
NLRP1 Inflammasome Activation in the Hippocampal Formation in Alzheimer’s Disease: Correlation with Neuropathological Changes and Unbiasedly Estimated Neuronal Loss



Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Brain Tissue Autopsy Samples
2.2. Immunohistochemical and Immunofluorescent Staining
2.3. Analysis of Immunohistochemically Stained Sections
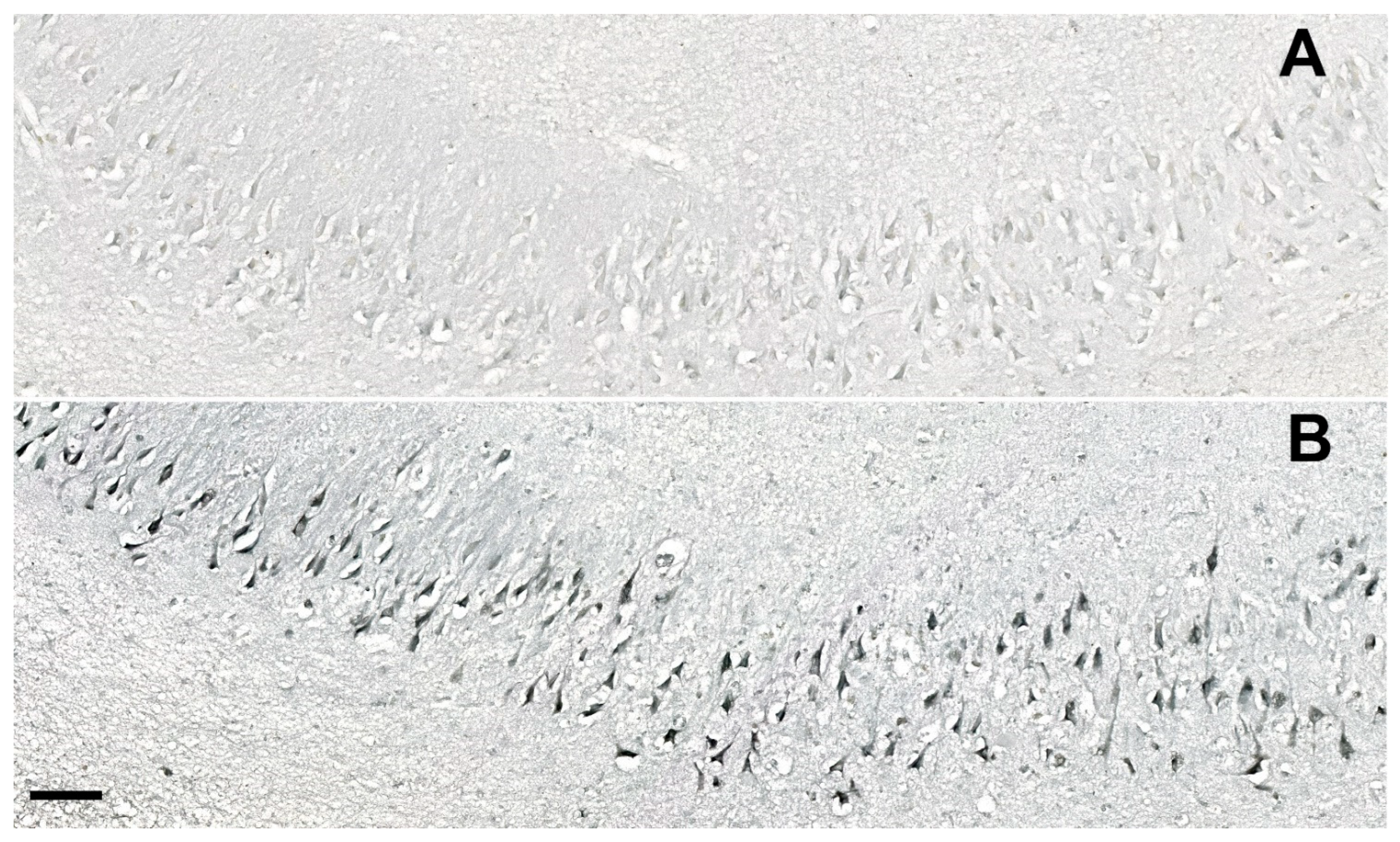
2.4. Unbiased Quantification of Neurons and Neurofibrillary Tangles
2.5. Statistical Analysis
3. Results
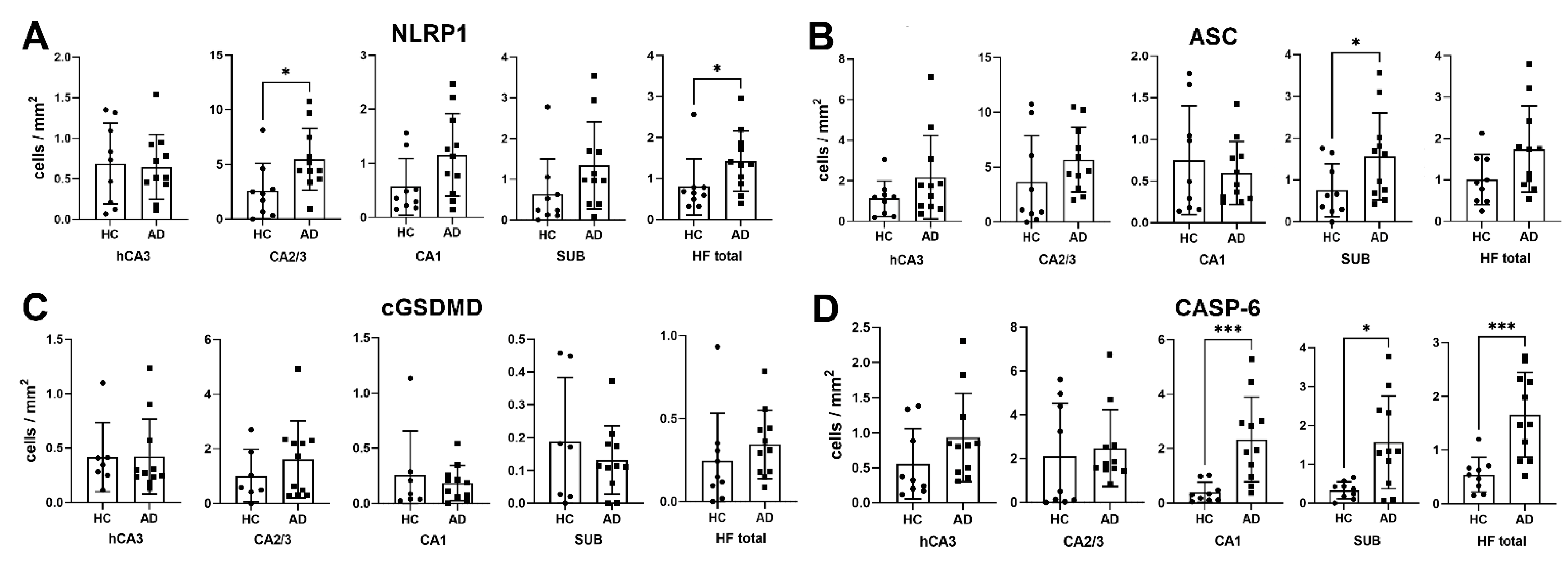
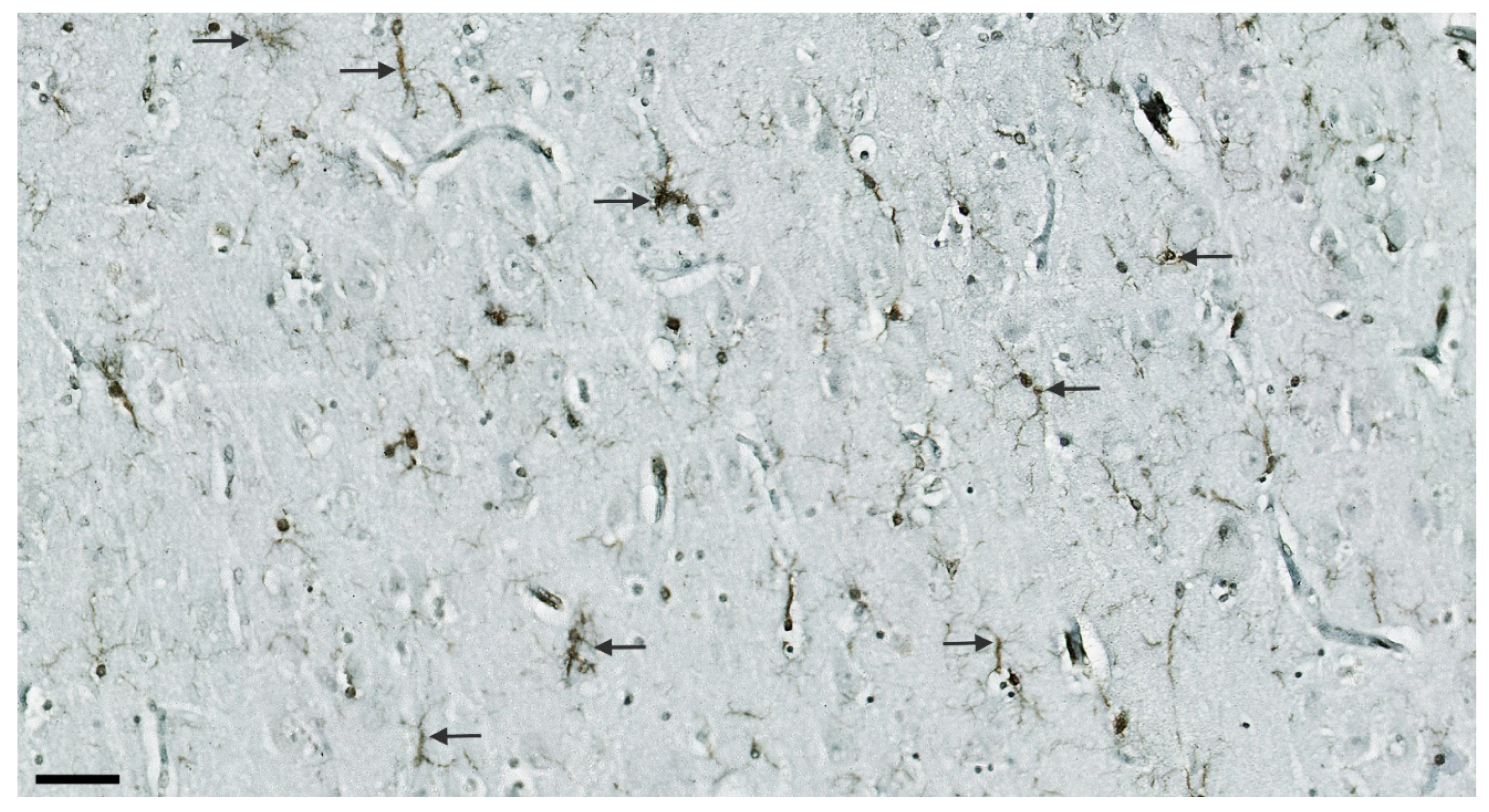
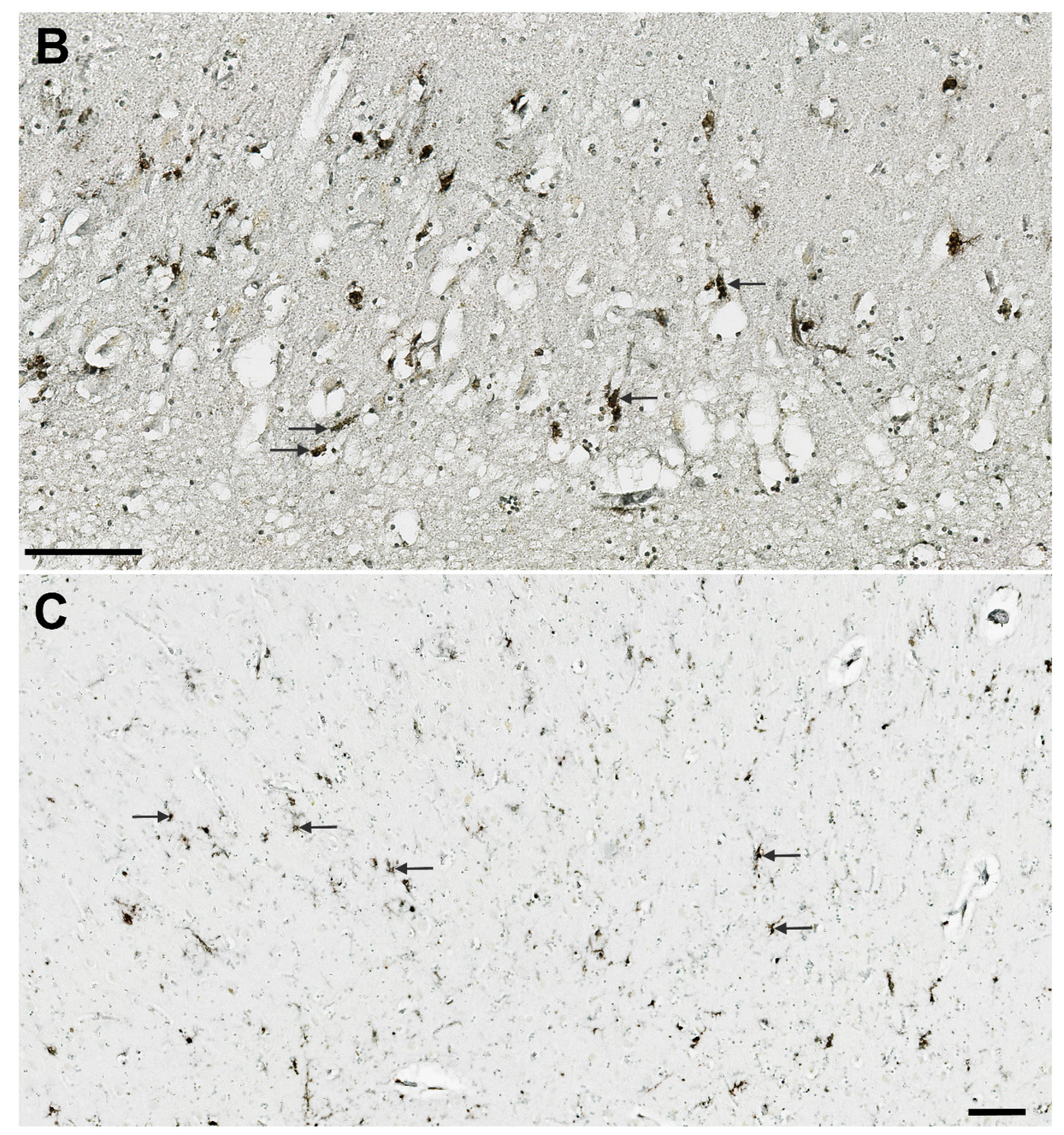
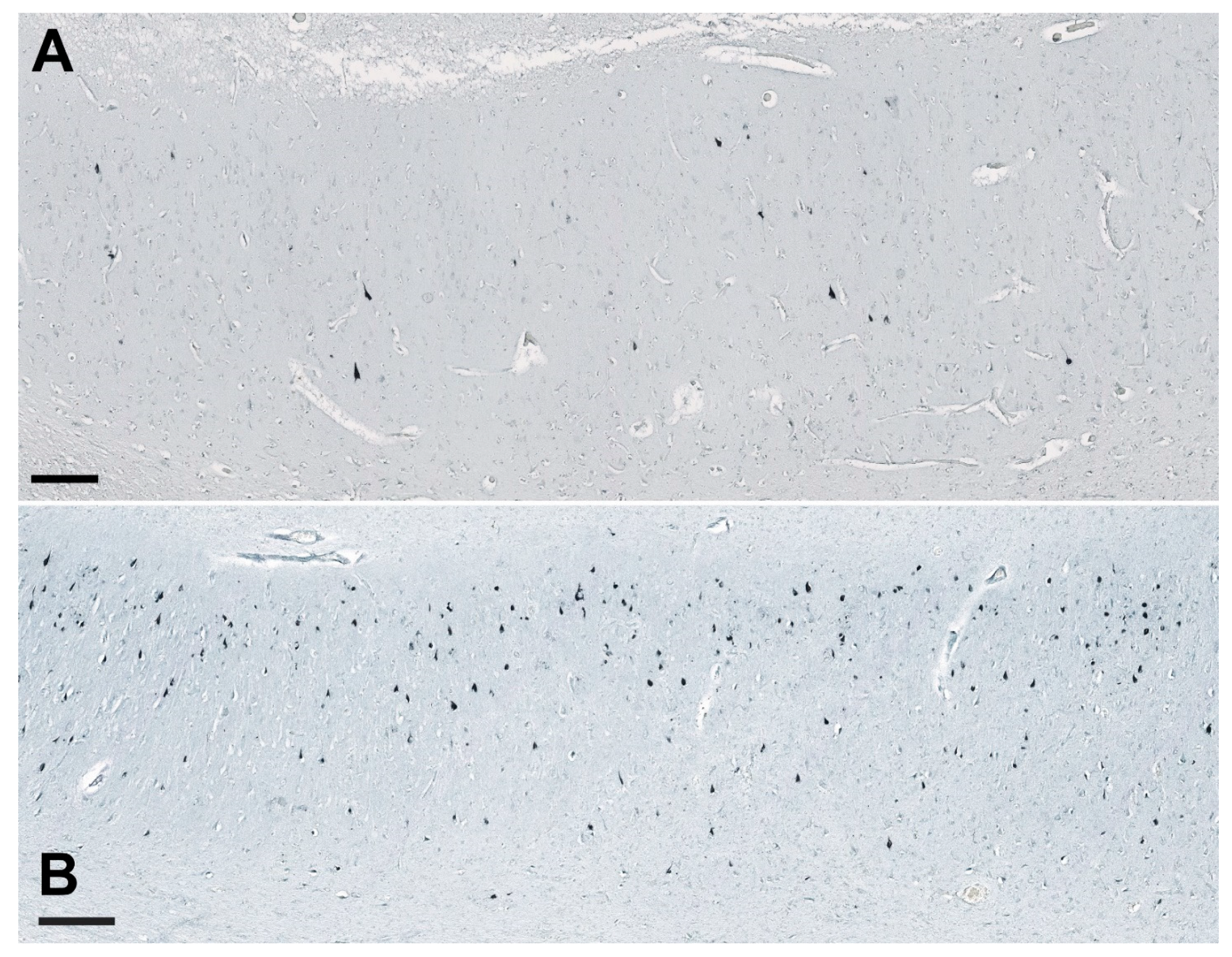
3.1. Analysis of NLRP1, ASC, cGSDMD, and Caspase-6 Immunostaining
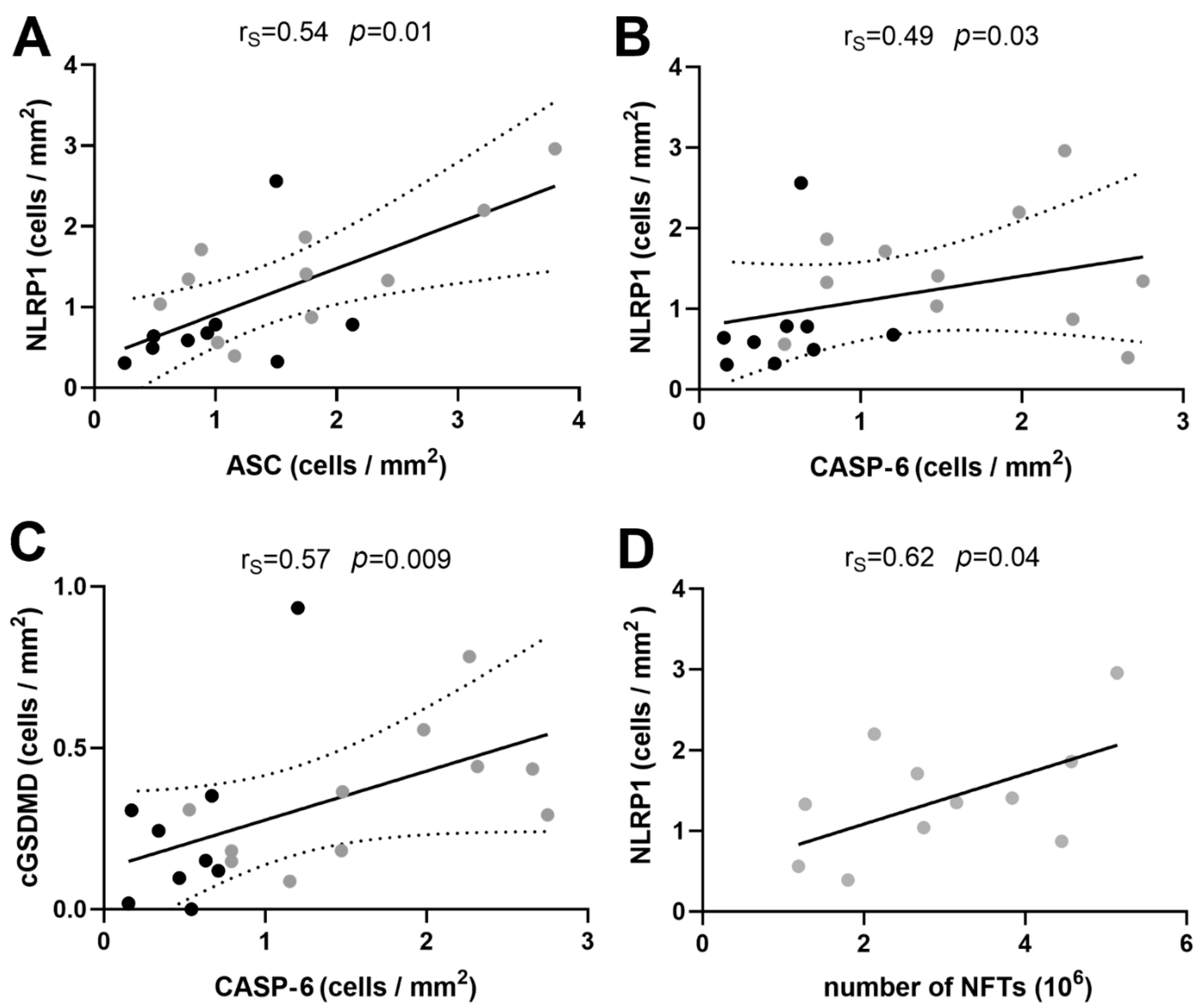
3.2. Correlations between Immunohistochemical Markers of Inflammasome Activation
3.3. Correlation of Immunohistochemical Markers with the Age of the Subjects and Duration of Disease
3.4. Correlations between NLRP1 Inflammasome, Neurofibrillary Tangles, Number of Neurons, and Disease Duration
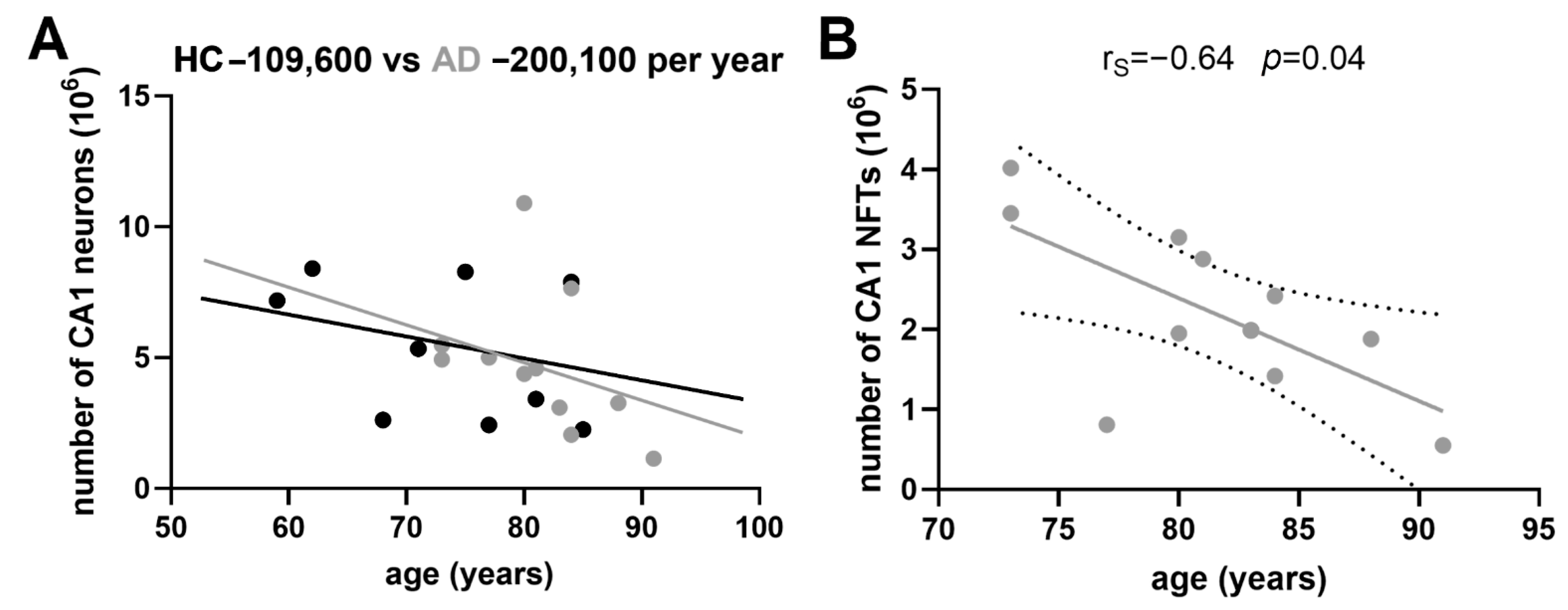
3.5. Age-Related Neuronal Loss and Number of Neurofibrillary Tangles
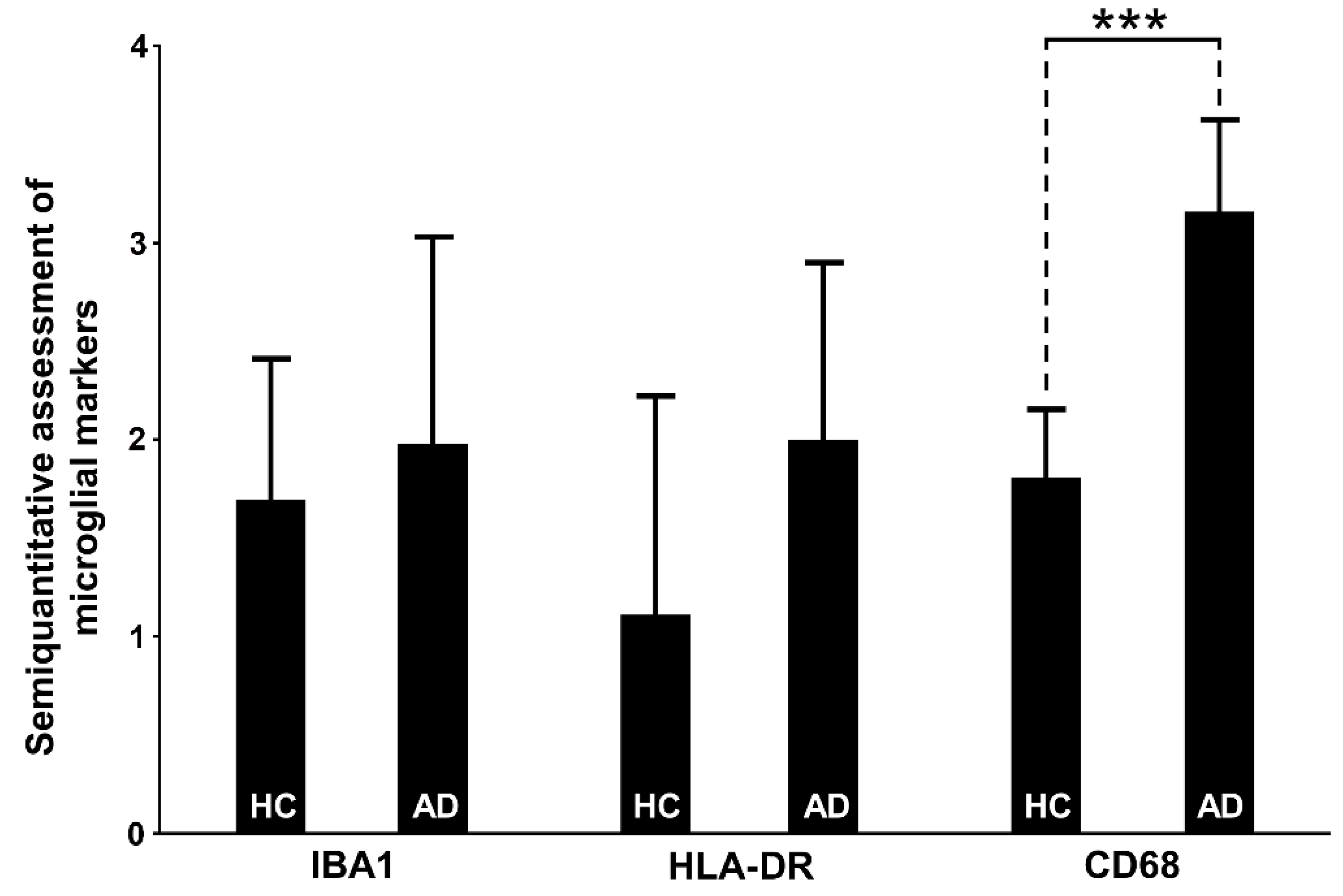
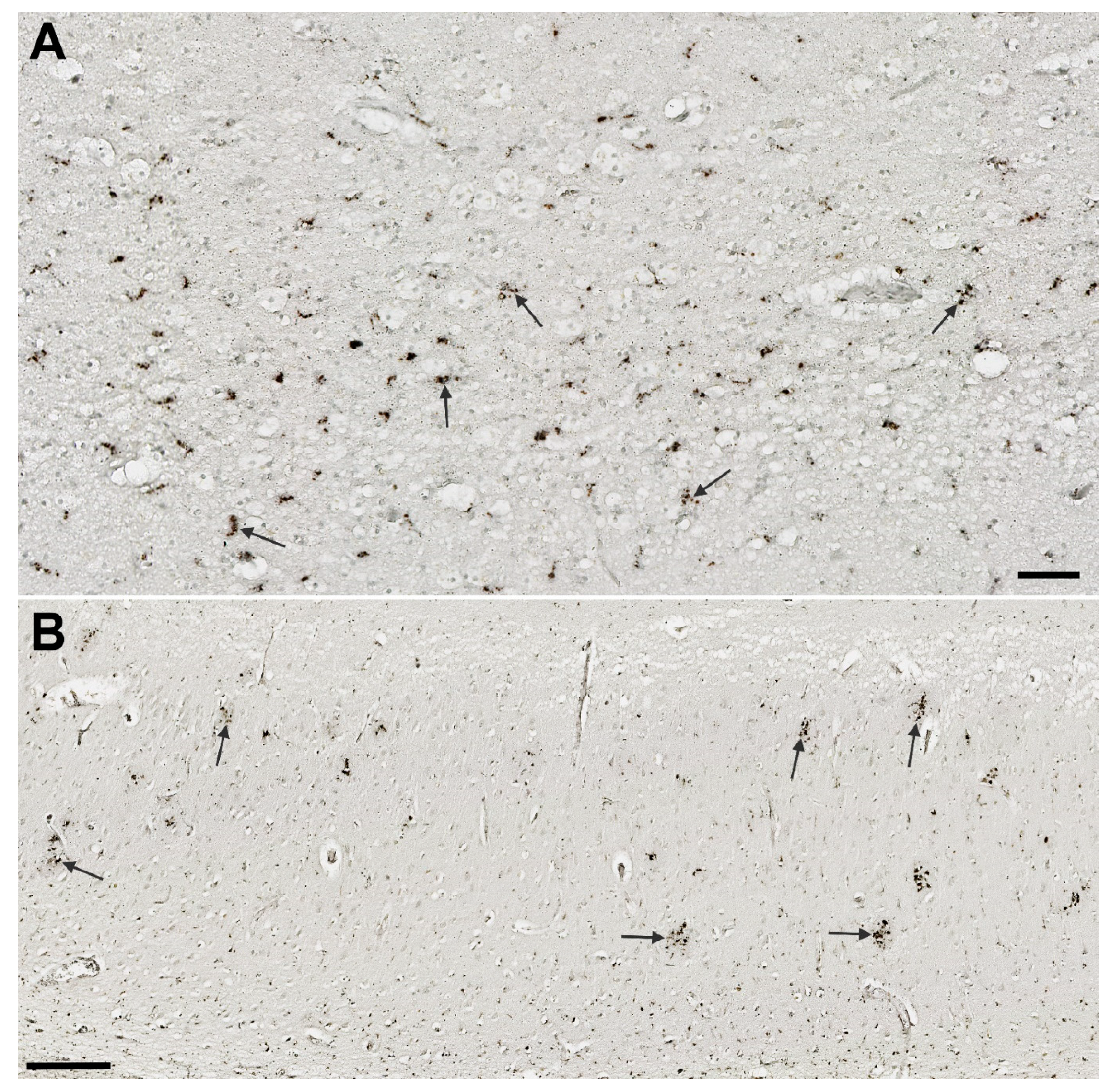
3.6. Assessment of Microglial Markers
3.6.1. IBA1
3.6.2. HLA-DR
3.6.3. CD68
3.7. Illustrations of the Most Significant Findings
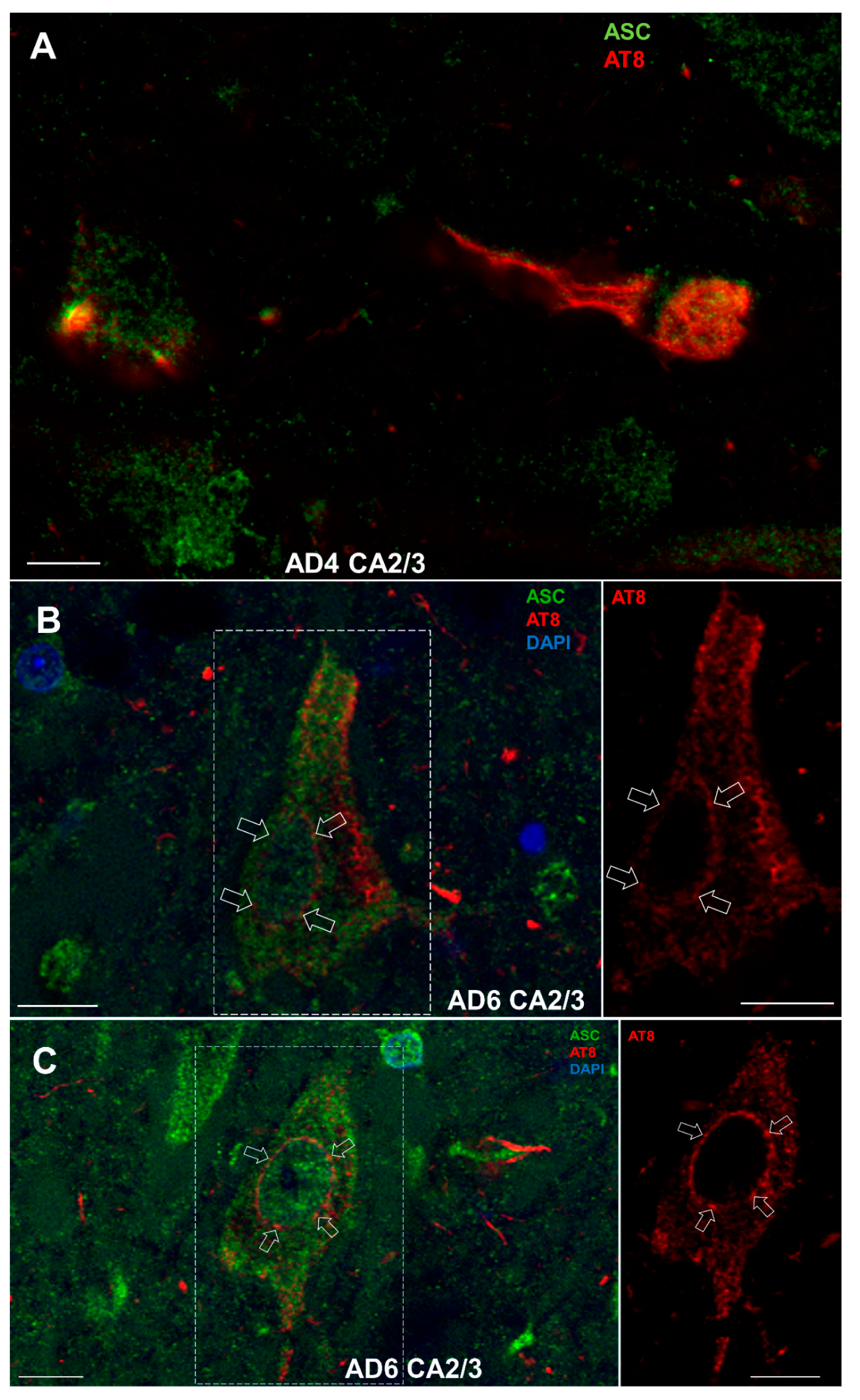
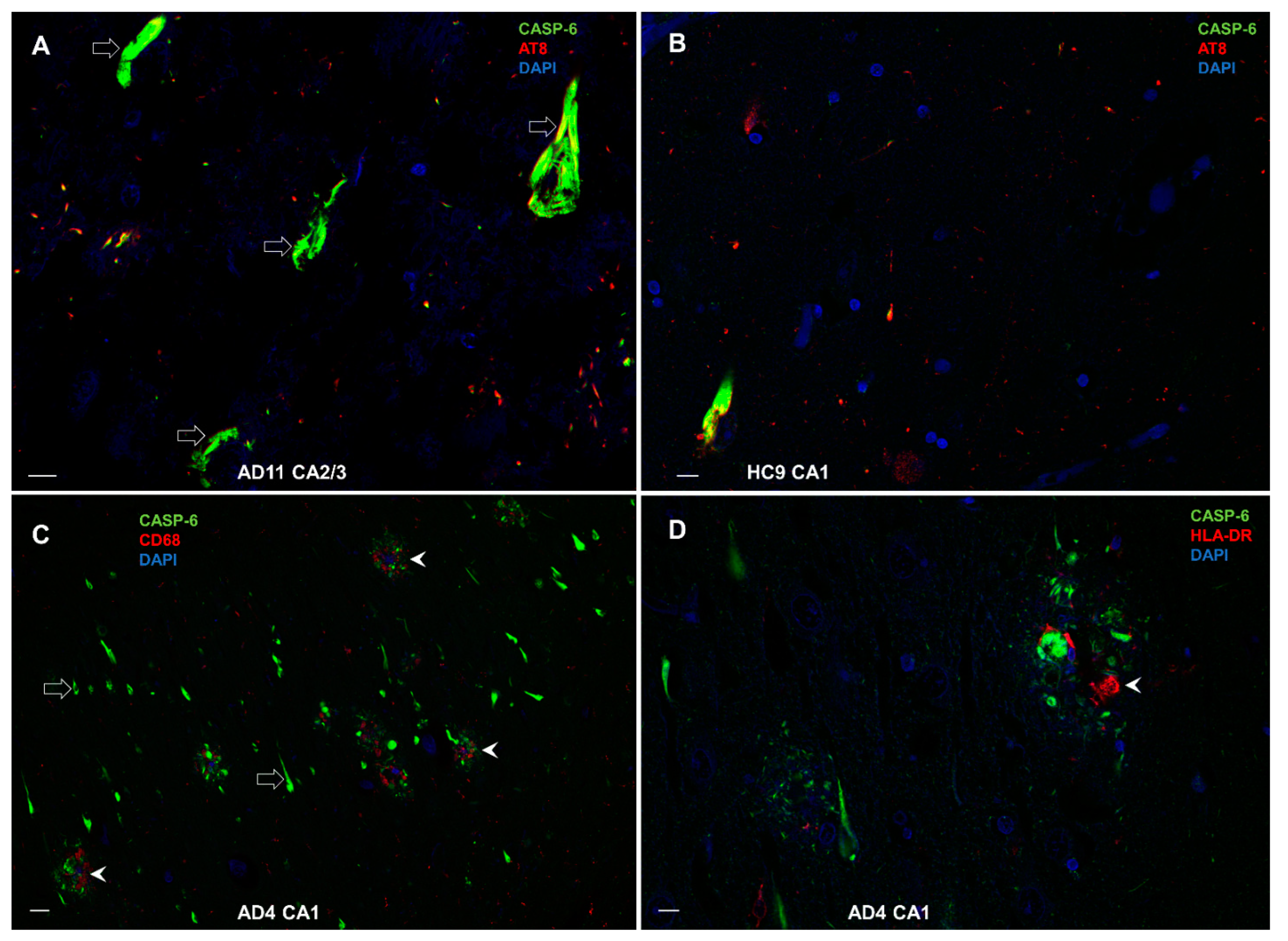
3.8. Double-Labeling Experiments
3.8.1. Colocalization of NLRP1 with ASC, AT8, CD68, and HLA-DR
3.8.2. Colocalization of ASC with AT8
3.8.3. Colocalization of CASP-6 with AT8, CD68, and HLA-DR
4. Discussion
4.1. NLRP1 and ASC Immunoreactivity
4.2. Cleaved Gasdermin and Caspase-6 Immunoreactivity
4.3. Relationship of Inflammasome Activation with Neuron Loss, Neurofibrillary Pathology, and Other Indices of Neurodegeneration
4.4. Relationship of Inflammasome Activation with Microglial Markers IBA1, HLA-DR, and CD68
4.5. Limitation of the Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4R tau | tau proteins with four microtubule-binding domain repeats |
| AD | Alzheimer’s disease |
| Aβ | amyloid β |
| APP | amyloid precursor protein |
| ASC | apoptosis-associated speck-like protein |
| CA | cornu Ammonis |
| CARD | caspase activation and recruitment domain |
| CASP-1 | caspase-1 |
| CASP-6 | caspase-6 |
| CD68 | cluster of differentiation 68 |
| CERAD | Consortium to Establish a Registry for Alzheimer’s Disease |
| cGSDMD | cleaved gasdermin |
| DSM-IV | Diagnostic and Statistical Manual of Mental Disorders, fourth edition |
