
L-Ornithine L-Aspartate Restores Mitochondrial Function and Modulates Intracellular Calcium Homeostasis in Parkinson’s Disease Models
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Determination of Mitochondrial Oxidative Activity
2.4. Confocal Microscopy and Mitochondrial Function
2.5. Western Blot Analysis
2.6. [Ca2+]i Measurement
2.7. Nitric Oxide Detection
2.8. Statistical Analysis
3. Results
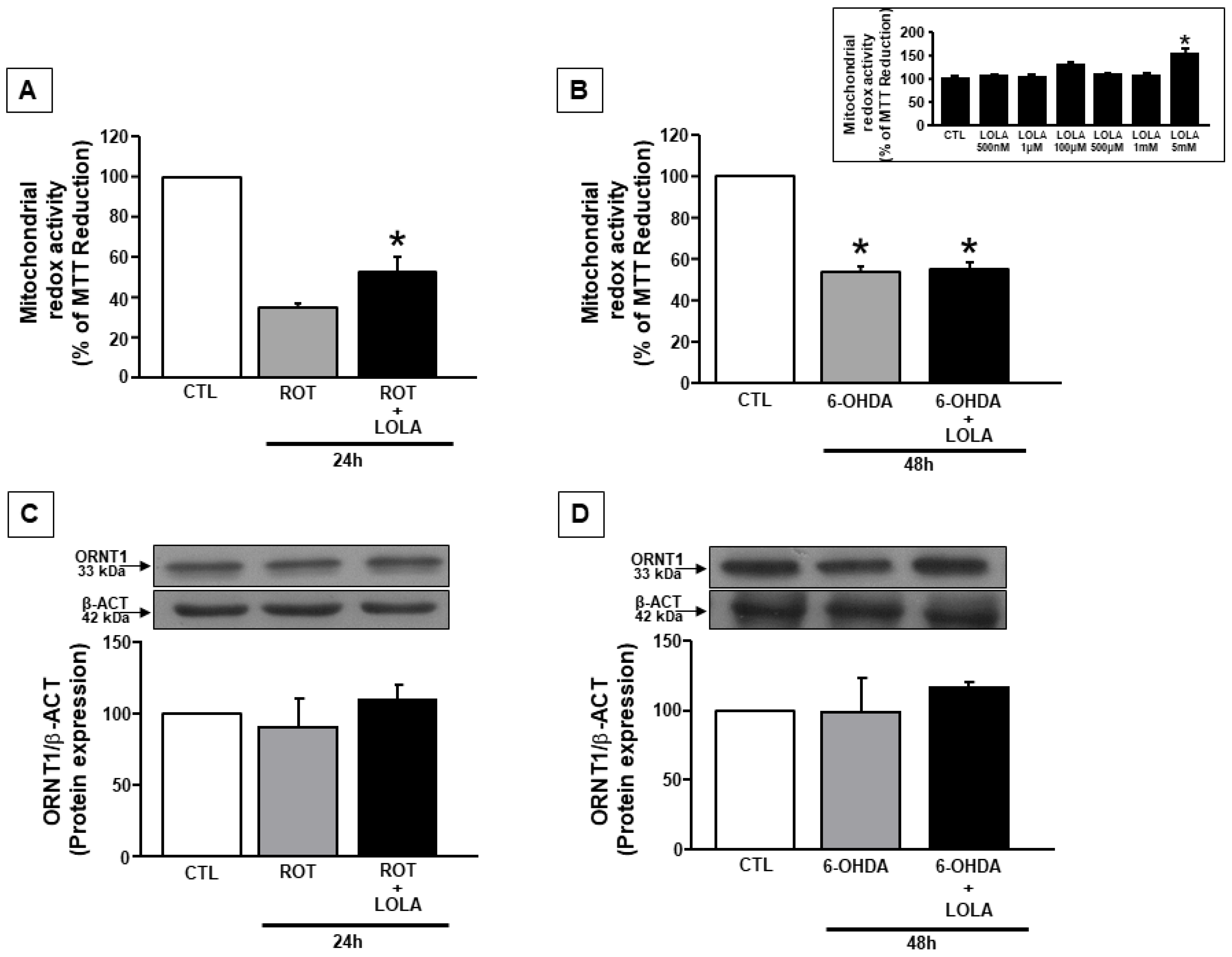
3.1. LOLA Treatment Improves Mitochondrial Redox Activity in SH-SY5Y Cells Treated with ROT and 6-OHDA
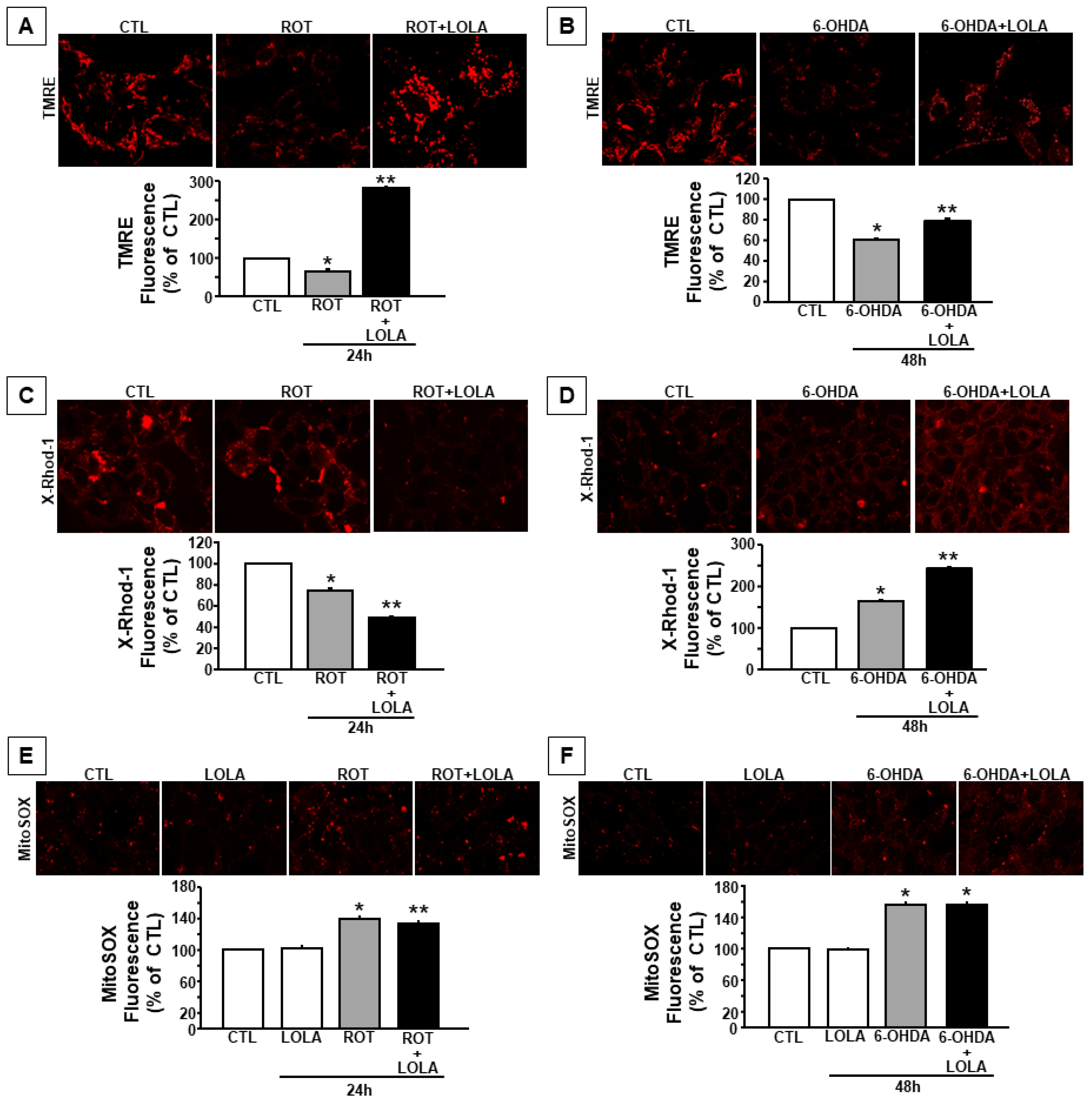
3.2. Mitochondrial Dysfunction Induced by ROT and 6-OHDA Exposure in SH-SY5Y Cells Is Counteracted by LOLA Treatment
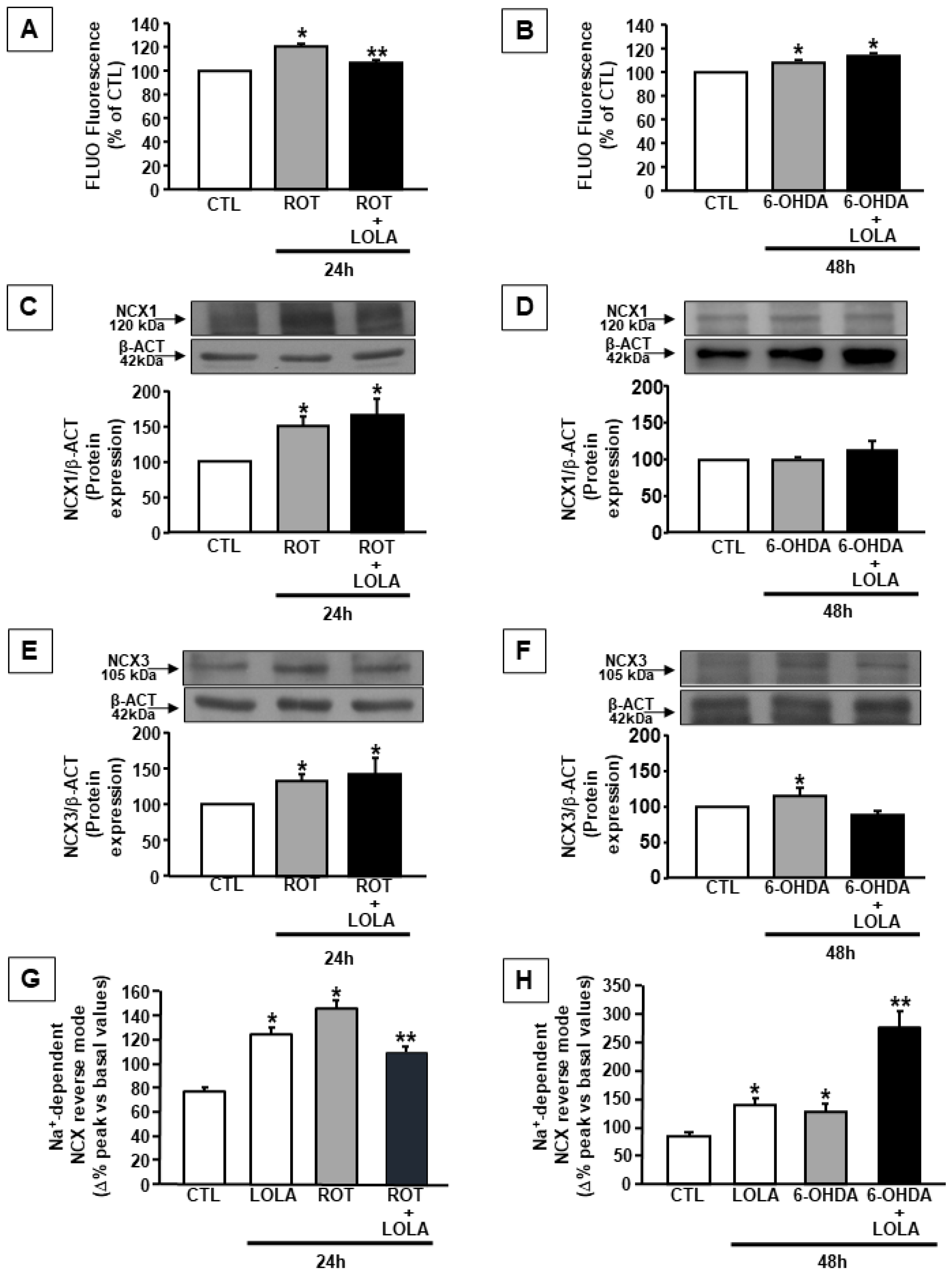
3.3. LOLA Treatment Differently Regulates the Effects of ROT- and 6-OHDA on NCX1 and NCX3 Expression and Activity in SH-SY5Y Cells
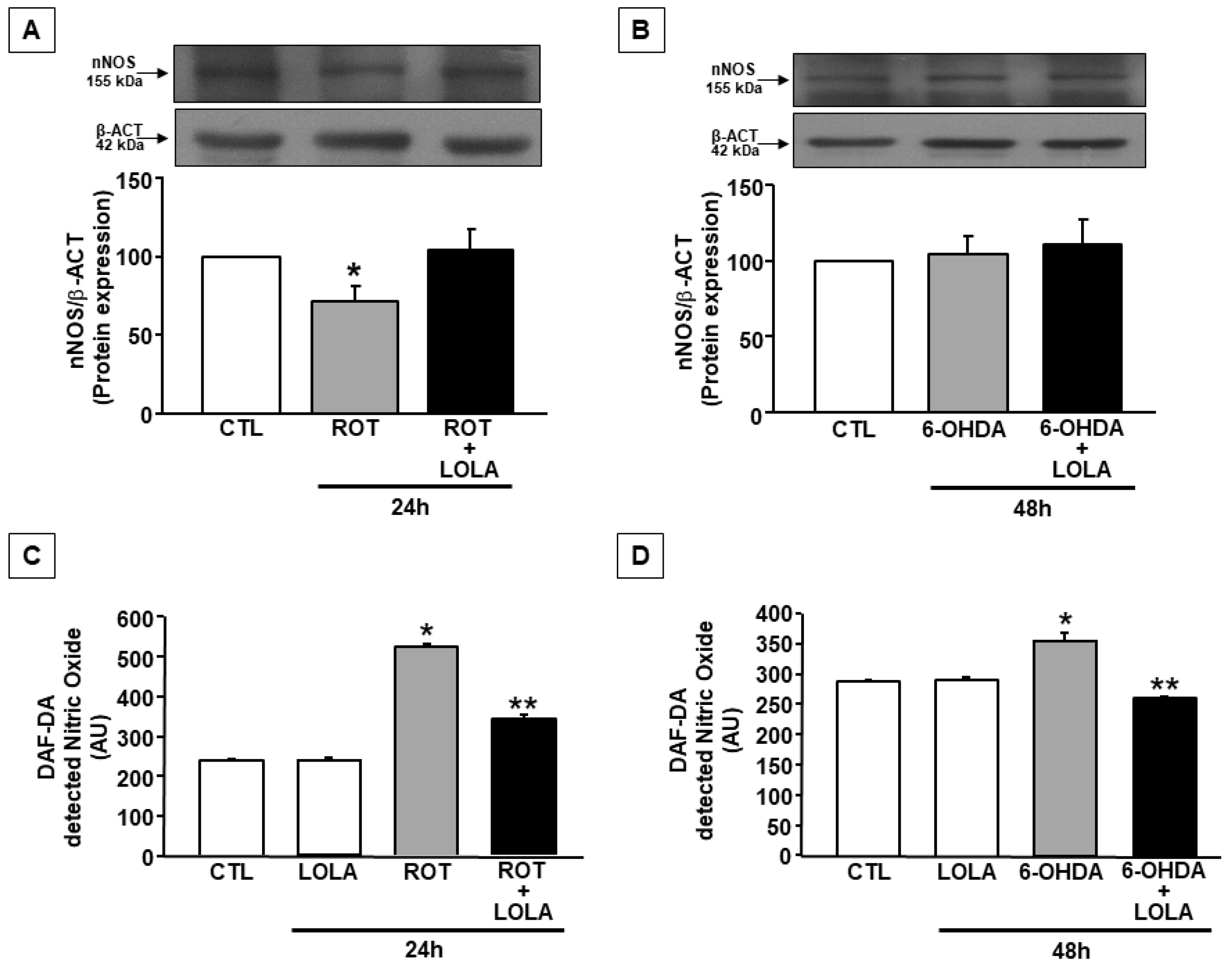
3.4. LOLA Treatment Reduces NO Production in SH-SY5Y Cells Exposed to ROT and 6-OHDA
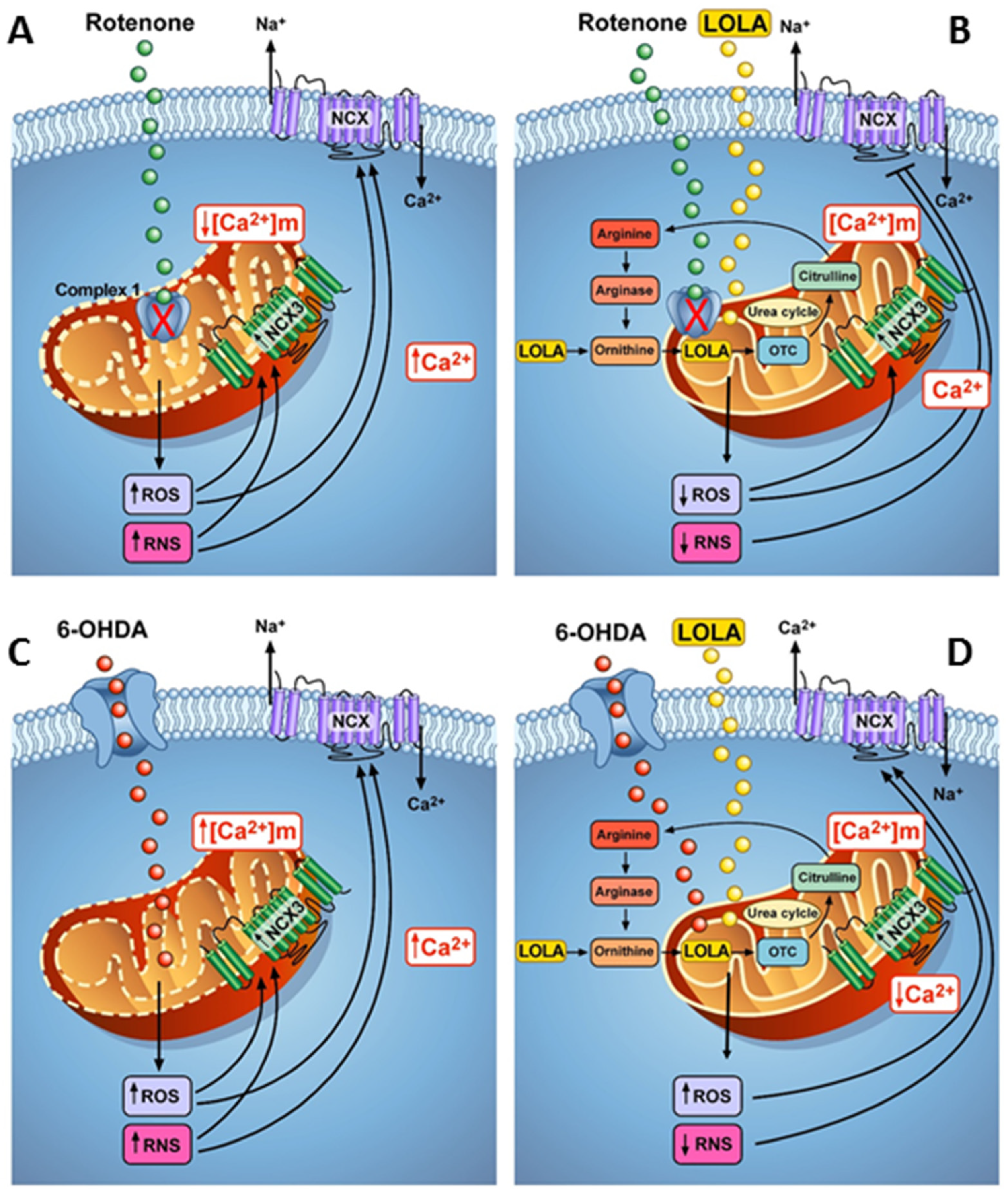
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.; Lees, A.; Stern, M. Milestones in Parkinson’s disease—Clinical and pathologic features. Mov. Disord. 2011, 26, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lan, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S. Past, present, and future of Parkinson’s disease: A special essay on the 200th anniversary of the shaking palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Caroline, M.