrossRef]
- Payne, J.A. Functional characterization of the neuronal-specific K-Cl cotransporter: Implications for [K+]o regulation. Am. J. Physiol. 1997, 273, C1516–C1525. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, K.J.; Hochman, S. Spinal cord injury causes plasticity in a subpopulation of lamina I GABAergic interneurons. J. Neurophysiol. 2008, 100, 212–223. [Google Scholar] [CrossRef]
- Ferrini, F.; Perez-Sanchez, J.; Ferland, S.; Lorenzo, L.E.; Godin, A.G.; Plasencia-Fernandez, I.; Cottet, M.; Castonguay, A.; Wang, F.; Salio, C.; et al. Differential chloride homeostasis in the spinal dorsal horn locally shapes synaptic metaplasticity and modality-specific sensitization. Nat. Commun. 2020, 11, 3935. [Google Scholar] [CrossRef]
- Abraham, W.C. Metaplasticity: Tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 2008, 9, 387. [Google Scholar] [CrossRef]
- Valeeva, G.; Tressard, T.; Mukhtarov, M.; Baude, A.; Khazipov, R. An Optogenetic Approach for Investigation of Excitatory and Inhibitory Network GABA Actions in Mice Expressing Channelrhodopsin-2 in GABAergic Neurons. J. Neurosci. 2016, 36, 5961–5973. [Google Scholar] [CrossRef] [PubMed]
- Kirmse, K.; Kummer, M.; Kovalchuk, Y.; Witte, O.W.; Garaschuk, O.; Holthoff, K. GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 2015, 6, 7750. [Google Scholar] [CrossRef]
- Lee-Hotta, S.; Uchiyama, Y.; Kametaka, S. Role of the BDNF-TrkB pathway in KCC2 regulation and rehabilitation following neuronal injury: A mini review. Neurochem. Int. 2019, 128, 32–38. [Google Scholar] [CrossRef]
- Ferrini, F.; De Koninck, Y. Microglia control neuronal network excitability via BDNF signalling. Neural. Plast. 2013, 2013, 429815. [Google Scholar] [CrossRef] [PubMed]
- Puskarjov, M.; Ahmad, F.; Khirug, S.; Sivakumaran, S.; Kaila, K.; Blaesse, P. BDNF is required for seizure-induced but not developmental up-regulation of KCC2 in the neonatal hippocampus. Neuropharmacology 2015, 88, 103–109. [Google Scholar] [CrossRef]
- Ohira, K.; Hayashi, M. A new aspect of the TrkB signaling pathway in neural plasticity. Curr. Neuropharmacol. 2009, 7, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, M.A.; Uvarov, P.; Mavrovic, M.; Poncer, J.C.; Kaila, K. The Multifaceted Roles of KCC2 in Cortical Development. Trends Neurosci. 2021, 44, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W. Tuning the regulator: Phosphorylation of KCC2 at two specific sites is critical for neurodevelopment. Sci. Signal 2019, 12, 603. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Deeb, T.Z.; Puskarjov, M.; Silayeva, L.; Liang, B.; Kaila, K.; Moss, S.J. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013, 36, 726–737. [Google Scholar] [CrossRef]
- Wake, H.; Watanabe, M.; Moorhouse, A.J.; Kanematsu, T.; Horibe, S.; Matsukawa, N.; Asai, K.; Ojika, K.; Hirata, M.; Nabekura, J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 2007, 27, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Ostroumov, A.; Thomas, A.M.; Kimmey, B.A.; Karsch, J.S.; Doyon, W.M.; Dani, J.A. Stress Increases Ethanol Self-Administration via a Shift toward Excitatory GABA Signaling in the Ventral Tegmental Area. Neuron 2016, 92, 493–504. [Google Scholar] [CrossRef]
- Hewitt, S.A.; Wamsteeker, J.I.; Kurz, E.U.; Bains, J.S. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 2009, 12, 438–443. [Google Scholar] [CrossRef]
- Pozzi, D.; Rasile, M.; Corradini, I.; Matteoli, M. Environmental regulation of the chloride transporter KCC2: Switching inflammation off to switch the GABA on? Transl. Psychiatry 2020, 10, 349. [Google Scholar] [CrossRef]
- Bilchak, J.N.; Yeakle, K.; Caron, G.; Malloy, D.; Côté, M.P. Enhancing KCC2 activity decreases hyperreflexia and spasticity after chronic spinal cord injury. Exp. Neurol. 2021, 338, 113605. [Google Scholar] [CrossRef]
- Tashiro, S.; Shinozaki, M.; Mukaino, M.; Renault-Mihara, F.; Toyama, Y.; Liu, M.; Nakamura, M.; Okano, H. BDNF Induced by Treadmill Training Contributes to the Suppression of Spasticity and Allodynia After Spinal Cord Injury via Upregulation of KCC2. Neurorehabil. Neural Repair 2015, 29, 677–689. [Google Scholar] [CrossRef]
- Kharod, S.C.; Kang, S.K.; Kadam, S.D. Off-Label Use of Bumetanide for Brain Disorders: An Overview. Front. Neurosci. 2019, 13, 310. [Google Scholar] [CrossRef] [Green Version]
- Klomjai, W.; Roche, N.; Lamy, J.C.; Bede, P.; Giron, A.; Bussel, B.; Bensmail, D.; Katz, R.; Lackmy-Vallée, A. Furosemide Unmasks Inhibitory Dysfunction after Spinal Cord Injury in Humans: Implications for Spasticity. J. Neurotrauma 2019, 36, 1469–1477. [Google Scholar] [CrossRef]
- Gagnon, M.; Bergeron, M.J.; Lavertu, G.; Castonguay, A.; Tripathy, S.; Bonin, R.P.; Perez-Sanchez, J.; Boudreau, D.; Wang, B.; Dumas, L.; et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 2013, 19, 1524–1528. [Google Scholar] [CrossRef]
- Delpire, E.; Days, E.; Lewis, L.M.; Mi, D.; Kim, K.; Lindsley, C.W.; Weaver, C.D. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. USA 2009, 106, 5383–5388. [Google Scholar] [CrossRef]
- American Association of Neurological Surgeons. Spasticity. 2019. Available online: https://www.aans.org/Patients/Neurosurgical-Conditions-and-Treatments/Spasticity#:~:text=Spasticity%20is%20a%20condition%20in,affecting%20movement%2C%20speech%20and%20gait (accessed on 19 September 2020).
