
The “Yin and Yang” of Unfolded Protein Response in Cancer and Immunogenic Cell Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
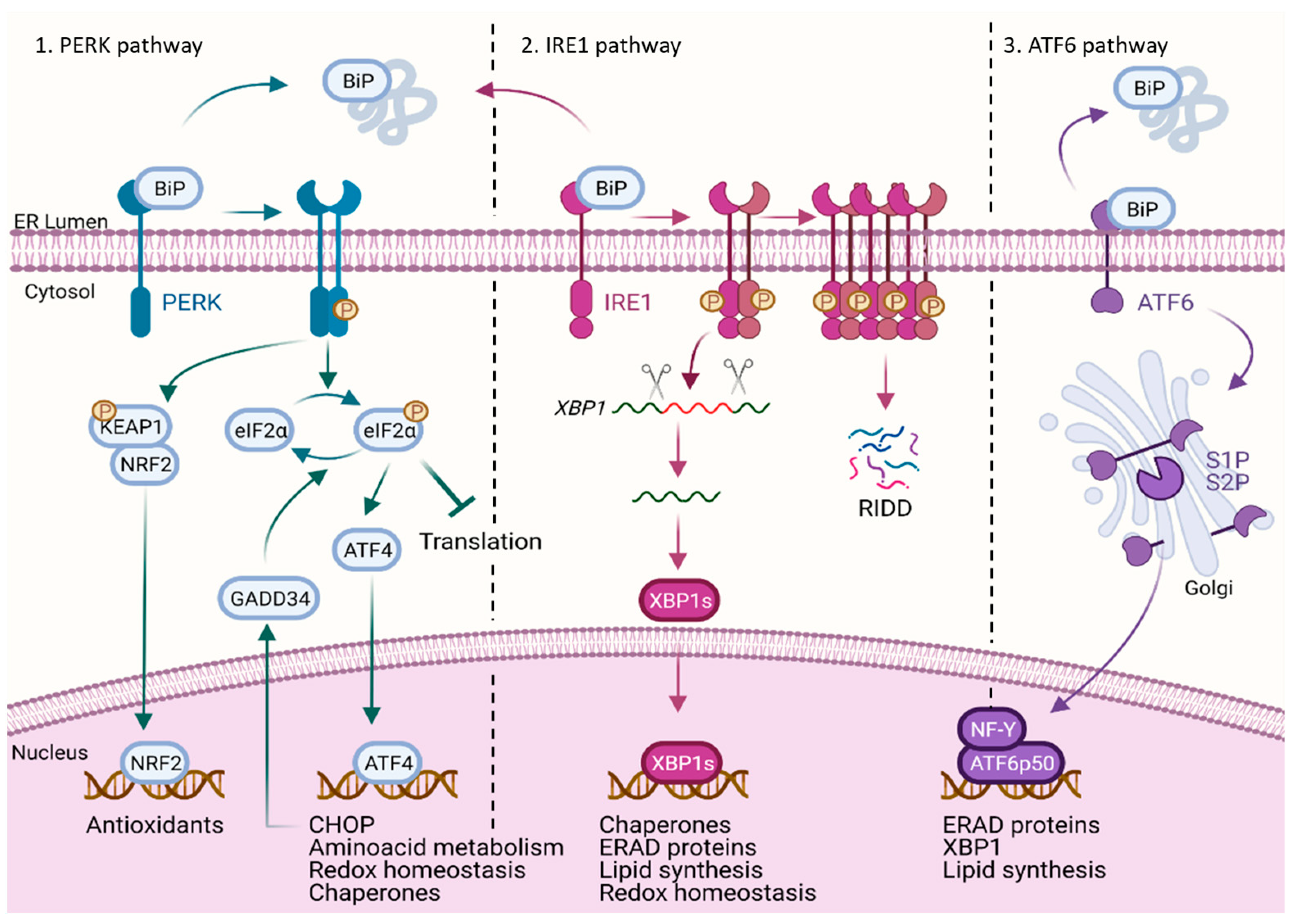
:1. The UPR and Its Main Branches
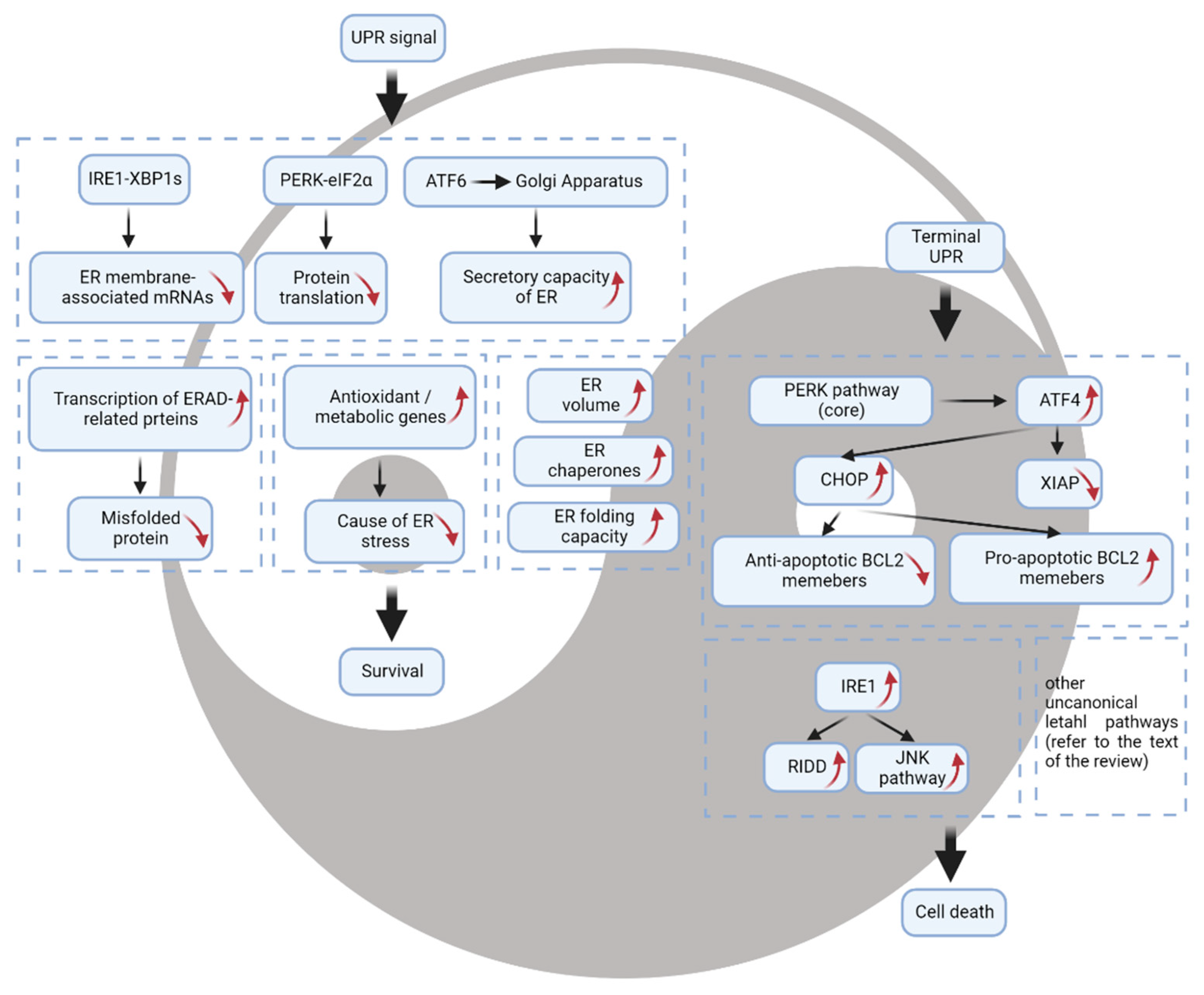
2. The Prosurvival Function of the UPR
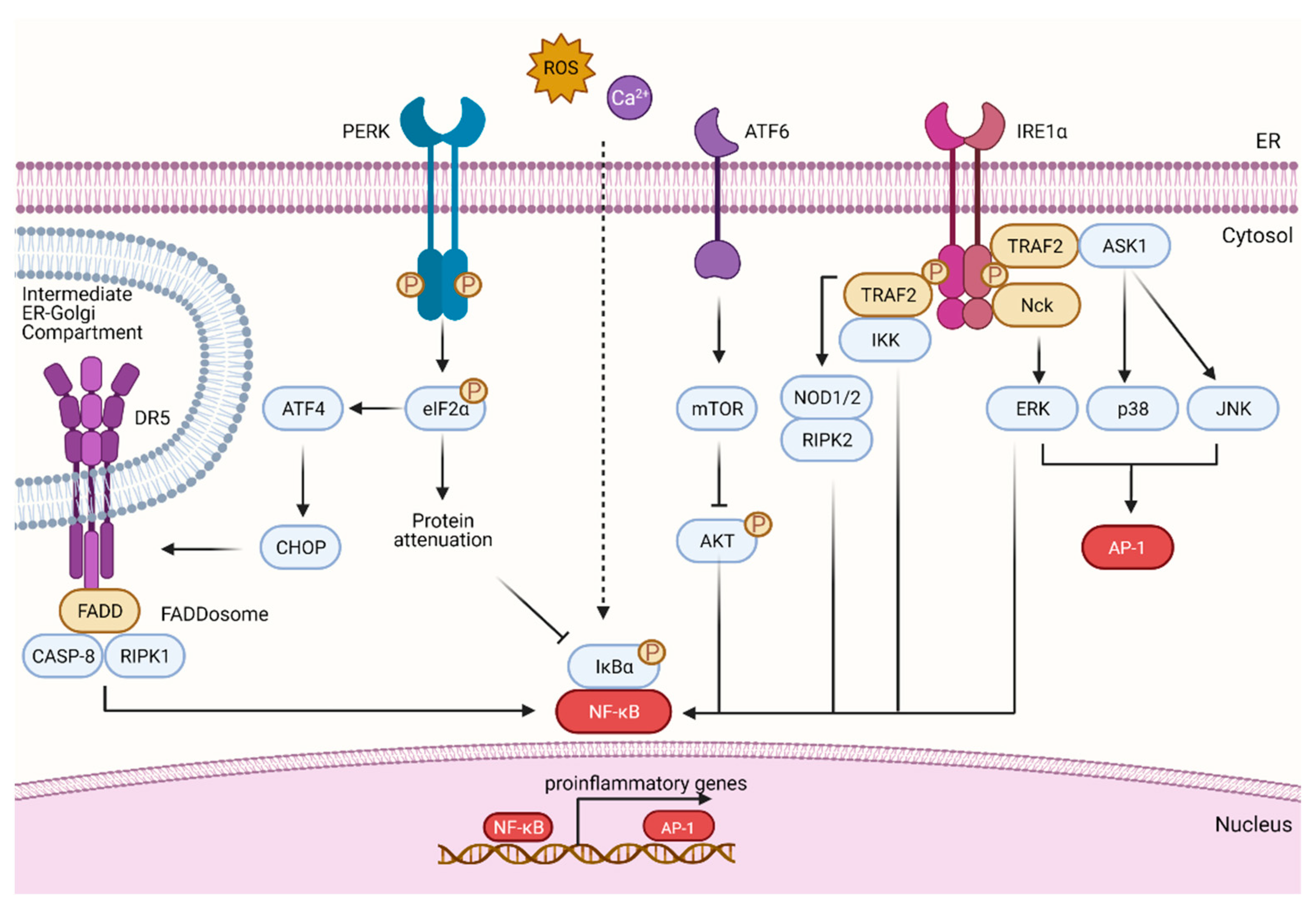
3. The UPR as a Mechanism of Cell Death
4. Proinflammatory Pathways Driven by the UPR
5. Autocrine Role of the UPR in Tumor Cells
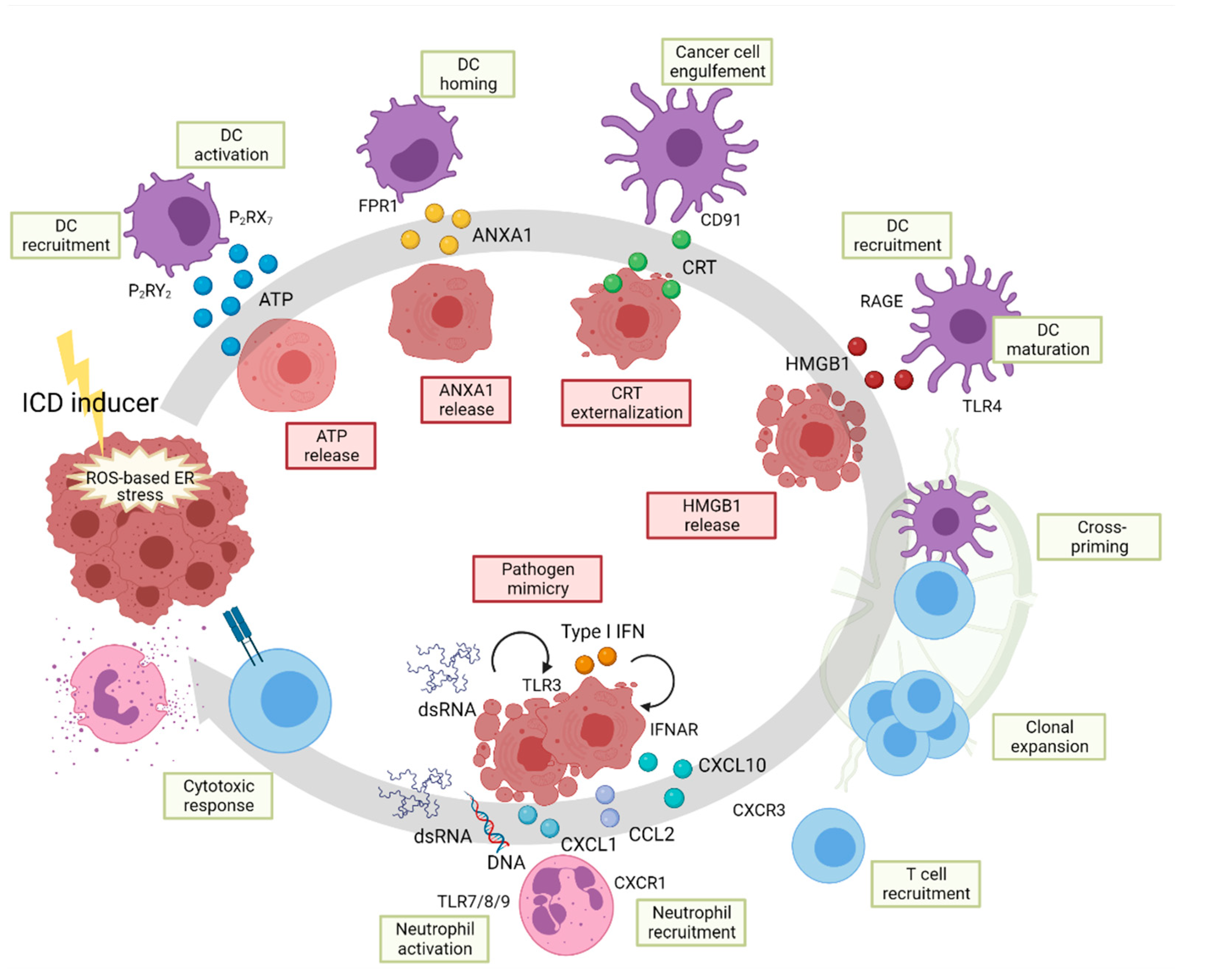
6. The UPR in Immunogenic Cell Death
7. Inflammation and ICD
8. Therapeutic Outlook and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Kimata, Y.; Kohno, K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol. 2011, 23, 135–142. [Google Scholar] [CrossRef]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar] [CrossRef]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Ali, M.M. Crystal structures reveal transient PERK luminal domain tetramerization in endoplasmic reticulum stress signaling. EMBO J. 2015, 34, 1589–1600. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- van Anken, E.; Bakunts, A.; Hu, C.-C.A.; Janssens, S.; Sitia, R. Molecular evaluation of endoplasmic reticulum homeostasis meets humoral immunity. Trends Cell Biol. 2021, 31, 529–541. [Google Scholar] [CrossRef]
- Brewer, J.W.; Diehl, J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.E.; Dalton, L.E.; Clarke, H.J.; Malzer, E.; Dominicus, C.S.; Patel, V.; Moorhead, G.; Ron, D.; Marciniak, S.J. Actin dynamics tune the integrated stress response by regulating eukaryotic initiation factor 2α dephosphorylation. eLife 2015, 4, e04872. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Rato, C.; Yan, Y.; Crespillo-Casado, A.; Clarke, H.J.; Harding, H.P.; Marciniak, S.J.; Read, R.J.; Ron, D. G-actin provides substrate-specificity to eukaryotic initiation factor 2α holophosphatases. eLife 2015, 4, e04871. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [Green Version]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [PubMed]
- B’chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, M.G.; Higuchi-Sanabria, R.; Garcia, G.; Tsui, C.K.; Dillin, A. Beyond the cell factory: Homeostatic regulation of and by the UPRER. Sci. Adv. 2020, 6, eabb9614. [Google Scholar] [CrossRef] [PubMed]
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef]