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Cookson, M.R. The biochemistry of Parkinson’s disease. Annu. Rev. Biochem. 2005, 74, 29–52. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef]
- Pang, S.Y.Y.; Ho, P.W.L.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 16, 8–23. [Google Scholar] [CrossRef]
- Costa, G.; Sisalli, M.J.; Simola, N.; Della Notte, S.; Casu, M.A.; Serra, M.; Pinna, A.; Feliciello, A.; Annunziato, L.; Scorziello, A.; et al. Gender Differences in Neurodegeneration, Neuroinflammation and Na+-Ca2+ Exchangers in the Female A53T Transgenic Mouse Model of Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 118. [Google Scholar] [CrossRef]
- Di Martino, R.; Sisalli, M.J.; Sirabella, R.; Della Notte, S.; Borzacchiello, D.; Feliciello, A.; Annunziato, L.; Scorziello, A. Ncx3-Induced Mitochondrial Dysfunction in Midbrain Leads to Neuroinflammation in Striatum of A53t-α-Synuclein Transgenic Old Mice. Int. J. Mol. Sci. 2021, 22, 8177. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Cur. Neurol. Neurosc. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [PubMed]
- Grünewalda, A.; Kumarc, K.R.; Suec, C. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobio. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.S.; Swerdlow, R.H.; Miller, S.W.; Sheeman, B.; Parker, W.D.; Davis, R.E. Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer’s disease and Parkinson’s disease. Ann. N. Y. Acad. Sci. 1999, 893, 176–191. [Google Scholar] [CrossRef]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta 2009, 1792, 651–663. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Dobrowolny, H.; Keilhoff, G.; Steiner, J. In human brain ornithine transcarbamylase (OTC) immunoreactivity is strongly expressed in a small number of nitrergic neurons. Metab. Brain Dis. 2017, 32, 2143–2147. [Google Scholar] [CrossRef]
- Zanatta, A.; Rodrigues, M.D.N.; Amaral, A.U.; Souza, D.G.; Quincozes-Santos, A.; Wajner, M. Ornithine and Homocitrulline Impair Mitochondrial Function, Decrease Antioxidant Defenses and Induce Cell Death in Menadione-Stressed Rat Cortical Astrocytes: Potential Mechanisms of Neurological Dysfunction in HHH Syndrome. Neurochem. Res. 2016, 41, 2190–2198. [Google Scholar] [CrossRef]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Meth. 1989, 119, 203–210. [Google Scholar] [CrossRef]
- Amoroso, S.; Gioielli, A.; Cataldi, M.; Di Renzo, G.; Annunziato, L. In the neuronal cell line SH-SY5Y, oxidative stress-induced free radical overproduction causes cell death without any participation of intracellular Ca2+ increase. Biochim. Biophys. Acta 1999, 1452, 151–160. [Google Scholar] [CrossRef]