- Elbasiouny, S.M.; Moroz, D.; Bakr, M.M.; Mushahwar, V.K. Management of spasticity after spinal cord injury: Current techniques and future directions. Neurorehabil. Neural Repair 2010, 24, 23–33. [Google Scholar] [CrossRef]
- Thibaut, A.; Chatelle, C.; Ziegler, E.; Bruno, M.A.; Laureys, S.; Gosseries, O. Spasticity after stroke: Physiology, assessment and treatment. Brain Inj. 2013, 27, 1093–1105. [Google Scholar] [CrossRef]
- Burke, D. Clinical uses of H reflexes of upper and lower limb muscles. Clin. Neurophysiol. Pr. 2016, 1, 9–17. [Google Scholar] [CrossRef]
- Watanabe, T. The role of therapy in spasticity management. Am. J. Phys. Med. Rehabil. 2004, 83, S45–S49. [Google Scholar] [CrossRef]
- Chang, E.; Ghosh, N.; Yanni, D.; Lee, S.; Alexandru, D.; Mozaffar, T. A Review of Spasticity Treatments: Pharmacological and Interventional Approaches. Crit. Rev. Phys. Rehabil. Med. 2013, 25, 11–22. [Google Scholar] [CrossRef]
- Shulga, A.; Thomas-Crusells, J.; Sigl, T.; Blaesse, A.; Mestres, P.; Meyer, M.; Yan, Q.; Kaila, K.; Saarma, M.; Rivera, C.; et al. Posttraumatic GABA(A)-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J. Neurosci. 2008, 28, 6996–7005. [Google Scholar] [CrossRef]
- Bos, R.; Sadlaoud, K.; Boulenguez, P.; Buttigieg, D.; Liabeuf, S.; Brocard, C.; Haase, G.; Bras, H.; Vinay, L. Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. USA 2013, 110, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Ishida, K.; Kiyama, H.; Yamashita, T.; Lee, S. Down-regulation of KCC2 expression and phosphorylation in motoneurons, and increases the number of in primary afferent projections to motoneurons in mice with post-stroke spasticity. PLoS ONE 2014, 9, e114328. [Google Scholar] [CrossRef]
- Mòdol, L.; Mancuso, R.; Alé, A.; Francos-Quijorna, I.; Navarro, X. Differential effects on KCC2 expression and spasticity of ALS and traumatic injuries to motoneurons. Front. Cell Neurosci. 2014, 8, 7. [Google Scholar] [CrossRef]
- Fauss, G.N.K.; Hudson, K.E.; Grau, J.W. Role of Descending Serotonergic Fibers in the Development of Pathophysiology after Spinal Cord Injury (SCI): Contribution to Chronic Pain, Spasticity, and Autonomic Dysreflexia. Biology 2022, 11, 234. [Google Scholar] [CrossRef]
- Khan, F.; Amatya, B.; Bensmail, D.; Yelnik, A. Non-pharmacological interventions for spasticity in adults: An overview of systematic reviews. Ann. Phys. Rehabil. Med. 2019, 62, 265–273. [Google Scholar] [CrossRef]
- Beverungen, H.; Klaszky, S.C.; Klaszky, M.; Côté, M.P. Rehabilitation Decreases Spasticity by Restoring Chloride Homeostasis through the Brain-Derived Neurotrophic Factor-KCC2 Pathway after Spinal Cord Injury. J. Neurotrauma 2020, 37, 846–859. [Google Scholar] [CrossRef]
- Li, X.; Song, X.; Fang, L.; Ding, J.; Qi, L.; Wang, Q.; Dong, C.; Wang, S.; Wu, J.; Wang, T.; et al. Body Weight-Supported Treadmill Training Ameliorates Motoneuronal Hyperexcitability by Increasing GAD-65/67 and KCC2 Expression via TrkB Signaling in Rats with Incomplete Spinal Cord Injury. Neurochem. Res. 2022, 47, 1679–1691. [Google Scholar] [CrossRef]
- Chen, B.; Li, Y.; Yu, B.; Zhang, Z.; Brommer, B.; Williams, P.R.; Liu, Y.; Hegarty, S.V.; Zhou, S.; Zhu, J.; et al. Reactivation of Dormant Relay Pathways in Injured Spinal Cord by KCC2 Manipulations. Cell 2018, 174, 521–535.e513. [Google Scholar] [CrossRef]
- Grau, J.W.; Huang, Y.J.; Turtle, J.D.; Strain, M.M.; Miranda, R.C.; Garraway, S.M.; Hook, M.A. When Pain Hurts: Nociceptive Stimulation Induces a State of Maladaptive Plasticity and Impairs Recovery after Spinal Cord Injury. J. Neurotrauma 2017, 34, 1873–1890. [Google Scholar] [CrossRef]
- Hasbargen, T.; Ahmed, M.M.; Miranpuri, G.; Li, L.; Kahle, K.T.; Resnick, D.; Sun, D. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Ann. N. Y. Acad. Sci. 2010, 1198, 168–172. [Google Scholar] [CrossRef]
- Johns Hopkins Medicine. What Is Pain/Types of Pain Treated. 2020. Available online: https://www.hopkinsmedicine.org/pain/blaustein_pain_center/patient_care/what_is_pain.html (accessed on 11 July 2020).
