
Investigation of Radiotracer Metabolic Stability In Vitro with CYP-Overexpressing Hepatoma Cell Lines

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Liquid Chromatography (LC)
2.3. Mass Spectrometry (MS)
2.4. Radio Thin-Layer Chromatography (Radio-TLC)
2.5. Cell Culture
2.6. Radiotracer Incubation
2.7. Testosterone Incubation
2.8. Microsome Assay
3. Results and Discussion
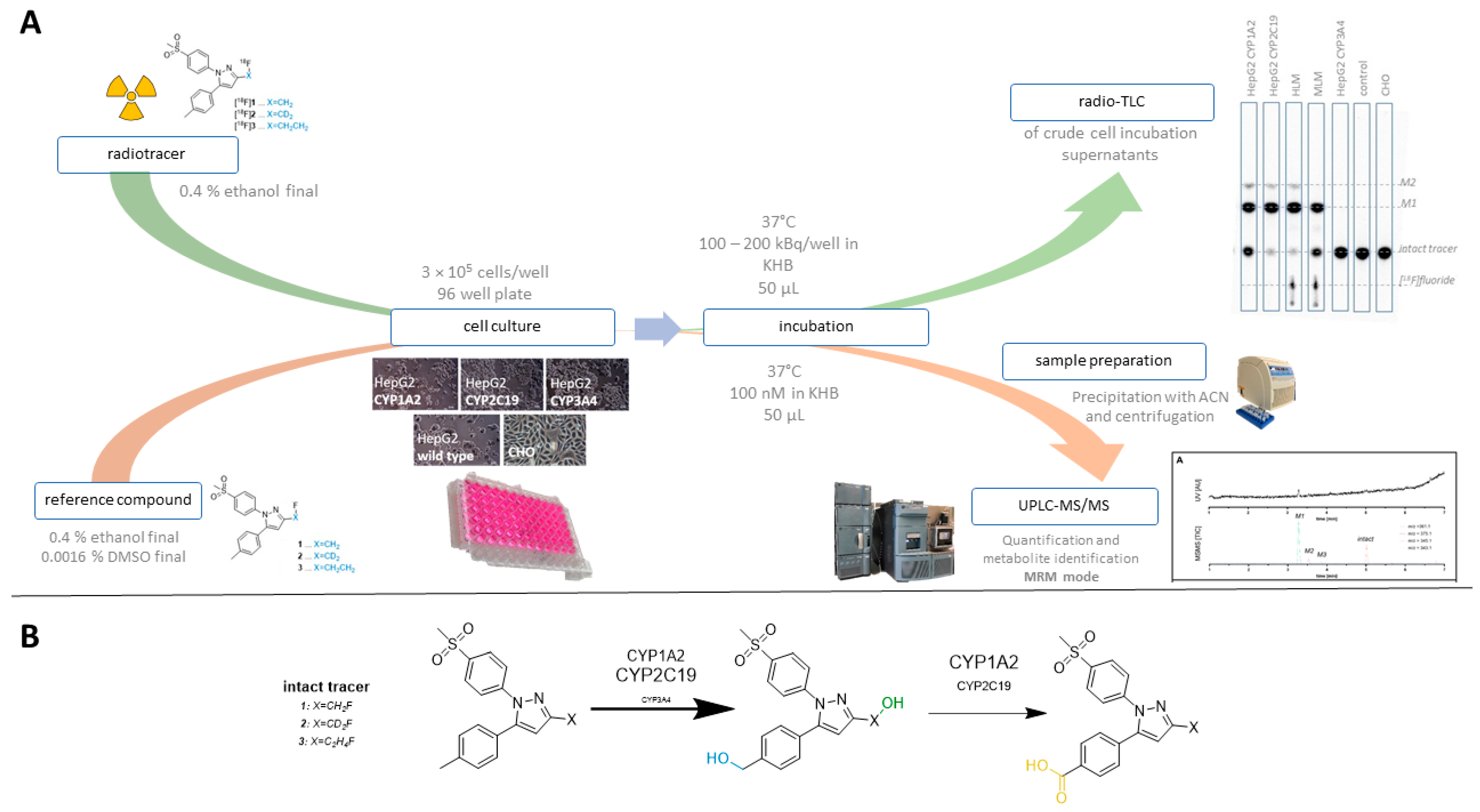
3.1. Method Development
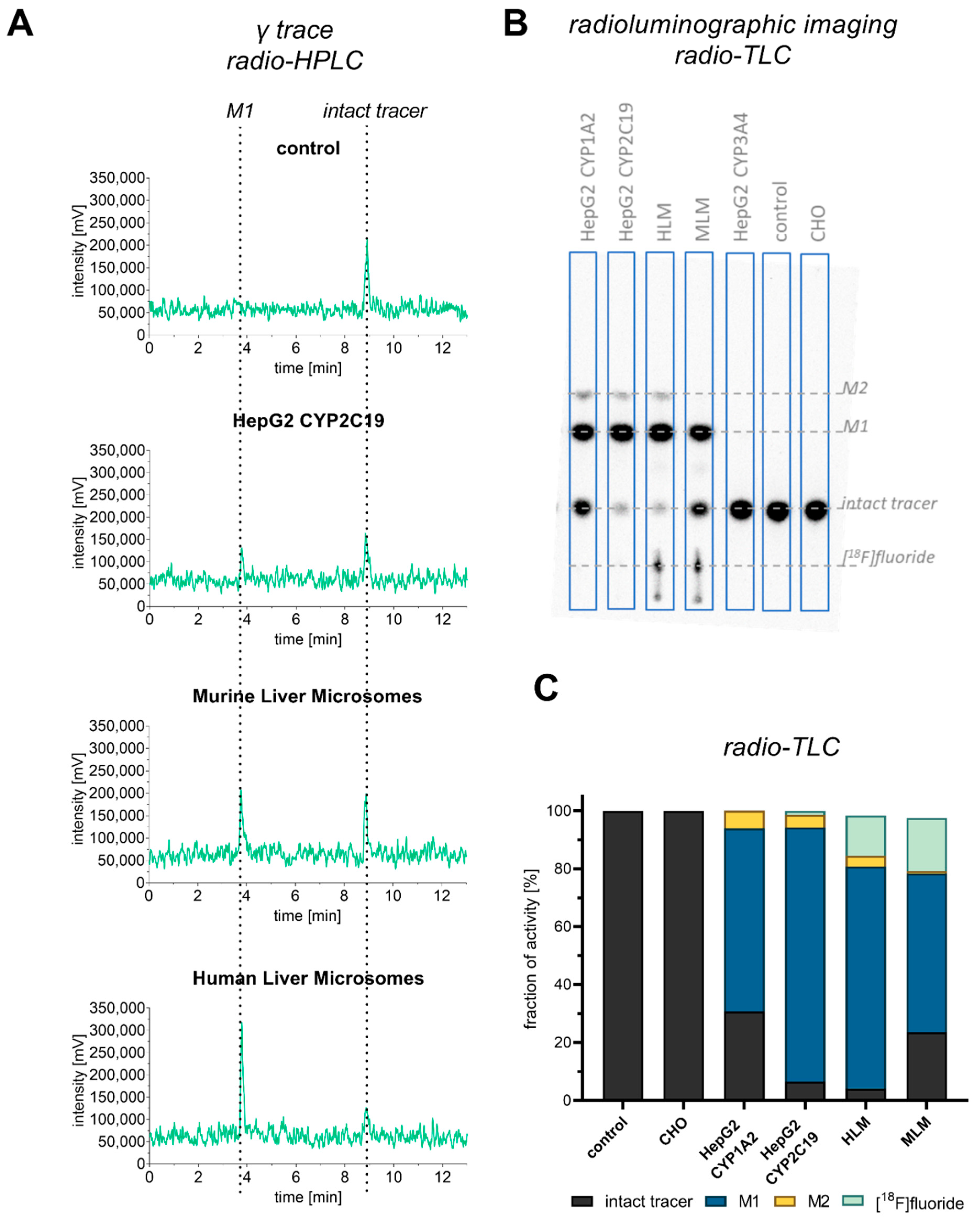
3.2. Comparison with Microsome Assay
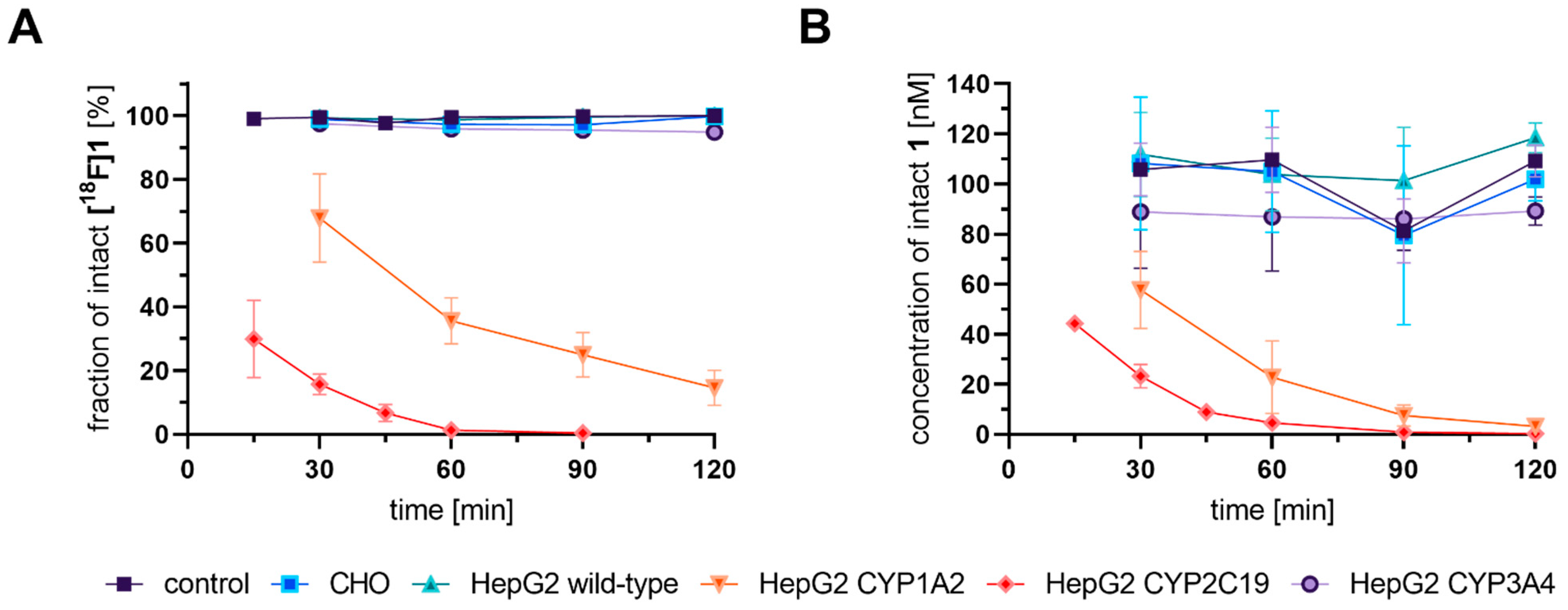
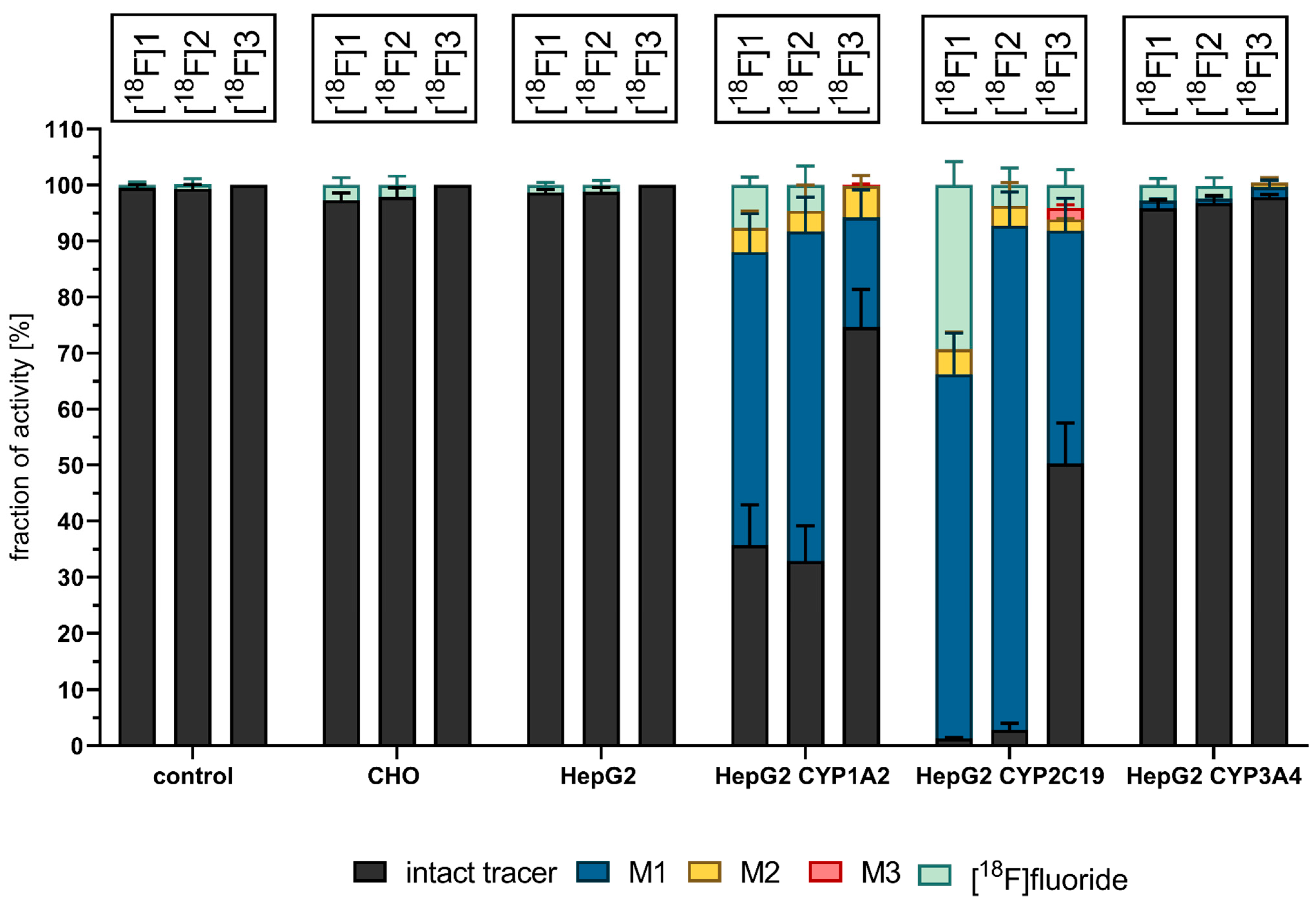
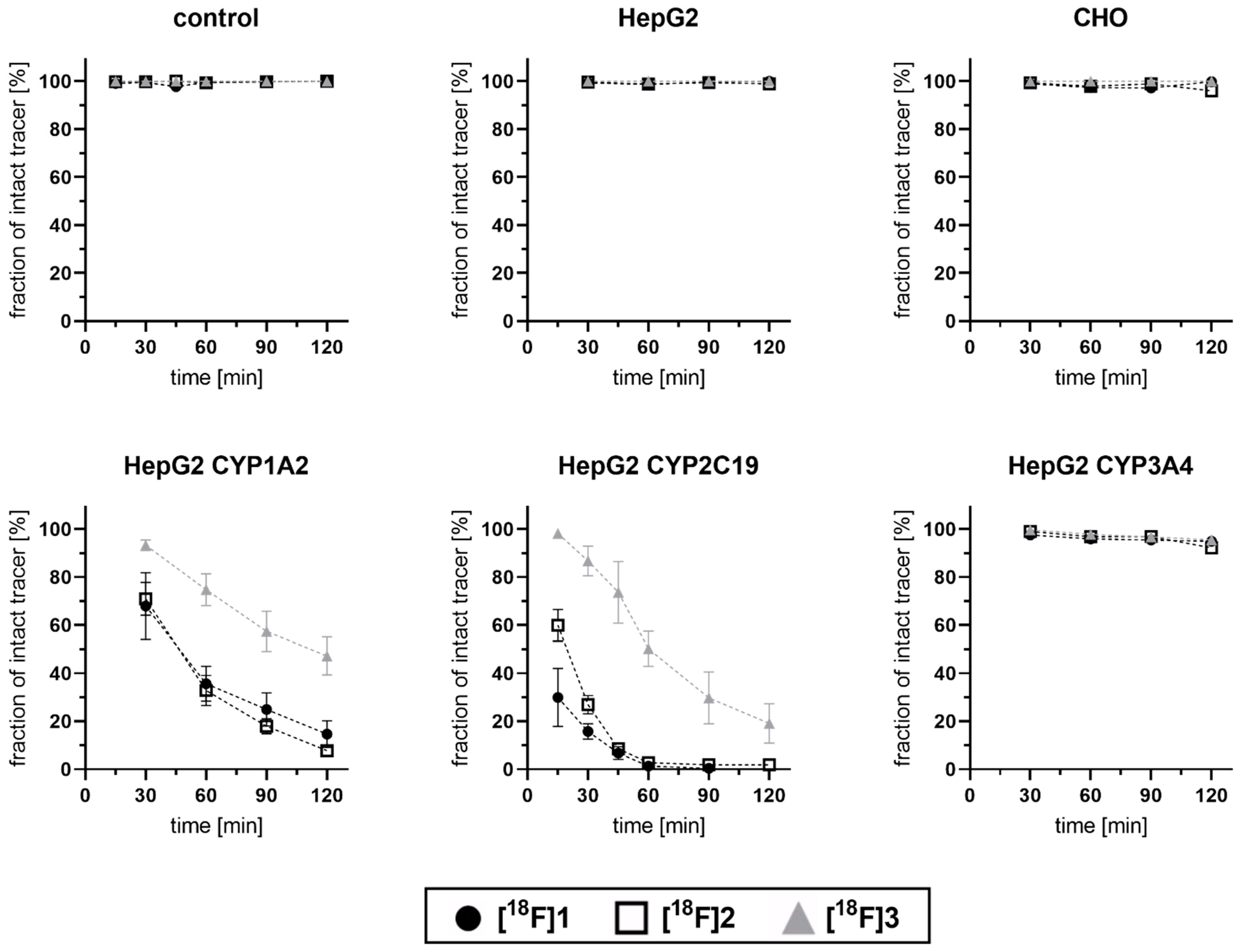
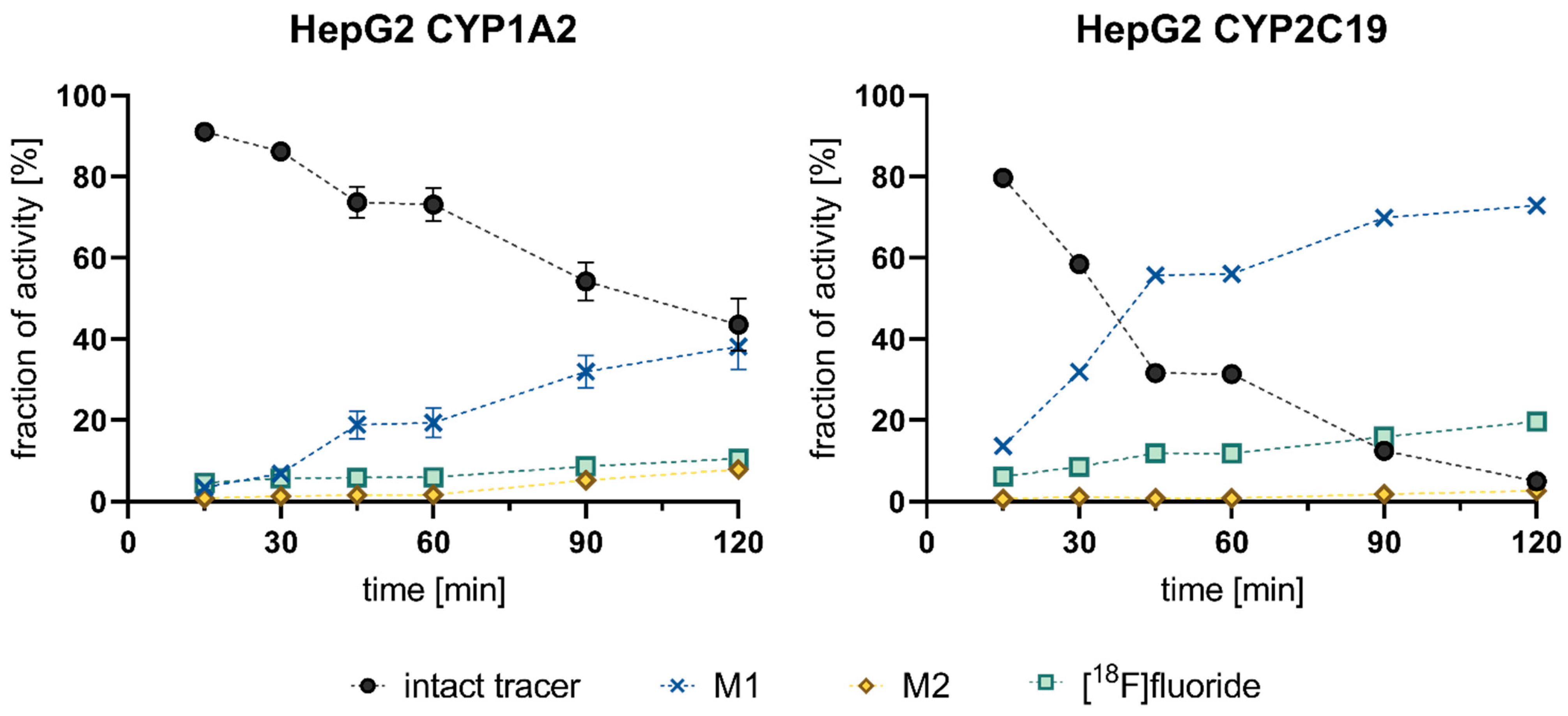
3.3. Cell-Based Radiotracer Metabolism of [18F]1, [18F]2, and [18F]3
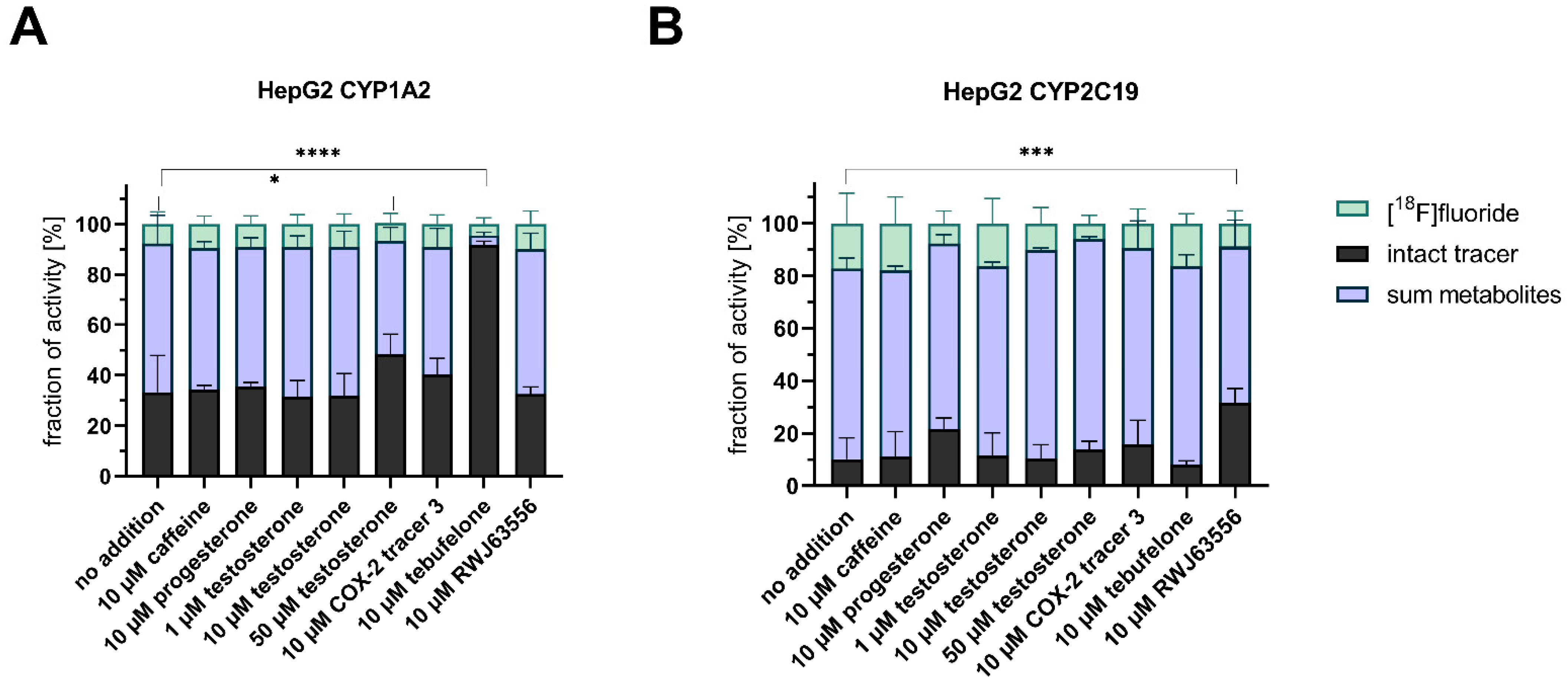
3.4. Competitive Incubations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuchar, M.; Mamat, C. Methods to Increase the Metabolic Stability of 18F-Radiotracers. Molecules 2015, 20, 16186–16220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agdeppa, E.D.; Spilker, M.E. A Review of Imaging Agent Development. AAPS J. 2009, 11, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Bier, D.; Holschbach, M.H.; Wutz, W.; Olsson, R.A.; Coenen, H.H. Metabolism of the A1 adenosine receptor positron emission tomography ligand [18F] 8-cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine ([18F] CPFPX) in rodents and humans. Drug Metab. Dispos. 2006, 34, 570–576. [Google Scholar] [CrossRef]
- Kharasch, E.D.; Thummel, K.E. Identification of cytochrome P450 2E1 as the predominant enzyme catalyzing human liver microsomal defluorination of sevoflurane, isoflurane, and methoxyflurane. J. Am. Soc. Anesthesiol. 1993, 79, 795–807. [Google Scholar] [CrossRef]