- Livigni, A.; Scorziello, A.; Agnese, S.; Adornetto, A.; Carlucci, A.; Garbi, A.; Castaldo, I.; Annunziato, L.; Avvedimento, V.E.; Feliciello, A. Mitochondrial AKAP121 links cAMP and src signalling to oxidative metabolism. Mol. Biol. Cell 2006, 17, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Sisalli, M.J.; Ianniello, G.; Savoia, C.; Cuomo, O.; Annunziato, L.; Scorziello, A. Knocking-out the Siah2 E3 ubiquitin ligase prevents mitochondrial NCX3 degradation, regulates mitochondrial fission and fusion, and restores mitochondrial function in hypoxic neurons. Cell Commun. Signal. 2020, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Testai, L.; Barrese, V.; Soldovieri, M.V.; Ambrosino, P.; Martelli, A.; Vinciguerra, I.; Miceli, F.; Greenwood, I.A.; Curtis, M.J.; Breschi, M.C.; et al. Expression and function of Kv7.4 channels in rat cardiac mitochondria: Possible targets for cardioprotection. Cardiovasc. Res. 2016, 110, 40–50. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, S.; Greco, F.; Patrizia Falanga, A.; Tedeschi, V.; Piccialli, I.; Marzano, M.; Terracciano, M.; Secondo, A.; Roviello, G.; Oliviero, G.; et al. Probing the Ca2+ mobilizing properties on primary cortical neurons of a new stable cADPR mimic. Bioorg. Chem. 2021, 117, 105401–105414. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Secondo, A.; Montoro, P.; Piacente, S.; Rimoli, M.G.; Minale, M.; de Caprariis, P.; Annunziato, L. Galactosyl Derivatives of L-Arginine and D-Arginine: Synthesis, Stability, Cell Permeation, and Nitric Oxide Production in Pituitary GH3 Cells. J. Med. Chem. 2006, 49, 4826–4833. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Llewelyn Roderick, H. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Denton, R.M.; McCormack, J.G. The role of calcium in the regulation of mitochondrial metabolism. Biochem. Soc. Trans. 1980, 8, 266–268. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef]
- Denton, R.; McCormack, J. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu. Rev. Physiol. 1990, 52, 451–466. [Google Scholar] [CrossRef]
- Scorziello, A.; Savoia, C.; Sisalli, M.J.; Adornetto, A.; Secondo, A.; Boscia, F. NCX3 regulates mitochondrial Ca2+ handling through the AKAP121-anchored signaling complex and prevents hypoxia-induced neuronal death. J. Cell Sci. 2013, 126, 5566–5577. [Google Scholar] [PubMed]
- Secondo, A.; Staiano, R.I.; Scorziello, A.; Sirabella, R.; Boscia, F.; Adornetto, A.; Valsecchi, V.; Molinaro, P.; Canzoniero, L.M.; Di Renzo, G.; et al. BHK cells transfected with NCX3 are more resistant to hypoxia followed by reoxygenation than those transfected with NCX1 and NCX2: Possible relationship with mitochondrial membrane potential. Cell Calcium 2007, 42, 521–535. [Google Scholar] [CrossRef] [PubMed]
- El-Bassossy, H.M.; El-Fawal, R.; Fahmy, A.; Watson, M.L. Arginase inhibition alleviates hypertension in the metabolic syndrome. Br. J. Pharmacol. 2013, 169, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, L.; Pignataro, G.; Di Renzo, G.F. Pharmacology of brain Na+/Ca2+ exchanger: From molecular biology to therapeutic perspectives. Pharmacol. Rev. 2004, 56, 633–654. [Google Scholar] [CrossRef]
- Elfering, S.L.; Sarkela, T.M.; Giulivi, C. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem. 2002, 277, 38079–38086. [Google Scholar] [CrossRef]
- Kanai, A.J.; Pearce, L.L.; Clemens, P.R.; Birder, L.A.; VanBibber, M.M.; Choi, S.Y.; de Groat, W.C.; Peterson, J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. USA 2001, 98, 14126–14131. [Google Scholar] [CrossRef]
- Finocchietto, O.; Barreyro, F.; Holod, S.; Peralta, J.; Franco, M.C.; Mendez, C.; Converso, D.P.; Estévez, A.; Carreras, M.C.; Poderoso, J.J. Control of muscle mitochondria by insulin entails activation of Akt2-mtNOS pathway: Imlpications for the metabolic syndrome. PLoS ONE 2008, 3, e1749. [Google Scholar] [CrossRef]
- Parihar, M.S.; Nazarewicz, R.R.; Kincaid, E.; Bringold, U.; Ghafourifar, P. Association of mitochondrial nitric oxide synthase activity with respiratory chain complex I. Biochem. Biophys. Res. Commun. 2008, 366, 23–28. [Google Scholar] [CrossRef]
- Bombicino, S.S.; Iglesias, D.E.; Zaobornyj, T.; Boveris, A.; Valdez, L.B. Mitochondrial nitric oxide production supported by reverse electron transfer. Arch. Biochem. Biophys. 2016, 607, 8–19. [Google Scholar] [CrossRef]