- Kent, M. Maladaptive Pain. In The Oxford Dictionary of Sports Science & Medicine, 3rd ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [Green Version]
- Kitayama, T. The Role of K(+)-Cl(−)-Cotransporter-2 in Neuropathic Pain. Neurochem. Res. 2018, 43, 110–115. [Google Scholar] [CrossRef]
- Gierthmühlen, J.; Baron, R. Neuropathic Pain. Semin Neurol. 2016, 36, 462–468. [Google Scholar] [CrossRef]
- Willis, W.D., Jr. Dorsal root potentials and dorsal root reflexes: A double-edged sword. Exp. Brain Res. 1999, 124, 395–421. [Google Scholar] [CrossRef]
- Markenson, J.A. Mechanisms of chronic pain. Am. J. Med. 1996, 101, 6s–18s. [Google Scholar] [CrossRef]
- Kandel, E.R.; Schwartz, J.H.; Jessell, T.M.; Siegelbaum, S.A.; Hudspeth, A.J.; Mack, S. Principles of Neural Science, 15th ed.; McGraw-Hill Education: New York, NY, USA, 2013. [Google Scholar]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef]
- Willis, W.D. Mechanisms of central sensitization of nociceptive dorsal horn neurons. In Spinal Cord Plasticity: Alterations in Reflex Function; Patterson, M.M., Grau, J.W., Eds.; Kluwer Academic Publishers: Norwell, MA, USA, 2001; pp. 127–161. [Google Scholar]
- Ji, R.R.; Kohno, T.; Moore, K.A.; Woolf, C.J. Central sensitization and LTP: Do pain and memory share similar mechanisms? Trends Neurosci. 2003, 26, 696–705. [Google Scholar] [CrossRef]
- Sandkühler, J. Learning and memory in pain pathways. Pain 2000, 88, 113–118. [Google Scholar] [CrossRef]
- Gebhart, G.F. Descending modulation of pain. Neurosci. Biobehav. Rev. 2004, 27, 729–737. [Google Scholar] [CrossRef]
- Gjerstad, J.; Tjølsen, A.; Hole, K. Induction of long-term potentiation of single wide dynamic range neurones in the dorsal horn is inhibited by descending pathways. Pain 2001, 91, 263–268. [Google Scholar] [CrossRef]
- Sandkühler, J.; Liu, X. Induction of long-term potentiation at spinal synapses by noxious stimulation or nerve injury. Eur. J. Neurosci. 1998, 10, 2476–2480. [Google Scholar] [CrossRef]
- Gao, Y.J.; Ji, R.R. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J. 2009, 2, 11–17. [Google Scholar] [CrossRef]
- Huang, Y.J.; Lee, K.H.; Grau, J.W. Complete spinal cord injury (SCI) transforms how brain derived neurotrophic factor (BDNF) affects nociceptive sensitization. Exp. Neurol. 2017, 288, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Garraway, S.M.; Hochman, S. Modulatory actions of serotonin, norepinephrine, dopamine, and acetylcholine in spinal cord deep dorsal horn neurons. J. Neurophysiol. 2001, 86, 2183–2194. [Google Scholar] [CrossRef]
- Crown, E.D.; Grau, J.W. Evidence that descending serotonergic systems protect spinal cord plasticity against the disruptive effect of uncontrollable stimulation. Exp. Neurol. 2005, 196, 164–176. [Google Scholar] [CrossRef]
- Grau, J.W. Learning from the spinal cord: How the study of spinal cord plasticity informs our view of learning. Neurobiol. Learn. Mem. 2014, 108, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Baumbauer, K.M.; Hoy, K.C., Jr.; Huie, J.R.; Hughes, A.J.; Woller, S.A.; Puga, D.A.; Setlow, B.; Grau, J.W. Timing in the absence of supraspinal input I: Variable, but not fixed, spaced stimulation of the sciatic nerve undermines spinally-mediated instrumental learning. Neuroscience 2008, 155, 1030–1047. [Google Scholar] [CrossRef]
- Grau, J.W.; Barstow, D.G.; Joynes, R.L. Instrumental learning within the spinal cord: I. Behavioral properties. Behav. Neurosci. 1998, 112, 1366–1386. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.R.; Crown, E.D.; Grau, J.W. Nociceptive plasticity inhibits adaptive learning in the spinal cord. Neuroscience 2006, 141, 421–431. [Google Scholar] [CrossRef]
- Hook, M.A.; Huie, J.R.; Grau, J.W. Peripheral inflammation undermines the plasticity of the isolated spinal cord. Behav. Neurosci. 2008, 122, 233–249. [Google Scholar] [CrossRef]
- Sivilotti, L.; Woolf, C.J. The contribution of GABAA and glycine receptors to central sensitization: Disinhibition and touch-evoked allodynia in the spinal cord. J. Neurophysiol. 1994, 72, 169–179. [Google Scholar] [CrossRef]
- Sorkin, L.S.; Eddinger, K.A.; Woller, S.A.; Yaksh, T.L. Origins of antidromic activity in sensory afferent fibers and neurogenic inflammation. Semin Immunopathol. 2018, 40, 237–247. [Google Scholar] [CrossRef]