- Yin, H.; Anders, M.W.; Jones, J.P. Metabolism of 1, 2-dichloro-1-fluoroethane and 1-fluoro-1, 2, 2-trichloroethane: Electronic factors govern the regioselectivity of cytochrome P450-dependent oxidation. Chem. Res. Toxicol. 1996, 9, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Liu, X. The conduct of drug metabolism studies considered good practice (II): In vitro experiments. Curr. Drug Metab. 2007, 8, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, N.J.; Lechón, M.J.; Houston, J.B.; Hallifax, D.; Brown, H.S.; Maurel, P.; Kenna, J.G.; Gustavsson, L.; Lohmann, C.; Skonberg, C.; et al. Primary hepatocytes: Current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab. Rev. 2007, 39, 159–234. [Google Scholar] [CrossRef]
- Ding, X.; Kaminsky, L.S. Human extrahepatic cytochromes P450: Function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 149–173. [Google Scholar] [CrossRef]
- Schneider, D.; Bier, D.; Bauer, A.; Holschbach, M.; Neumaier, B.; Oskamp, A. Relevance of In Vitro Metabolism Models to PET Radiotracer Development: Prediction of In Vivo Clearance in Rats from Microsomal Stability Data. Pharmaceuticals 2019, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Spielmann, H.; Castle, J.V.; Gomez, M.J. In Vitro Methods in Pharmaceutical Research; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Tolosa, L.; Gómez-Lechón, M.J.; López, S.; Guzmán, C.; Castell, J.V.; Donato, M.T.; Jover, R. Human Upcyte Hepatocytes: Characterization of the Hepatic Phenotype and Evaluation for Acute and Long-Term Hepatotoxicity Routine Testing. Toxicol. Sci. 2016, 152, 214–229. [Google Scholar] [CrossRef] [Green Version]