- Wu, Y.N.; Sudarshan, V.K.; Zhu, S.C.; Shao, Y.F.; Kim, S.J.; Zhang, Y.H. Functional interactions between complex I and complex II with nNOS in regulating cardiac mitochondrial activity in sham and hypertensive rat hearts. Pflugers Arch. -Eur. J. Physiol. 2020, 472, 1743–1755. [Google Scholar] [CrossRef]
- Sirabella, R.; Secondo, A.; Pannaccione, A.; Scorziello, A.; Valsecchi, V.; Adornetto, A.; Bilo, L.; Di Renzo, G.F.; Annunziato, L. Anoxia-induced NF-kappaB-dependent upregulation of NCX1 contributes to Ca2+ refilling into endoplasmic reticulum in cortical neurons. Stroke 2009, 40, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Sisalli, M.J.; Feliciello, A.; Della Notte, S.; Di Martino, R.; Borzacchiello, D.; Annunziato, L.; Scorziello, A. Nuclear-encoded NCX3 and AKAP121: Two novel modulators of mitochondrial calcium efflux in normoxic and hypoxic neurons. Cell Calcium 2020, 87, 102193. [Google Scholar] [CrossRef]
- Pignataro, G.; Gala, R.; Cuomo, O.; Tortiglione, A.; Giaccio, L.; Castaldo, P.; Sirabella, R.; Matrone, C.; Canitano, A.; Amoroso, S.; et al. Two sodium/calcium exchanger gene products, NCX1 and NCX3, play a major role in the development of permanent focal cerebral ischemia. Stroke 2004, 35, 2566–2570. [Google Scholar] [CrossRef] [PubMed]