- Ferguson, A.R.; Washburn, S.N.; Crown, E.D.; Grau, J.W. GABA(A) receptor activation is involved in noncontingent shock inhibition of instrumental conditioning in spinal rats. Behav. Neurosci. 2003, 117, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Lee, K.H.; Murphy, L.; Garraway, S.M.; Grau, J.W. Acute spinal cord injury (SCI) transforms how GABA affects nociceptive sensitization. Exp. Neurol. 2016, 285, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Boyce, V.S.; Mendell, L.M. Neurotrophins and spinal circuit function. Front. Neural. Circuits. 2014, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Pinilla, F.; Huie, J.R.; Ying, Z.; Ferguson, A.R.; Crown, E.D.; Baumbauer, K.M.; Edgerton, V.R.; Grau, J.W. BDNF and learning: Evidence that instrumental training promotes learning within the spinal cord by up-regulating BDNF expression. Neuroscience 2007, 148, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Grau, J.W.; Huie, J.R.; Garraway, S.M.; Hook, M.A.; Crown, E.D.; Baumbauer, K.M.; Lee, K.H.; Hoy, K.C.; Ferguson, A.R. Impact of behavioral control on the processing of nociceptive stimulation. Front. Physiol. 2012, 3, 262. [Google Scholar] [CrossRef] [PubMed]
- Vaynman, S.; Gomez-Pinilla, F. License to run: Exercise impacts functional plasticity in the intact and injured central nervous system by using neurotrophins. Neurorehabil. Neural. Repair 2005, 19, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Huie, J.R.; Garraway, S.M.; Baumbauer, K.M.; Hoy, K.C., Jr.; Beas, B.S.; Montgomery, K.S.; Bizon, J.L.; Grau, J.W. Brain-derived neurotrophic factor promotes adaptive plasticity within the spinal cord and mediates the beneficial effects of controllable stimulation. Neuroscience 2012, 200, 74–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Grau, J.W. Ionic plasticity and pain: The loss of descending serotonergic fibers after spinal cord injury transforms how GABA affects pain. Exp. Neurol. 2018, 306, 105–116. [Google Scholar] [CrossRef]
- King, T.; Vera-Portocarrero, L.; Gutierrez, T.; Vanderah, T.W.; Dussor, G.; Lai, J.; Fields, H.L.; Porreca, F. Unmasking the tonic-aversive state in neuropathic pain. Nat. Neurosci. 2009, 12, 1364–1366. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Brualla, I.; Boulenguez, P.; Brocard, C.; Liabeuf, S.; Viallat-Lieutaud, A.; Navarro, X.; Udina, E.; Brocard, F. Activation of 5-HT(2A) Receptors Restores KCC2 Function and Reduces Neuropathic Pain after Spinal Cord Injury. Neuroscience 2018, 387, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zarepour, L.; Gharaylou, Z.; Hadjighassem, M.; Shafaghi, L.; Majedi, H.; Behzad, E.; Hosseindoost, S.; Ramezani, F.; Nasirinezhad, F. Preliminary study of analgesic effect of bumetanide on neuropathic pain in patients with spinal cord injury. J. Clin. Neurosci. 2020, 81, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.R.; Huie, J.R.; Crown, E.D.; Grau, J.W. Central nociceptive sensitization vs. spinal cord training: Opposing forms of plasticity that dictate function after complete spinal cord injury. Front. Physiol. 2012, 3, 396. [Google Scholar] [CrossRef] [PubMed]
- Huie, J.R.; Baumbauer, K.M.; Lee, K.H.; Bresnahan, J.C.; Beattie, M.S.; Ferguson, A.R.; Grau, J.W. Glial tumor necrosis factor alpha (TNFα) generates metaplastic inhibition of spinal learning. PLoS ONE 2012, 7, e39751. [Google Scholar] [CrossRef] [PubMed]
- Huie, J.R.; Stuck, E.D.; Lee, K.H.; Irvine, K.A.; Beattie, M.S.; Bresnahan, J.C.; Grau, J.W.; Ferguson, A.R. AMPA Receptor Phosphorylation and Synaptic Colocalization on Motor Neurons Drive Maladaptive Plasticity below Complete Spinal Cord Injury. eNeuro 2015, 2, ENEURO.0091-15.2015. [Google Scholar] [CrossRef] [PubMed]
- Nomura, H.; Sakai, A.; Nagano, M.; Umino, M.; Suzuki, H. Expression changes of cation chloride cotransporters in the rat spinal cord following intraplantar formalin. Neurosci. Res. 2006, 56, 435–440. [Google Scholar] [CrossRef]
- Tsuruga, K.; Hashimoto, T.; Kato, R.; Kato, R.; Uchida, Y.; Hase, T.; Morimoto, Y. Plantar injection of formalin in rats reduces the expression of a potassium chroride cotransporter KCC2 in the spinal cord and a kinase inhibitor suppresses this reduction. Biomed. Res. 2016, 37, 243–249. [Google Scholar] [CrossRef]
- Funk, K.; Woitecki, A.; Franjic-Würtz, C.; Gensch, T.; Möhrlen, F.; Frings, S. Modulation of chloride homeostasis by inflammatory mediators in dorsal root ganglion neurons. Mol. Pain 2008, 4, 32. [Google Scholar] [CrossRef]