| HC | healthy controls |
| HF | hippocampal formation |
| hCA3 | hilar part of the CA3 field |
| HLA-DR | Human Leukocyte Antigen–DR isotype |
| IBA1 | Ionized calcium-binding adapter molecule 1 |
| NFTs | neurofibrillary tangles |
| NINCDS-ADRDA | National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer’s Disease and Related Disorders Association |
| NLRs | NOD-like leucine-rich repeat receptors |
| NLRP1 | nucleotide-binding domain and leucine-rich repeat-containing receptor family, pyrin domain-containing protein 1 |
| NLRP3 | nucleotide-binding domain and leucine-rich repeat-containing receptor family, pyrin domain-containing protein 3 |
| NN | number of neurons |
| NOD | nucleotide-binding and oligomerization domain |
| PBS | phosphate buffered saline |
| rP | Pearson’s correlation coefficient |
| rS | Spearman’s correlation coefficient |
| SD | standard deviation |
| SUB | subiculum |
References
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Španić, E.; Langer Horvat, L.; Hof, P.R. Blood-brain barrier and innate immunity in the pathogenesis of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2019, 168, 99–145. [Google Scholar] [PubMed]
- Bellucci, A.; Westwood, A.J.; Ingram, E.; Casamenti, F.; Goedert, M.; Spillantini, M.G. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am. J. Pathol. 2004, 165, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, A.; Kawarabayashi, T.; Murakami, T.; Matsubara, E.; Ikeda, M.; Hagiwara, H.; Westaway, D.; George-Hyslop, P.S.; Shoji, M.; Nakazato, Y. Microglial activation in brain lesions with tau deposits: Comparison of human tauopathies and tau transgenic mice TgTauP301L. Brain Res. 2008, 1214, 159–168. [Google Scholar] [CrossRef]
- Zotova, E.; Nicoll, J.A.R.; Kalaria, R.; Holmes, C.; Boche, D. Inflammation in Alzheimer’s disease: Relevance to pathogenesis and therapy. Alzheimer’s Res. Ther. 2010, 2, 1–9. [Google Scholar] [CrossRef]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.W. Amyloid-β protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Španić, E.; Langer Horvat, L.; Hof, P.R.; Šimić, G. Role of microglial cells in Alzheimer’s Disease tau propagation. Front. Aging Neurosci. 2019, 11, 271. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pei, L.; Zang, D.; Xue, Y.; Wang, X.; Chen, Y.; Li, J.; Yu, J.; Gao, Q.; Di, W.; et al. Gender differences of NLRP1 inflammasome in mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2020, 12, 512097. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Yin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R. Glial cell dysregulation: A new perspective on Alzheimer’s disease. Neurotox. Res. 2007, 12, 215–232. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Denver, P.; McClean, P.L. Distinguishing normal brain aging from the development of Alzheimer’s disease: Inflammation, insulin signaling and cognition. Neural Regen. Res. 2018, 13, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Chiroma, S.M.; Baharuldin, M.T.H.; Taib, C.N.M.; Amom, Z.; Jagadeesan, S.; Moklas, M.A.M. Inflammation in Alzheimer’s disease: A friend or foe? Biomed. Res. Ther. 2018, 5, 2552–2564. [Google Scholar] [CrossRef]
- Albornoz, E.A.; Woodruff, T.M.; Gordon, R. Inflammasomes in CNS diseases. Exp. Suppl. 2018, 108, 41–60. [Google Scholar]
- Alcocer-Gómez, E.; Castejón-Vega, B.; López-Sánchez, M.; Cordero, M.D. Inflammasomes in clinical practice: A brief introduction. Exp. Suppl. 2018, 108, 1–8. [Google Scholar]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Pe´trin, D.; Zhang, Y.; Bergeron, C.; Goodyer, C.G.; LeBlanc, A.C. Caspase-1 activation of caspase-6 in human apoptotic neurons. Cell Death Differ. 2006, 13, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Albrecht, S.; Bourdeau, M.; Petzke, T.; Bergeron, C.; LeBlanc, A.C. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am. J. Pathol. 2004, 165, 523–531. [Google Scholar] [CrossRef]
- Albrecht, S.; Bourdeau, M.; Bennett, D.; Mufson, E.J.; Bhattacharjee, M.; LeBlanc, A.C. Activation of caspase-6 in aging and mild cognitive impairment. Am. J. Pathol. 2007, 170, 1200–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theofilas, P.; Ehrenberg, A.J.; Nguy, A.; Thackrey, J.M.; Dunlop, S.; Mejia, M.B.; Alho, T.A.; Leite, R.E.P.; Rodriguez, R.D.; Suemoto, C.K.; et al. Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: A quantitative study in humans. Neurobiol. Aging 2018, 61, 1–12. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Pontillo, A.; Catamo, E.; Arosio, B.; Mari, D.; Crovella, S. NALP1/NLRP1 genetic variants are associated with Alzheimer’s disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 277–281. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Tan, M.S.; Tan, L.; Jiang, T.; Zhu, X.C.; Wang, H.F.; Jia, C.D.; Yu, J.T. Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieria-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks crossseed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Dansokho, C.; Heneka, M.T. Neuroinflammatory responses in Alzheimer’s disease. J. Neural. Transm. 2018, 125, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Pickard, B.S.; Chan, E.W.L.; Gan, S.Y. The role of neuronal NLRP1 inflammasome in Alzheimer’s disease: Bringing neurons into the neuroinflammation game. Mol. Neurobiol. 2019, 56, 7741–7753. [Google Scholar] [CrossRef] [PubMed]