- Stanley, L.A.; Wolf, C.R. Through a glass, darkly? HepaRG and HepG2 cells as models of human phase I drug metabolism. Drug Metab. Rev. 2022, 54, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, S. Three-Dimensional Liver Culture Systems to Maintain Primary Hepatic Properties for Toxicological Analysis In Vitro. Int. J. Mol. Sci. 2021, 22, 214. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Lahoz, A.; Castell, J.V.; Gómez-Lechón, M.J. Cell lines: A tool for in vitro drug metabolism studies. Curr. Drug Metab. 2008, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Asha, S.; Vidyavathi, M. Role of Human Liver Microsomes in In Vitro Metabolism of Drugs—A Review. Appl. Biochem. Biotechnol. 2010, 160, 1699–1722. [Google Scholar] [CrossRef]
- Castell, J.V.; Jover, R.; Martnez-Jimnez, C.P.; Gmez-Lechn, M.J. Hepatocyte cell lines: Their use, scope and limitations in drug metabolism studies. Expert Opin. Drug Metab. Toxicol. 2006, 2, 183–212. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Antona, C.; Donato, M.T.; Boobis, A.; Edwards, R.J.; Watts, P.S.; Castell, J.V.; Gómez-Lechón, M.-J. Cytochrome P450 expression in human hepatocytes and hepatoma cell lines: Molecular mechanisms that determine lower expression in cultured cells. Xenobiotica 2002, 32, 505–520. [Google Scholar] [CrossRef]
- Steinbrecht, S.; Pfeifer, N.; Herzog, N.; Katzenberger, N.; Schulz, C.; Kammerer, S.; Küpper, J.-H. HepG2-1A2 C2 and C7: Lentivirus vector-mediated stable and functional overexpression of cytochrome P450 1A2 in human hepatoblastoma cells. Toxicol. Lett. 2020, 319, 155–159. [Google Scholar] [CrossRef]
- Satoh, D.; Iwado, S.; Abe, S.; Kazuki, K.; Wakuri, S.; Oshimura, M.; Kazuki, Y. Establishment of a novel hepatocyte model that expresses four cytochrome P450 genes stably via mammalian-derived artificial chromosome for pharmacokinetics and toxicity studies. PLoS ONE 2017, 12, e0187072. [Google Scholar] [CrossRef] [Green Version]