- Coull, J.A.; Boudreau, D.; Bachand, K.; Prescott, S.A.; Nault, F.; Sík, A.; De Koninck, P.; De Koninck, Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 2003, 424, 938–942. [Google Scholar] [CrossRef]
- Coull, J.A.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef]
- Miletic, G.; Miletic, V. Loose ligation of the sciatic nerve is associated with TrkB receptor-dependent decreases in KCC2 protein levels in the ipsilateral spinal dorsal horn. Pain 2008, 137, 532–539. [Google Scholar] [CrossRef]
- Galan, A.; Cervero, F. Painful stimuli induce in vivo phosphorylation and membrane mobilization of mouse spinal cord NKCC1 co-transporter. Neuroscience 2005, 133, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, M.H.; Cervero, F. Role of the NKCC1 co-transporter in sensitization of spinal nociceptive neurons. Pain 2010, 151, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.C.; Wang, Y.Y.; Tian, L.X.; Yan, X.J.; Guo, Y.X.; Zhao, Y.L.; Baba, S.S.; Jia, H.; Wang, H.S.; Li, M.; et al. Spinal 5-HT(2A) receptor is involved in electroacupuncture inhibition of chronic pain. Mol. Pain 2022, 18, 17448069221087583. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, F.; Wang, L.; Xu, Y.; Lv, L.; Duan, W.; Bai, R.; Meng, Z.; Shao, X. Involvement of the BDNF-TrkB-KCC2 pathway in neuropathic pain after brachial plexus avulsion. Brain Behav. 2022, 12, e2464. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Chen, Y.; Jiang, C.; Chen, G.; Wang, K.; Chandra, S.; Bortsov, A.; Lioudyno, M.; Zeng, Q.; Wang, P.; et al. Repurposing cancer drugs identifies kenpaullone which ameliorates pathologic pain in preclinical models via normalization of inhibitory neurotransmission. Nat. Commun. 2021, 12, 6208. [Google Scholar] [CrossRef] [PubMed]
- Machelska, H.; Celik, M. Recent advances in understanding neuropathic pain: Glia, sex differences, and epigenetics. F1000Res 2016, 5, 2743. [Google Scholar] [CrossRef] [PubMed]
- Mapplebeck, J.C.S.; Lorenzo, L.E.; Lee, K.Y.; Gauthier, C.; Muley, M.M.; De Koninck, Y.; Prescott, S.A.; Salter, M.W. Chloride Dysregulation through Downregulation of KCC2 Mediates Neuropathic Pain in Both Sexes. Cell Rep. 2019, 28, 590–596.e594. [Google Scholar] [CrossRef]
- Dedek, A.; Xu, J.; Lorenzo, L.; Godin, A.G.; Kandegedara, C.M.; Glavina, G.; Landrigan, J.A.; Lombroso, P.J.; De Koninck, Y.; Tsai, E.C.; et al. Sexual dimorphism in a neuronal mechanism of spinal hyperexcitability across rodent and human models of pathological pain. Brain 2022, 145, 1124–1138. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, F.; Trang, T.; Mattioli, T.A.; Laffray, S.; Del’sGuidice, T.; Lorenzo, L.E.; Castonguay, A.; Doyon, N.; Zhang, W.; Godin, A.G.; et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl⁻ homeostasis. Nat. Neurosci. 2013, 16, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrini, F.; Lorenzo, L.E.; Godin, A.G.; Quang, M.L.; De Koninck, Y. Enhancing KCC2 function counteracts morphine-induced hyperalgesia. Sci. Rep. 2017, 7, 3870. [Google Scholar] [CrossRef] [PubMed]
- Hook, M.A.; Liu, G.T.; Washburn, S.N.; Ferguson, A.R.; Bopp, A.C.; Huie, J.R.; Grau, J.W. The impact of morphine after a spinal cord injury. Behav. Brain Res. 2007, 179, 281–293. [Google Scholar] [CrossRef]
- Hook, M.A.; Moreno, G.; Woller, S.; Puga, D.; Hoy, K., Jr.; Balden, R.; Grau, J.W. Intrathecal morphine attenuates recovery of function after a spinal cord injury. J. Neurotrauma 2009, 26, 741–752. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Morgado, C.; Pinto-Ribeiro, F.; Tavares, I. Diabetes affects the expression of GABA and potassium chloride cotransporter in the spinal cord: A study in streptozotocin diabetic rats. Neurosci. Lett. 2008, 438, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Jolivalt, C.G.; Lee, C.A.; Ramos, K.M.; Calcutt, N.A. Allodynia and hyperalgesia in diabetic rats are mediated by GABA and depletion of spinal potassium-chloride co-transporters. Pain 2008, 140, 48–57. [Google Scholar] [CrossRef]
- Gwak, Y.S.; Hulsebosch, C.E. GABA and central neuropathic pain following spinal cord injury. Neuropharmacology 2011, 60, 799–808. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. About Epilepsy. Available online: https://www.cdc.gov/epilepsy/about/fast-facts.htm (accessed on 3 October 2021).