- Misra, J.; Kim, D.-K.; Choi, W.; Koo, S.-H.; Lee, C.-H.; Back, S.-H.; Kaufman, R.J.; Choi, H.-S. Transcriptional cross talk between orphan nuclear receptor ERRγ and transmembrane transcription factor ATF6α coordinates endoplasmic reticulum stress response. Nucleic Acids Res. 2013, 41, 6960–6974. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Agostinis, P. Mitochondria-Associated Membranes and ER Stress. Curr. Top. Microbiol. Immunol. 2018, 414, 73–102. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Giordano, F.; Gerlo, S.; Segura, I.; Van Eygen, S.; Molenberghs, G.; Rocha, S.; Houcine, A.; Derua, R.; Verfaillie, T.; et al. The ER Stress Sensor PERK Coordinates ER-Plasma Membrane Contact Site Formation through Interaction with Filamin-A and F-Actin Remodeling. Mol. Cell 2017, 65, 885–899.e6. [Google Scholar] [CrossRef]
- Tanwar, J.; Motiani, R.K. Role of SOCE architects STIM and Orai proteins in Cell Death. Cell Calcium 2018, 69, 19–27. [Google Scholar] [CrossRef]
- Bhat, T.A.; Chaudhary, A.K.; Kumar, S.; O’Malley, J.; Inigo, J.R.; Kumar, R.; Yadav, N.; Chandra, D. Endoplasmic reticulum-mediated unfolded protein response and mitochondrial apoptosis in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Brewer, J.W.; Diehl, J.A.; Hendershot, L.M. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- León-Annicchiarico, C.L.; Ramírez-Peinado, S.; Domínguez-Villanueva, D.; Gonsberg, A.; Lampidis, T.J.; Muñoz-Pinedo, C. ATF4 mediates necrosis induced by glucose deprivation and apoptosis induced by 2-deoxyglucose in the same cells. FEBS J. 2015, 282, 3647–3658. [Google Scholar] [CrossRef]
- Martín-Pérez, R.; Palacios, C.; Yerbes, R.; Cano-González, A.; Iglesias-Serret, D.; Gil, J.; Reginato, M.J.; López-Rivas, A. Activated ERBB2/HER2 licenses sensitivity to apoptosis upon endoplasmic reticulum stress through a PERK-dependent pathway. Cancer Res. 2014, 74, 1766–1777. [Google Scholar] [CrossRef]
- Glab, J.A.; Doerflinger, M.; Nedeva, C.; Jose, I.; Mbogo, G.W.; Paton, J.C.; Paton, A.W.; Kueh, A.J.; Herold, M.J.; Huang, D.C.; et al. DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 2017, 24, 944–950. [Google Scholar] [CrossRef]
- Lam, M.; Marsters, S.A.; Ashkenazi, A.; Walter, P. Misfolded proteins bind and activate death receptor 5 to trigger apoptosis during unresolved endoplasmic reticulum stress. eLife 2020, 9, e52291. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Messah, C.; Han, J.; LaVail, M.M.; Kaufman, R.J.; Lin, J.H. Translational and posttranslational regulation of XIAP by eIF2α and ATF4 promotes ER stress-induced cell death during the unfolded protein response. Mol. Biol. Cell 2014, 25, 1411–1420. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Nagashima, S.; Shiiba, I.; Uda, A.; Tokuyama, T.; Ito, N.; Fukuda, T.; Matsushita, N.; Ishido, S.; Iwawaki, T.; et al. MITOL prevents ER stress-induced apoptosis by IRE1α ubiquitylation at ER-mitochondria contact sites. EMBO J. 2019, 38, e100999. [Google Scholar] [CrossRef] [PubMed]
- Yamani, L.; Latreille, M.; Larose, L. Interaction of Nck1 and PERK phosphorylated at Y561 negatively modulates PERK activity and PERK regulation of pancreatic β-cell proinsulin content. Mol. Biol. Cell 2014, 25, 702–711. [Google Scholar] [CrossRef]
- Pinkaew, D.; Chattopadhyay, A.; King, M.D.; Chunhacha, P.; Liu, Z.; Stevenson, H.L.; Chen, Y.; Sinthujaroen, P.; McDougal, O.M.; Fujise, K. Fortilin binds IRE1α and prevents ER stress from signaling apoptotic cell death. Nat. Commun. 2017, 8, 18. [Google Scholar] [CrossRef]
- Lisbona, F.; Rojas-Rivera, D.; Thielen, P.; Zamorano, S.; Todd, D.; Martinon, F.; Glavic, A.; Kress, C.; Lin, J.H.; Walter, P.; et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol. Cell 2009, 33, 679–691. [Google Scholar] [CrossRef]
- Rodriguez, D.A.; Zamorano, S.; Lisbona, F.; Rojas-Rivera, D.; Urra, H.; Cubillos-Ruiz, J.R.; Armisen, R.; Henriquez, D.R.; Cheng, E.H.; Letek, M.; et al. BH3-only proteins are part of a regulatory network that control the sustained signalling of the unfolded protein response sensor IRE1α. EMBO J. 2012, 31, 2322–2335. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Agostinis, P. PERK and filamin A in actin cytoskeleton remodeling at ER-plasma membrane contact sites. Mol. Cell. Oncol. 2017, 4, e1340105. [Google Scholar] [CrossRef]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J.M. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Zhao, Y.; Wang, J.; Yang, X.; Li, S.; Wang, Y.; Yang, X.; Fei, J.; Hao, X.; Zhao, Y.; et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int. J. Biol. Sci. 2021, 17, 2703–2717. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J. Exp. Clin. Cancer Res. 2019, 38, 402. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef]
- Nguyên, D.T.; Kebache, S.; Fazel, A.; Wong, H.N.; Jenna, S.; Emadali, A.; Lee, E.-H.; Bergeron, J.J.M.; Kaufman, R.J.; Larose, L.; et al. Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol. Biol. Cell 2004, 15, 4248–4260. [Google Scholar] [CrossRef]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chávez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef]