- Severini, C.; Barbato, C.; Di Certo, M.G.; Gabanella, F.; Petrella, C.; Stadio, A.D.; de Vincentiis, M.; Polimeni, A.; Ralli, M.; Greco, A. Alzheimer’s Disease: New concepts on the role of autoimmunity and NLRP3 inflammasome in the pathogenesis of the disease. Curr. Neuropharmacol. 2021, 19, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef]
- Wang, H.; Yang, F.; Xin, R.; Cui, D.; He, J.; Zhang, S.; Sun, Y. The gut microbiota attenuate neuroinflammation in manganese exposure by inhibiting cerebral NLRP3 inflammasome. Biomed. Pharmacother. 2020, 129, 110449. [Google Scholar] [CrossRef]
- Kummer, J.A.; Broekhuizen, R.; Everett, H.; Agostini, L.; Kuijk, L.; Martinon, F.; van Bruggen, R.; Tschopp, J. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J. Histochem. Cytochem. 2007, 55, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Ding, N.; Wang, K.; Li, Z. Electroacupuncture could influence the expression of IL-1 β and NLRP3 inflammasome in hippocampus of Alzheimer’s Disease animal model. Evid. Based Complement. Altern. Med. 2018, 2018, 8296824. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.J.; Lin, C.H.; Lee, M.C.; Hsieh-Li, H.M.; Chen, C.M.; Wu, Y.R.; Chang, K.H.; Lee-Chen, G.J. Formulated Chinese medicine Shaoyao Gancao Tang reduces NLRP1 and NLRP3 in Alzheimer’s disease cell and mouse models for neuroprotection and cognitive improvement. Aging 2021, 13, 15620–15637. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Q.; Guan, H.; Zhou, Y.; Liu, L. Schisandrin inhibits NLRP1 inflammasome-mediated neuronal pyroptosis in mouse models of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2021, 17, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Guan, B.; Tan, S.; Zhu, H.; Ren, D.; Li, R.; Xiao, L. Bushen Huoxue acupuncture inhibits NLRP1 inflammasomemediated neuronal pyroptosis in SAMP8 mouse model of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2021, 17, 339–346. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hirano, A. A comparative study of modified Bielschowsky, Bodian and thioflavin S stain on Alzheimer’s neurofibrillary tangles. Neuropathol. Appl. Neurobiol. 1986, 12, 3–9. [Google Scholar] [CrossRef]
- Šimić, G.; Kostović, I.; Winblad, B.; Bogdanović, N. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J. Comp. Neurol. 1997, 379, 482–494. [Google Scholar] [CrossRef]
- West, M.J.; Gundersen, H.J. Unbiased stereological estimation of the number of neurons in the human hippocampus. J. Comp. Neurol. 1990, 296, 1–22. [Google Scholar] [CrossRef]