- World Health Organization. about Epilepsy. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 3 October 2020).
- Begley, C.E.; Famulari, M.; Annegers, J.F.; Lairson, D.R.; Reynolds, T.F.; Coan, S.; Dubinsky, S.; Newmark, M.E.; Leibson, C.; So, E.L.; et al. The cost of epilepsy in the United States: An estimate from population-based clinical and survey data. Epilepsia 2000, 41, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Duy, P.Q.; David, W.B.; Kahle, K.T. Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front. Cell Neurosci. 2019, 13, 515. [Google Scholar] [CrossRef]
- Wood, J.D. The role of gamma-aminobutyric acid in the mechanism of seizures. Prog. Neurobiol. 1975, 5, 77–95. [Google Scholar] [CrossRef]
- Moore, Y.E.; Kelley, M.R.; Brandon, N.J.; Deeb, T.Z.; Moss, S.J. Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci. 2017, 40, 555–571. [Google Scholar] [CrossRef]
- Moore, Y.E.; Deeb, T.Z.; Chadchankar, H.; Brandon, N.J.; Moss, S.J. Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. USA 2018, 115, 10166–10171. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O.; Scharfman, H.E. Temporal Lobe Epilepsy and the BDNF Receptor, TrkB. In Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Kahle, K.T.; Khanna, A.R.; Duan, J.; Staley, K.J.; Delpire, E.; Poduri, A. The KCC2 Cotransporter and Human Epilepsy: Getting Excited About Inhibition. Neuroscientist 2016, 22, 555–562. [Google Scholar] [CrossRef]
- Stödberg, T.; McTague, A.; Ruiz, A.J.; Hirata, H.; Zhen, J.; Long, P.; Farabella, I.; Meyer, E.; Kawahara, A.; Vassallo, G.; et al. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 2015, 6, 8038. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Palma, E.; Roseti, C.; Lauro, C.; Cipriani, R.; de Groot, M.; Aronica, E.; Limatola, C. Anomalous levels of Cl- transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia 2011, 52, 1635–1644. [Google Scholar] [CrossRef]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Cresto, N.; Baulac, M.; Duyckaerts, C.; Kourdougli, N.; Chazal, G.; et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014, 6, 244ra289. [Google Scholar] [CrossRef]
- Sullivan, B.J.; Kipnis, P.A.; Carter, B.M.; Shao, L.R.; Kadam, S.D. Targeting ischemia-induced KCC2 hypofunction rescues refractory neonatal seizures and mitigates epileptogenesis in a mouse model. Sci. Signal 2021, 14, eabg2648. [Google Scholar] [CrossRef]
- Eftekhari, S.; Mehvari Habibabadi, J.; Najafi Ziarani, M.; Hashemi Fesharaki, S.S.; Gharakhani, M.; Mostafavi, H.; Joghataei, M.T.; Beladimoghadam, N.; Rahimian, E.; Hadjighassem, M.R. Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia 2013, 54, e9–e12. [Google Scholar] [CrossRef]
- Rahmanzadeh, R.; Mehrabi, S.; Barati, M.; Ahmadi, M.; Golab, F.; Kazmi, S.; Joghataei, M.T.; Seifi, M.; Gholipourmalekabadi, M. Effect of Co-administration of Bumetanide and Phenobarbital on Seizure Attacks in Temporal Lobe Epilepsy. Basic Clin. Neurosci. 2018, 9, 408–416. [Google Scholar] [CrossRef]
- Ziobro, J.M.; Eschbach, K.; Shellhaas, R.A. Novel Therapeutics for Neonatal Seizures. Neurotherapeutics 2021, 18, 1564–1581. [Google Scholar] [CrossRef]
- Deidda, G.; Parrini, M.; Naskar, S.; Bozarth, I.F.; Contestabile, A.; Cancedda, L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015, 21, 318–326. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Nomura, T.; Xu, J.; Contractor, A. The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. 2014, 34, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.T.; Armstrong, J.; Roche, A.; Ortez, C.; Pérez, A.; O’Callaghan Mdel, M.; Pereira, A.; Sanmartí, F.; Ormazábal, A.; Artuch, R.; et al. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS ONE 2013, 8, e68851. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.; Gage, F.H.; et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef]
- National Institute on Drug Abuse. Trends and Statistics; Costs Substance Abuse. Available online: https://www.drugabuse.gov/drug-topics/trends-statistics/costs-substance-abuse (accessed on 17 August 2020).