- Kim, S.; Joe, Y.; Kim, H.J.; Kim, Y.-S.; Jeong, S.O.; Pae, H.-O.; Ryter, S.W.; Surh, Y.-J.; Chung, H.T. Endoplasmic reticulum stress-induced IRE1α activation mediates cross-talk of GSK-3β and XBP-1 to regulate inflammatory cytokine production. J. Immunol. 2015, 194, 4498–4506. [Google Scholar] [CrossRef]
- Yamazaki, H.; Hiramatsu, N.; Hayakawa, K.; Tagawa, Y.; Okamura, M.; Ogata, R.; Huang, T.; Nakajima, S.; Yao, J.; Paton, A.W.; et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 2009, 183, 1480–1487. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef]
- Zeng, L.; Liu, Y.-P.; Sha, H.; Chen, H.; Qi, L.; Smith, J.A. XBP-1 couples endoplasmic reticulum stress to augmented IFN-beta induction via a cis-acting enhancer in macrophages. J. Immunol. 2010, 185, 2324–2330. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Suganami, T.; Hachiya, R.; Shirakawa, I.; Kim-Saijo, M.; Tanaka, M.; Hamaguchi, M.; Takai-Igarashi, T.; Nakai, M.; Miyamoto, Y.; et al. Activating transcription factor 4 links metabolic stress to interleukin-6 expression in macrophages. Diabetes 2014, 63, 152–161. [Google Scholar] [CrossRef]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S.H. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef]
- Sullivan, G.P.; O’Connor, H.; Henry, C.M.; Davidovich, P.; Clancy, D.M.; Albert, M.L.; Cullen, S.P.; Martin, S.J. TRAIL Receptors Serve as Stress-Associated Molecular Patterns to Promote ER-Stress-Induced Inflammation. Dev. Cell 2020, 52, 714–730.e5. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.K.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef]
- Li, X.; Zhang, K.; Li, Z. Unfolded protein response in cancer: The physician’s perspective. J. Hematol. Oncol. 2011, 4, 8. [Google Scholar] [CrossRef]
- Davies, M.P.A.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef]
- Garg, A.D.; De Ruysscher, D.; Agostinis, P. Immunological metagene signatures derived from immunogenic cancer cell death associate with improved survival of patients with lung, breast or ovarian malignancies: A large-scale meta-analysis. Oncoimmunology 2016, 5, e1069938. [Google Scholar] [CrossRef]
- Bi, M.; Naczki, C.; Koritzinsky, M.; Fels, D.; Blais, J.; Hu, N.; Harding, H.; Novoa, I.; Varia, M.; Raleigh, J.; et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005, 24, 3470–3481. [Google Scholar] [CrossRef]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.-T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.-H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 2012, 122, 4621–4634. [Google Scholar] [CrossRef]