- Sisalli, M.J.; Secondo, A.; Esposito, A.; Valsecchi, V.; Savoia, C.; Di Renzo, G.F.; Annunziato, L.; Scorziello, A. Endoplasmic reticulum refilling and mitochondrial calcium extrusion promoted in neurons by NCX1 and NCX3 in ischemic preconditioning are determinant for neuroprotection. Cell Death Differ. 2014, 21, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Sirabella, R.; Sisalli, M.J.; Costa, G.; Omura, K.; Ianniello, G.; Pinna, A. NCX1 and NCX3 as potential factors contributing to neurodegeneration and neuroinflammation in the A53T transgenic mouse model of Parkinson’s disease. Cell Death Dis. 2018, 9, 725. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sisalli, M.J.; Della Notte, S.; Secondo, A.; Ventra, C.; Annunziato, L.; Scorziello, A. L-Ornithine L-Aspartate Restores Mitochondrial Function and Modulates Intracellular Calcium Homeostasis in Parkinson’s Disease Models. Cells 2022, 11, 2909. https://doi.org/10.3390/cells11182909
Sisalli MJ, Della Notte S, Secondo A, Ventra C, Annunziato L, Scorziello A. L-Ornithine L-Aspartate Restores Mitochondrial Function and Modulates Intracellular Calcium Homeostasis in Parkinson’s Disease Models. Cells. 2022; 11(18):2909. https://doi.org/10.3390/cells11182909
Chicago/Turabian StyleSisalli, Maria Josè, Salvatore Della Notte, Agnese Secondo, Carmelo Ventra, Lucio Annunziato, and Antonella Scorziello. 2022. "L-Ornithine L-Aspartate Restores Mitochondrial Function and Modulates Intracellular Calcium Homeostasis in Parkinson’s Disease Models" Cells 11, no. 18: 2909. https://doi.org/10.3390/cells11182909