- Steinbrecht, S.; König, R.; Schmidtke, K.-U.; Herzog, N.; Scheibner, K.; Krüger-Genge, A.; Jung, F.; Kammerer, S.; Küpper, J.-H. Metabolic activity testing can underestimate acute drug cytotoxicity as revealed by HepG2 cell clones overexpressing cytochrome P450 2C19 and 3A4. Toxicology 2019, 412, 37–47. [Google Scholar] [CrossRef]
- Herzog, N.; Katzenberger, N.; Martin, F.; Schmidtke, K.-U.; Küpper, J.-H. Generation of cytochrome P450 3A4-overexpressing HepG2 cell clones for standardization of hepatocellular testosterone 6β-hydroxylation activity. J. Cell. Biotechnol. 2015, 1, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Steinbrecht, S.; Kammerer, S.; Küpper, J.-H. HepG2 cells with recombinant cytochrome P450 enzyme overexpression: Their use and limitation as in vitro liver model. J. Cell. Biotechnol. 2019, 5, 55–64. [Google Scholar] [CrossRef]
- Steinbrecht, S.; Kiebist, J.; König, R.; Thiessen, M.; Schmidtke, K.-U.; Kammerer, S.; Küpper, J.-H.; Scheibner, K. Synthesis of cyclophosphamide metabolites by a peroxygenase from Marasmius rotula for toxicological studies on human cancer cells. AMB Express 2020, 10, 128. [Google Scholar] [CrossRef]
- Tietz, O.; Marshall, A.; Wuest, M.; Wang, M.; Wuest, F. Radiotracers for molecular imaging of cyclooxygenase-2 (COX-2) enzyme. Curr. Med. Chem. 2013, 20, 4350–4369. [Google Scholar] [CrossRef]
- Laube, M.; Kniess, T.; Pietzsch, J. Radiolabeled COX-2 inhibitors for non-invasive visualization of COX-2 expression and activity—A critical update. Molecules 2013, 18, 6311–6355. [Google Scholar] [CrossRef] [Green Version]
- Dagallier, C.; Avry, F.; Touchefeu, Y.; Buron, F.; Routier, S.; Chérel, M.; Arlicot, N. Development of PET Radioligands Targeting COX-2 for Colorectal Cancer Staging, a Review of in vitro and Preclinical Imaging Studies. Front. Med. 2021, 8, 675209. [Google Scholar] [CrossRef]