- Horiguchi, M.; Koyanagi, S.; Okamoto, A.; Suzuki, S.O.; Matsunaga, N.; Ohdo, S. Stress-regulated transcription factor ATF4 promotes neoplastic transformation by suppressing expression of the INK4a/ARF cell senescence factors. Cancer Res. 2012, 72, 395–401. [Google Scholar] [CrossRef]
- Chitnis, N.S.; Pytel, D.; Bobrovnikova-Marjon, E.; Pant, D.; Zheng, H.; Maas, N.L.; Frederick, B.; Kushner, J.A.; Chodosh, L.A.; Koumenis, C.; et al. miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol. Cell 2012, 48, 353–364. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.A.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef]
- Santamaría, P.G.; Mazón, M.J.; Eraso, P.; Portillo, F. UPR: An upstream signal to EMT induction in cancer. J. Clin. Med. 2019, 8, 624. [Google Scholar] [CrossRef]
- Thorpe, J.A.; Schwarze, S.R. IRE1alpha controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress Chaperones 2010, 15, 497–508. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [Green Version]
- Binet, F.; Sapieha, P. ER stress and angiogenesis. Cell Metab. 2015, 22, 560–575. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Zanetti, M. Tumor stress inside out: Cell-extrinsic effects of the unfolded protein response in tumor cells modulate the immunological landscape of the tumor microenvironment. J. Immunol. 2011, 187, 4403–4409. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib induces anti-multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov. 2021, 2, 468–483. [Google Scholar] [CrossRef]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef]
- Tatsuno, K.; Yamazaki, T.; Hanlon, D.; Han, P.; Robinson, E.; Sobolev, O.; Yurter, A.; Rivera-Molina, F.; Arshad, N.; Edelson, R.L.; et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis. 2019, 10, 578. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Hsieh, S.-Y.; Fan, Y.-T.; Wei, W.-C.; Hsiao, P.-W.; Tsai, D.-H.; Wu, T.-S.; Yang, N.-S. Necroptosis promotes autophagy-dependent upregulation of DAMP and results in immunosurveillance. Autophagy 2018, 14, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Mónaco, A.; Chilibroste, S.; Yim, L.; Chabalgoity, J.A.; Moreno, M. Inflammasome activation, NLRP3 engagement and macrophage recruitment to tumor microenvironment are all required for Salmonella antitumor effect. Cancer Immunol. Immunother. 2022, 71, 2141–2150. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Énée, E.; Plo, I.; et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol. Cell 2020, 77, 748–760.e9. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Kyte, J.A.; Andresen, N.K.; Russnes, H.G.; Fretland, S.Ø.; Falk, R.S.; Lingjærde, O.C.; Naume, B. ICON: A randomized phase IIb study evaluating immunogenic chemotherapy combined with ipilimumab and nivolumab in patients with metastatic hormone receptor positive breast cancer. J. Transl. Med. 2020, 18, 269. [Google Scholar] [CrossRef]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.-C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.M.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef]
- Giglio, P.; Gagliardi, M.; Tumino, N.; Antunes, F.; Smaili, S.; Cotella, D.; Santoro, C.; Bernardini, R.; Mattei, M.; Piacentini, M.; et al. PKR and GCN2 stress kinases promote an ER stress-independent eIF2α phosphorylation responsible for calreticulin exposure in melanoma cells. Oncoimmunology 2018, 7, e1466765. [Google Scholar] [CrossRef]
- Gomes-da-Silva, L.C.; Zhao, L.; Bezu, L.; Zhou, H.; Sauvat, A.; Liu, P.; Durand, S.; Leduc, M.; Souquere, S.; Loos, F.; et al. Photodynamic therapy with redaporfin targets the endoplasmic reticulum and Golgi apparatus. EMBO J. 2018, 37, e98354. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Ma, Y.; Mattarollo, S.R.; Adjemian, S.; Yang, H.; Aymeric, L.; Hannani, D.; Portela Catani, J.P.; Duret, H.; Teng, M.W.L.; Kepp, O.; et al. CCL2/CCR2-dependent recruitment of functional antigen-presenting cells into tumors upon chemotherapy. Cancer Res. 2014, 74, 436–445. [Google Scholar] [CrossRef]
- Sukkurwala, A.Q.; Martins, I.; Wang, Y.; Schlemmer, F.; Ruckenstuhl, C.; Durchschlag, M.; Michaud, M.; Senovilla, L.; Sistigu, A.; Ma, Y.; et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014, 21, 59–68. [Google Scholar] [CrossRef]
- Dudek-Perić, A.M.; Ferreira, G.B.; Muchowicz, A.; Wouters, J.; Prada, N.; Martin, S.; Kiviluoto, S.; Winiarska, M.; Boon, L.; Mathieu, C.; et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res. 2015, 75, 1603–1614. [Google Scholar] [CrossRef]
- Garg, A.D.; Vandenberk, L.; Fang, S.; Fasche, T.; Van Eygen, S.; Maes, J.; Van Woensel, M.; Koks, C.; Vanthillo, N.; Graf, N.; et al. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ. 2017, 24, 832–843. [Google Scholar] [CrossRef]