- Šimić, G.; Gnjidić, M.; Kostović, I. Cytoskeletal changes as an alternative view on pathogenesis of Alzheimer’s disease. Period. Biol. 1998, 100, 165–173. [Google Scholar]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.-C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammatome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Defining microglial states and nomenclature: A roadmap to 2030. Cell 2022. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsu, B.V.; Beierschmitt, C.; Ryan, A.P.; Agarwal, R.; Mitchell, P.S.; Daugherty, M.D. Diverse viral proteases activate the NLRP1 inflammasome. Elife 2021, 10, 1–76. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, P.; Cella, C.; Locker, N.; Ravindran, K.K.G.; Mendis, A.; Wafford, K.; Gilmour, G.; Dijk, D.-J.; Winsky-Sommerer, R. Improved sleep, memory, and cellular pathological features of tauopathy, including the NLRP3 inflammasome, after chronic administration of trazodone in rTg450 mice. Neurobiol. Dis. 2022, 42, 3494–3509. [Google Scholar]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, C.J. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef]
- Šimić, G.; Lucassen, P.J.; Krsnik, Ž.; Krušlin, B.; Kostović, I.; Winblad, B.; Bogdanović, N. nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer’s disease. Exp. Neurol. 2000, 165, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Frisoni, G.B.; Ganzola, R.; Canu, E.; Rüb, U.; Pizzini, F.B.; Alessandrini, F.; Zoccatelli, G.; Beltramelo, A.; Caltagirone, C.; Thompson, P.M. Mapping local hippocampal changes in Alzheimer’s disease and normal ageing with MRI at 3 Tesla. Brain 2008, 131, 3266–3276. [Google Scholar] [CrossRef] [Green Version]
- Ishizawa, T.; Ko, L.; Cookson, N.; Davies, P.; Espinoza, M.; Dickson, D.W. Selective neurofibrillary degeneration of the hippocampal CA2 sector is associated with four-repeat tauopathies. J. Neuropathol. Exp. Neurol. 2002, 61, 1040–1047. [Google Scholar] [CrossRef] [Green Version]
- Šimić, G. Pathological tau proteins in argyrophilic grain disease. Lancet Neurol. 2002, 1, 276. [Google Scholar] [CrossRef]
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef]
- Liu, A.K.L.; Chau, T.W.; Lim, E.J.; Ahmed, I.; Chang, R.C.-C.; Kalaitzakis, M.E.; Graeber, M.B.; Gentleman, S.M.; Pearce, R.K.B. Hippocampal CA2 Lewy pathology is associated with cholinergic degeneration in Parkinson’s disease with cognitive decline. Acta Neuropathol. Commun. 2019, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.Z.; Tan, L.; Bi, Y.L.; Shen, X.N.; Xu, W.; Ma, Y.H.; Li, H.Q.; Dong, Q.; Yu, J.T. Dynamic changes of CSF sTREM2 in preclinical Alzheimer’s disease: The CABLE study. Mol. Neurodegener. 