- Prabhakaran, J.; Molotkov, A.; Mintz, A.; Mann, J.J. Progress in PET Imaging of Neuroinflammation Targeting COX-2 Enzyme. Molecules 2021, 26, 3208. [Google Scholar] [CrossRef]
- Laube, M.; Gassner, C.; Neuber, C.; Wodtke, R.; Ullrich, M.; Haase-Kohn, C.; Löser, R.; Köckerling, M.; Kopka, K.; Kniess, T.; et al. Deuteration versus ethylation—Strategies to improve the metabolic fate of an 18 F-labeled celecoxib derivative. RSC Adv. 2020, 10, 38601–38611. [Google Scholar] [CrossRef]
- Gong, L.; Thorn, C.F.; Bertagnolli, M.M.; Grosser, T.; Altman, R.B.; Klein, T.E. Celecoxib pathways: Pharmacokinetics and pharmacodynamics. Pharm. Genom. 2012, 22, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Laube, M.; Wodtke, R.; Kopka, K.; Kniess, T.; Pietzsch, J. 18F-Chemistry in HPLC vials—A microliter scale radiofluorination approach. Nucl. Med. Biol. 2021, 96–97, S61. [Google Scholar] [CrossRef]
- Makhdoumi, P.; Zarghi, A.; Daraei, B.; Karimi, G. Evaluation of Cytotoxicity Effects of Chalcone Epoxide Analogues as a Selective COX-II Inhibitor in the Human Liver Carcinoma Cell Line. J. Pharmacopunct. 2017, 20, 207–212. [Google Scholar] [CrossRef]
- Morisaki, T.; Umebayashi, M.; Kiyota, A.; Koya, N.; Tanaka, H.; Onishi, H.; Katano, M. Combining celecoxib with sorafenib synergistically inhibits hepatocellular carcinoma cells in vitro. Anticancer Res. 2013, 33, 1387–1395. [Google Scholar] [PubMed]
- Xu, Z.; Liu, J.; Jianxin, C.; Yongliang, Z.; Pan, X. 17β-Estradiol inhibits testosterone-induced cell proliferation in HepG2 by modulating the relative ratios of 3 estrogen receptor isoforms to the androgen receptor. J. Cell. Biochem. 2018, 119, 8659–8671. [Google Scholar] [CrossRef] [PubMed]