- Rufo, N.; Korovesis, D.; Van Eygen, S.; Derua, R.; Garg, A.D.; Finotello, F.; Vara-Perez, M.; Rožanc, J.; Dewaele, M.; de Witte, P.A.; et al. Stress-induced inflammation evoked by immunogenic cell death is blunted by the IRE1α kinase inhibitor KIRA6 through HSP60 targeting. Cell Death Differ. 2022, 29, 230–245. [Google Scholar] [CrossRef]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef]
- Korbecki, J.; Kojder, K.; Simińska, D.; Bohatyrewicz, R.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of the Ligands of Receptors CCR1, CCR2, CCR3, and CCR4. Int. J. Mol. Sci. 2020, 21, 8412. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef]
- Bagratuni, T.; Wu, P.; Gonzalez de Castro, D.; Davenport, E.L.; Dickens, N.J.; Walker, B.A.; Boyd, K.; Johnson, D.C.; Gregory, W.; Morgan, G.J.; et al. XBP1s levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood 2010, 116, 250–253. [Google Scholar] [CrossRef] [Green Version]
- Lhomond, S.; Avril, T.; Dejeans, N.; Voutetakis, K.; Doultsinos, D.; McMahon, M.; Pineau, R.; Obacz, J.; Papadodima, O.; Jouan, F.; et al. Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 2018, 10, e15622. [Google Scholar] [CrossRef]
- Feng, Y.-X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Liu, M.; Wu, C.; Luo, S.; Hua, Q.; Chen, H.-T.; Weng, Y.; Xu, J.; Lin, H.; Wang, L.; Li, J.; et al. PERK reprograms hematopoietic progenitor cells to direct tumor-promoting myelopoiesis in the spleen. J. Exp. Med. 2022, 219, e20211498. [Google Scholar] [CrossRef]
- Wang, P.; Han, L.; Yu, M.; Cao, Z.; Li, X.; Shao, Y.; Zhu, G. The prognostic value of PERK in cancer and its relationship with immune cell infiltration. Front. Mol. Biosci. 2021, 8, 648752. [Google Scholar] [CrossRef]
- Pytel, D.; Gao, Y.; Mackiewicz, K.; Katlinskaya, Y.V.; Staschke, K.A.; Paredes, M.C.G.; Yoshida, A.; Qie, S.; Zhang, G.; Chajewski, O.S.; et al. PERK Is a Haploinsufficient Tumor Suppressor: Gene Dose Determines Tumor-Suppressive Versus Tumor Promoting Properties of PERK in Melanoma. PLoS Genet. 2016, 12, e1006518. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Vanacker, H.; Vetters, J.; Moudombi, L.; Caux, C.; Janssens, S.; Michallet, M.-C. Emerging role of the unfolded protein response in tumor immunosurveillance. Trends Cancer 2017, 3, 491–505. [Google Scholar] [CrossRef]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.-Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [Green Version]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.-H.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2013, 2, e00498. [Google Scholar] [CrossRef] [PubMed]