2020, 15, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Shen, H.; Sheng, Y.; Guan, Q. ADMSC Exo-MicroRNA-22 improve neurological function and neuroinflammation in mice with Alzheimer’s disease. J. Cell Mol. Med. 2021, 25, 7513–7523. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhang, X.; Xu, G.; Zeng, X.; Li, L. Thioredoxin-1 inhibits amyloid-β25–35-induced activation of NLRP1/caspase-1/GSDMD pyroptotic pathway in PC12 cells. Mol. Biol. Rep. 2022, 49, 3445–3452. [Google Scholar] [CrossRef]
- McKenzie, B.A.; Dixit, V.M.; Power, C. Fiery cell death: Pyroptosis in the central nervous system. Trends Neurosci. 2020, 43, 55–73. [Google Scholar] [CrossRef]
- Chan, A.H.; Schroeder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K.; Hosojima, S.; Hara, H.; Kushiyama, H.; Mahib, M.R.; Kinoshita, T.; Suda, T. Gasdermin D mediates the maturation and release of IL-1α downstream of inflammasomes. Cell Rep. 2021, 34, 108887. [Google Scholar] [CrossRef]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 2013, 33, 5053. [Google Scholar] [CrossRef]
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Goodyer, C.; LeBlanc, A. Selective and protracted apoptosis in human primary neurons microinjected with active caspase-3, -6, -7, and -8. J. Neurosci. 2000, 20, 8384–8389. [Google Scholar] [CrossRef]
- Raina, A.K.; Hochman, A.; Zhu, X.; Rottkamp, C.A.; Nunomura, A.; Siedlak, S.L.; Boux, H.; Castellani, R.J.; Perry, G.; Smith, M.A. Abortive apoptosis in Alzheimer’s disease. Acta Neuropathol. 2001, 101, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Klaiman, G.; Petzke, T.L.; Hammond, J.; LeBlanc, A.C. Targets of caspase-6 activity in human neurons and Alzheimer’s disease. Mol. Cell Proteom. 2008, 7, 1541–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, A.; Liu, H.; Goodyer, C.; Bergeron, C.; Hammond, J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem. 1999, 274, 23426–23436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramcharitar, J.M.; Afonso, V.; Albrecht, S.A.; Bennett, D.; LeBlanc, A.C. Caspase-6 activity predicts lower episodic memory ability in aged individuals. Neurobiol. Aging 2013, 34, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Noël, A.; Foveau, B.; LeBlanc, A.C. Caspase-6-cleaved tau fails to induce tau hyperphosphorylation and aggregation, neurodegeneration, glial inflammation, and cognitive deficits. Cell Death Dis. 2021, 12, 227. [Google Scholar] [CrossRef]
- Godefroy, N.; Foveau, B.; Albrecht, S.; Goodyer, C.G.; LeBlanc, A.C. Expression and activation of Caspase-6 in human fetal and adult tissues. PLoS ONE 2013, 8, e79313. [Google Scholar] [CrossRef] [Green Version]