- Zal, F.; Miladpour, B.; Taheri, R.; Heidari, I.; Mostafavi-Pour, Z. Estrogen and/or Progesterone Effects on HepG2 Human Cell Lines; Oxidant or Antioxidant? J. Adv. Med. Sci. Appl. Technol. 2015, 1, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Paini, A.; Mennecozzi, M.; Horvat, T.; Gerloff, K.; Palosaari, T.; Sala Benito, J.V.; Worth, A. Practical use of the Virtual Cell Based Assay: Simulation of repeated exposure experiments in liver cell lines. Toxicol. Vitr. 2017, 45, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Wodtke, R.; Hauser, C.; Ruiz-Gómez, G.; Jäckel, E.; Bauer, D.; Lohse, M.; Wong, A.; Pufe, J.; Ludwig, F.-A.; Fischer, S.; et al. Nε-Acryloyllysine Piperazides as Irreversible Inhibitors of Transglutaminase 2: Synthesis, Structure–Activity Relationships, and Pharmacokinetic Profiling. J. Med. Chem. 2018, 61, 4528–4560. [Google Scholar] [CrossRef] [PubMed]
- Busby, W.F.; Ackermann, J.M.; Crespi, C.L. Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab. Dispos. Biol. Fate Chem. 1999, 27, 246–249. [Google Scholar]
- Li, D.; Han, Y.; Meng, X.; Sun, X.; Yu, Q.; Li, Y.; Wan, L.; Huo, Y.; Guo, C. Effect of Regular Organic Solvents on Cytochrome P450-Mediated Metabolic Activities in Rat Liver Microsomes. Drug Metab. Dispos. 2010, 38, 1922–1925. [Google Scholar] [CrossRef]
- Jóźwik, I.K.; Kiss, F.M.; Gricman, Ł.; Abdulmughni, A.; Brill, E.; Zapp, J.; Pleiss, J.; Bernhardt, R.; Thunnissen, A.-M.W.H. Structural basis of steroid binding and oxidation by the cytochrome P450 CYP109E1 from Bacillus megaterium. FEBS J. 2016, 283, 4128–4148. [Google Scholar] [CrossRef]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef]
- Tang, C.; Shou, M.; Mei, Q.; Rushmore, T.H.; Rodrigues, A.D. Major Role of Human Liver Microsomal Cytochrome P450 2C9 (CYP2C9) in the Oxidative Metabolism of Celecoxib, a Novel Cyclooxygenase-II Inhibitor. J. Pharmacol. Exp. Ther. 2000, 293, 453–459. [Google Scholar]