- Suh, D.H.; Kim, M.-K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann. N. Y. Acad. Sci. 2012, 1271, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Garg, A.D.; Agostinis, P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Larkin, D.; Lara-Lemus, R.; Ramos-Castañeda, J.; Liu, M.; Arvan, P. Endoplasmic reticulum (ER) chaperone regulation and survival of cells compensating for deficiency in the ER stress response kinase, PERK. J. Biol. Chem. 2008, 283, 17020–17029. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.-L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef]
- Sato, H.; Shiba, Y.; Tsuchiya, Y.; Saito, M.; Kohno, K. 4μ8c inhibits insulin secretion independent of ire1α rnase activity. Cell Struct. Funct. 2017, 42, 61–70. [Google Scholar] [CrossRef]
- Chan, S.M.H.; Lowe, M.P.; Bernard, A.; Miller, A.A.; Herbert, T.P. The inositol-requiring enzyme 1 (IRE1α) RNAse inhibitor, 4µ8C, is also a potent cellular antioxidant. Biochem. J. 2018, 475, 923–929. [Google Scholar] [CrossRef]
- Mahameed, M.; Wilhelm, T.; Darawshi, O.; Obiedat, A.; Tommy, W.-S.; Chintha, C.; Schubert, T.; Samali, A.; Chevet, E.; Eriksson, L.A.; et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 2019, 10, 300. [Google Scholar] [CrossRef]
- Osorio, F.; Tavernier, S.J.; Hoffmann, E.; Saeys, Y.; Martens, L.; Vetters, J.; Delrue, I.; De Rycke, R.; Parthoens, E.; Pouliot, P.; et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat. Immunol. 2014, 15, 248–257. [Google Scholar] [CrossRef]
- Tavernier, S.J.; Osorio, F.; Vandersarren, L.; Vetters, J.; Vanlangenakker, N.; Van Isterdael, G.; Vergote, K.; De Rycke, R.; Parthoens, E.; van de Laar, L.; et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat. Cell Biol. 2017, 19, 698–710. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rufo, N.; Yang, Y.; De Vleeschouwer, S.; Agostinis, P. The “Yin and Yang” of Unfolded Protein Response in Cancer and Immunogenic Cell Death. Cells 2022, 11, 2899. https://doi.org/10.3390/cells11182899
Rufo N, Yang Y, De Vleeschouwer S, Agostinis P. The “Yin and Yang” of Unfolded Protein Response in Cancer and Immunogenic Cell Death. Cells. 2022; 11(18):2899. https://doi.org/10.3390/cells11182899
Chicago/Turabian StyleRufo, Nicole, Yihan Yang, Steven De Vleeschouwer, and Patrizia Agostinis. 2022. "The “Yin and Yang” of Unfolded Protein Response in Cancer and Immunogenic Cell Death" Cells 11, no. 18: 2899. https://doi.org/10.3390/cells11182899