- Hof, P.R.; Bussière, T.; Gold, G.; Kövari, E.; Giannakopoulos, P.; Bouras, C.; Perl, D.P.; Morrison, J.H. Stereological evidence for persistence of viable neurons in layer II of the entorhinal cortex and the CA1 field in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 2003, 62, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jiang, Y.; Zhu, J.; Liang, H.; He, X.; Qian, J.; Lin, H.; Tao, Y.; Zhu, K. Quantitative assessment of hippocampal tau pathology in AD and PART. J. Mol. Neurosci. 2020, 70, 1808–1811. [Google Scholar] [CrossRef]
- Cherry, J.D.; Esnault, C.D.; Baucom, Z.H.; Tripodis, Y.; Huber, B.R.; Alvarez, V.E.; Stein, T.D.; Dickson, D.W.; McKee, A.C. Tau isoforms are differentially expressed across the hippocampus in chronic traumatic encephalopathy and Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 86. [Google Scholar] [CrossRef]
- Leblanc, A.C.; Ramcharitar, J.; Afonso, V.; Hamel, E.; Bennett, D.A.; Pakavathkumar, P.; Albrecht, S. Caspase-6 activity in the CA1 region of the hippocampus induces age-dependent memory impairment. Cell Death Differ. 2014, 21, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Boscia, F.; Esposito, C.L.; Di Crisci, A.; de Franciscis, V.; Annunziato, L.; Cerchia, L. GDNF selectively induces microglial activation and neuronal survival in CA1/CA3 hippocampal regions exposed to NMDA insult through Ret/ERK signalling. PLoS ONE 2009, 4, e6486. [Google Scholar] [CrossRef] [PubMed]
- Rayamajhi, M.; Miao, E.A. Just say NO to NLRP3. Nat. Immunol. 2013, 14, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wang, H.; Liu, Z.; Lin, L.; Wang, L.; Xie, M.; Li, D.; Zhang, J.; Zhang, R. Endoplasmic reticulum stress-dependent activation of iNOS/NO-NF-κB signaling and NLRP3 inflammasome contributes to endothelial inflammation and apoptosis associated with microgravity. FASEB J. 2020, 34, 10835–10849. [Google Scholar] [CrossRef]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S.; et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Rathinam, V.A.K.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; Fitzgerald, K.A.; Sassetti, C.M. Nitric oxide controls tuberculosis immunopathology by inhibiting NLRP3 inflammasome-dependent IL-1β processing. Nat. Immunol. 2013, 14, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brone, B.; Huaux, F.; Octave, J.N.; et al. Aggregated tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [Green Version]
- Poitelon, Y.; Johnson, L.; Liddelow, S.A.; Giovannini, M.G.; Lana, D.; Ugolini, F. An Overview on the differential interplay among neurons-astrocytes-microglia in CA1 and CA3 hippocampus in hypoxia/ischemia. Front. Cell Neurosci. 2020, 14, 585833. [Google Scholar]