- Ridderström, M.; Zamora, I.; Fjellström, O.; Andersson, T.B. Analysis of selective regions in the active sites of human cytochromes P450, 2C8, 2C9, 2C18, and 2C19 homology models using GRID/CPCA. J. Med. Chem. 2001, 44, 4072–4081. [Google Scholar] [CrossRef] [PubMed]
- Kaffenberger, R.M.; Eichhold, T.H.; Doyle, M.J. Determination of tebufelone (a new anti-inflammatory drug) strength and stability in bulk drug, dosage formations and feed admixtures by reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1990, 505, 349–356. [Google Scholar] [CrossRef]
- Kirchner, T.; Argentieri, D.C.; Barbone, A.G.; Singer, M.; Steber, M.; Ansell, J.; Beers, S.A.; Wachter, M.P.; Wu, W.; Malloy, E.; et al. Evaluation of the Antiinflammatory Activity of a Dual Cyclooxygenase-2 Selective/5-Lipoxygenase Inhibitor, RWJ 63556, in a Canine Model of Inflammation. J. Pharmacol. Exp. Ther. 1997, 282, 1094–1101. [Google Scholar]
- Yamazaki, H.; Shimada, T. Progesterone and Testosterone Hydroxylation by Cytochromes P450 2C19, 2C9, and 3A4 in Human Liver Microsomes. Arch. Biochem. Biophys. 1997, 346, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Drug Development and Drug Interactions|Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 26 June 2021).
- Sumsakul, W.; Chaijaroenkul, W.; Na-Bangchang, K. In vitro inhibitory effects of plumbagin, the promising antimalarial candidate, on human cytochrome P450 enzymes. Asian Pac. J. Trop. Med. 2015, 8, 914–918. [Google Scholar] [CrossRef] [Green Version]
- Tassaneeyakul, W.; Guo, L.-Q.; Fukuda, K.; Ohta, T.; Yamazoe, Y. Inhibition Selectivity of Grapefruit Juice Components on Human Cytochromes P450. Arch. Biochem. Biophys. 2000, 378, 356–363. [Google Scholar] [CrossRef]