- CDC, N.C.f.H.S. Drug Overdose Deaths in the U.S. Top 100,000 Annually. 2021. Available online: https://www.cdc.gov/nchs/pressroom/nchs_press_releases/2021/20211117.htm (accessed on 19 July 2022).
- National Institute on Drug Abuse. Trends and Statistics; Overdose Death Rates. 2020. Available online: https://www.drugabuse.gov/drug-topics/trends-statistics/overdose-death-rates (accessed on 17 August 2020).
- American Psychiatric Association. Substance Use Disorders. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Berridge, K.C.; Robinson, T.E. Parsing reward. Trends Neurosci. 2003, 26, 507–513. [Google Scholar] [CrossRef]
- Laviolette, S.R.; Gallegos, R.A.; Henriksen, S.J.; van der Kooy, D. Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat. Neurosci. 2004, 7, 160–169. [Google Scholar] [CrossRef]
- Churchill, L.; Dilts, R.P.; Kalivas, P.W. Autoradiographic localization of gamma-aminobutyric acidA receptors within the ventral tegmental area. Neurochem. Res. 1992, 17, 101–106. [Google Scholar] [CrossRef]
- Klitenick, M.A.; DeWitte, P.; Kalivas, P.W. Regulation of somatodendritic dopamine release in the ventral tegmental area by opioids and GABA: An in vivo microdialysis study. J. Neurosci. 1992, 12, 2623–2632. [Google Scholar] [CrossRef]
- Taylor, A.M.; Castonguay, A.; Ghogha, A.; Vayssiere, P.; Pradhan, A.A.; Xue, L.; Mehrabani, S.; Wu, J.; Levitt, P.; Olmstead, M.C.; et al. Neuroimmune Regulation of GABAergic Neurons Within the Ventral Tegmental Area During Withdrawal from Chronic Morphine. Neuropsychopharmacology 2016, 41, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Vihavainen, T.; Relander, T.R.; Leiviskä, R.; Airavaara, M.; Tuominen, R.K.; Ahtee, L.; Piepponen, T.P. Chronic nicotine modifies the effects of morphine on extracellular striatal dopamine and ventral tegmental GABA. J. Neurochem. 2008, 107, 844–854. [Google Scholar] [CrossRef]
- Doyon, W.M.; Dong, Y.; Ostroumov, A.; Thomas, A.M.; Zhang, T.A.; Dani, J.A. Nicotine decreases ethanol-induced dopamine signaling and increases self-administration via stress hormones. Neuron 2013, 79, 530–540. [Google Scholar] [CrossRef]
- Ostroumov, A.; Wittenberg, R.E.; Kimmey, B.A.; Taormina, M.B.; Holden, W.M.; McHugh, A.T.; Dani, J.A. Acute Nicotine Exposure Alters Ventral Tegmental Area Inhibitory Transmission and Promotes Diazepam Consumption. eNeuro 2020, 7, ENEURO.0348-19.2020. [Google Scholar] [CrossRef]
- Thomas, A.M.; Ostroumov, A.; Kimmey, B.A.; Taormina, M.B.; Holden, W.M.; Kim, K.; Brown-Mangum, T.; Dani, J.A. Adolescent Nicotine Exposure Alters GABA(A) Receptor Signaling in the Ventral Tegmental Area and Increases Adult Ethanol Self-Administration. Cell Rep. 2018, 23, 68–77. [Google Scholar] [CrossRef]
- Ostchega, Y.F.C.; Nwankwo, T.; Nguyen, D.T. Hypertension prevalence among adults aged 18 and over: United States, 2017–2018. In NCHS Data Brief; National Center for Health Statistics: Hyattsville, MD, USA, 2018; Volume 364. [Google Scholar]
- Ye, Z.Y.; Li, D.P.; Byun, H.S.; Li, L.; Pan, H.L. NKCC1 upregulation disrupts chloride homeostasis in the hypothalamus and increases neuronal activity-sympathetic drive in hypertension. J. Neurosci. 2012, 32, 8560–8568. [Google Scholar] [CrossRef]
- Kim, Y.B.; Kim, Y.S.; Kim, W.B.; Shen, F.Y.; Lee, S.W.; Chung, H.J.; Kim, J.S.; Han, H.C.; Colwell, C.S.; Kim, Y.I. GABAergic excitation of vasopressin neurons: Possible mechanism underlying sodium-dependent hypertension. Circ. Res. 2013, 113, 1296–1307. [Google Scholar] [CrossRef]
- He, D.; Chen, H.; Zeng, M.; Xia, C.; Wang, J.; Shen, L.; Zhu, D.; Chen, Y.; Wang, J. Asthmatic Airway Vagal Hypertonia Involves Chloride Dyshomeostasis of Preganglionic Neurons in Rats. Front. Neurosci. 2020, 14, 31. [Google Scholar] [CrossRef]