- Scott, S.A.; Sangkuhl, K.; Gardner, E.E.; Stein, C.M.; Hulot, J.S.; Johnson, J.A.; Roden, D.M.; Klein, T.E.; Shuldiner, A.R.; Clinical Pharmacogenetics Implementation, C. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin. Pharmacol. Ther. 2011, 90, 328–332. [Google Scholar] [CrossRef]
- Consortium, P.V. CYP2C19. Available online: https://www.pharmvar.org/gene/CYP2C19 (accessed on 11 May 2022).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | CHO | HepG2 | CYP1A2 | CYP2C19 | CYP3A4 | ||
|---|---|---|---|---|---|---|---|
| Supernatant | MBq/well at t0 | 1.11 | 0.95 | 0.91 | 1.10 | 1.14 | 0.81 |
| (%) | 87.8 | 74.8 | 71.7 | 86.9 | 90.7 | 64.2 | |
| Wash | MBq/well at t0 | 0.15 | 0.16 | 0.18 | 0.11 | 0.11 | 0.23 |
| (%) | 11.7 | 12.8 | 14.4 | 8.9 | 8.9 | 18.4 | |
| Cell lysate | MBq/well at t0 | - | 0.09 | 0.14 | 0.03 | 0.02 | 0.20 |
| (%) | - | 7.4 | 10.8 | 2.5 | 1.6 | 15.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemm, S.; Köhler, S.; Wodtke, R.; Jung, F.; Küpper, J.-H.; Pietzsch, J.; Laube, M. Investigation of Radiotracer Metabolic Stability In Vitro with CYP-Overexpressing Hepatoma Cell Lines. Cells 2022, 11, 2447. https://doi.org/10.3390/cells11152447
Lemm S, Köhler S, Wodtke R, Jung F, Küpper J-H, Pietzsch J, Laube M. Investigation of Radiotracer Metabolic Stability In Vitro with CYP-Overexpressing Hepatoma Cell Lines. Cells. 2022; 11(15):2447. https://doi.org/10.3390/cells11152447
Chicago/Turabian StyleLemm, Sandy, Susanne Köhler, Robert Wodtke, Friedrich Jung, Jan-Heiner Küpper, Jens Pietzsch, and Markus Laube. 2022. "Investigation of Radiotracer Metabolic Stability In Vitro with CYP-Overexpressing Hepatoma Cell Lines" Cells 11, no. 15: 2447. https://doi.org/10.3390/cells11152447