- Wang, S.; Xiang, Y.Y.; Ellis, R.; Wattie, J.; Feng, M.; Inman, M.D.; Lu, W.Y. Effects of furosemide on allergic asthmatic responses in mice. Clin. Exp. Allergy 2011, 41, 1456–1467. [Google Scholar] [CrossRef]
- Polosa, R.; Rajakulasingam, K.; Prosperini, G.; Church, M.K.; Holgate, S.T. Relative potencies and time course of changes in adenosine 5′-monophosphate airway responsiveness with inhaled furosemide and bumetanide in asthma. J. Allergy Clin. Immunol. 1993, 92, 288–297. [Google Scholar] [CrossRef]
- Inokuchi, R.; Aoki, A.; Aoki, Y.; Yahagi, N. Effectiveness of inhaled furosemide for acute asthma exacerbation: A meta-analysis. Crit. Care 2014, 18, 621. [Google Scholar] [CrossRef] [Green Version]
- Hungin, A.P.; Whorwell, P.J.; Tack, J.; Mearin, F. The prevalence, patterns and impact of irritable bowel syndrome: An international survey of 40,000 subjects. Aliment. Pharm. 2003, 17, 643–650. [Google Scholar] [CrossRef]
- Tang, D.; Qian, A.H.; Song, D.D.; Ben, Q.W.; Yao, W.Y.; Sun, J.; Li, W.G.; Xu, T.L.; Yuan, Y.Z. Role of the potassium chloride cotransporter isoform 2-mediated spinal chloride homeostasis in a rat model of visceral hypersensitivity. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G767–G778. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 2012, 18, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Grau, J.W.; Hudson, K.E.; Tarbet, M.M.; Strain, M.M. Behavioral Studies of Spinal Conditioning: The Spinal Cord Is Smarter Than You Think It Is. Exp. Psychol. Anim. Learn. Cogn. 2022; advance online publication. [Google Scholar]
- Ko, M.C.; Lee, M.C.; Amstislavskaya, T.G.; Tikhonova, M.A.; Yang, Y.L.; Lu, K.T. Inhibition of NKCC1 attenuated hippocampal LTP formation and inhibitory avoidance in rat. PLoS ONE 2014, 9, e106692. [Google Scholar] [CrossRef]
- Doyon, N.; Prescott, S.A.; De Koninck, Y. Mild KCC2 Hypofunction Causes Inconspicuous Chloride Dysregulation that Degrades Neural Coding. Front. Cell Neurosci. 2015, 9, 516. [Google Scholar] [CrossRef]
- Beggs, S.; Salter, M.W. The known knowns of microglia-neuronal signalling in neuropathic pain. Neurosci. Lett. 2013, 557, 37–42. [Google Scholar] [CrossRef]
- Lu, V.B.; Biggs, J.E.; Stebbing, M.J.; Balasubramanyan, S.; Todd, K.G.; Lai, A.Y.; Colmers, W.F.; Dawbarn, D.; Ballanyi, K.; Smith, P.A. Brain-derived neurotrophic factor drives the changes in excitatory synaptic transmission in the rat superficial dorsal horn that follow sciatic nerve injury. J. Physiol. 2009, 587, 1013–1032. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Cherubini, E. The GABA Polarity Shift and Bumetanide Treatment: Making Sense Requires Unbiased and Undogmatic Analysis. Cells 2022, 11, 396. [Google Scholar] [CrossRef]
- Löscher, W.; Kaila, K. Reply to the commentary by Ben-Ari and Delpire: Bumetanide and neonatal seizures: Fiction versus reality. Epilepsia 2021, 62, 941–946. [Google Scholar] [CrossRef]
- Tang, B.L. The Expanding Therapeutic Potential of Neuronal KCC2. Cells 2020, 9, 240. [Google Scholar] [CrossRef] [Green Version]
- Hegarty, S.V.; Stanicka, J. K(+)/Cl(−) co-transporter-2 upmodulation: A multi-modal therapy to treat spinal cord injury. Neural. Regen Res. 2022, 17, 1984–1986. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hudson, K.E.; Grau, J.W. Ionic Plasticity: Common Mechanistic Underpinnings of Pathology in Spinal Cord Injury and the Brain. Cells 2022, 11, 2910. https://doi.org/10.3390/cells11182910
Hudson KE, Grau JW. Ionic Plasticity: Common Mechanistic Underpinnings of Pathology in Spinal Cord Injury and the Brain. Cells. 2022; 11(18):2910. https://doi.org/10.3390/cells11182910
Chicago/Turabian StyleHudson, Kelsey E., and James W. Grau. 2022. "Ionic Plasticity: Common Mechanistic Underpinnings of Pathology in Spinal Cord Injury and the Brain" Cells 11, no. 18: 2910. https://doi.org/10.3390/cells11182910