- Sun, D.; Gao, G.; Zhong, B.; Zhang, H.; Ding, S.; Sun, Z.; Zhang, Y.; Li, W. NLRP1 inflammasome involves in learning and memory impairments and neuronal damages during aging process in mice. Behav. Brain Funct. 2021, 17, 11. [Google Scholar] [CrossRef]
- Šimić, G.; Bexheti, S.; Kelović, Z.; Kos, M.; Grbić, K.; Hof, P.R.; Kostović, I. Hemispheric asymmetry, modular variability and age-related changes in the human entorhinal cortex. Neuroscience 2005, 130, 911–925. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Van Eldik, L.J.; Schmitt, F.A.; Neltner, J.H.; Ighodaro, E.T.; Webster, S.J.; Patel, E.; Abner, E.L.; Kryscio, R.; Nelson, P.T. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol. Commun. 2015, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Minett, T.; Classey, J.; Matthews, F.E.; Fahrenhold, M.; Taga, M.; Brayne, C.; Ince, P.G.; Nicoll, J.A.R.; Boche, D.; MRC CFAS. Microglial immunophenotype in dementia with Alzheimer’s pathology. J. Neuroinflammation 2016, 13, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrickx, D.A.E.; van Eden, C.G.; Schuurman, K.G.; Hamann, J.; Huitinga, I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J. Neuroimmunol. 2017, 309, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; Brochado, P.; Robinson, A.; Reis, I.; Costa, P.; Mann, D.M.; Pires, M.M.; Sousa, N. Patterns of microglial cell activation in Alzheimer’s disease and frontotemporal lobar degeneration. Neurodegener. Dis. 2017, 17, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau protein disrupts nucleoplasmic transport in Alzheimer’s disease. Neuron 2019, 99, 925–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Hu, Y.; Fryatt, G.L.; Ghorbani, M.; Obst, J.; Menassa, D.A.; Martin-Estebane, M.; Muntslag, T.A.O.; Olmos-Alonso, A.; Guerrero-Carrasco, M.; Thomas, D.; et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep. 2021, 35, 109228. [Google Scholar] [CrossRef]
- Edler, M.K.; Sherwood, C.C.; Meindl, R.S.; Munger, E.L.; Hopkins, W.D.; Ely, J.J.; Erwin, J.M.; Perl, D.P.; Mufson, E.J.; Hof, P.R.; et al. Microglia changes associated to Alzheimer’s disease pathology in aged chimpanzees. J. Comp. Neurol. 2018, 526, 2921–2930. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | HC | ||||
| Age | Gender | Cause of Death | |||
| HC1 | 59 | M | Car accident | ||
| HC2 | 62 | F | Car accident | ||
| HC3 | 68 | M | Car accident | ||
| HC4 | 71 | F | Myocardial infarction | ||
| HC5 | 75 | M | Car accident | ||
| HC6 | 77 | F | Myocardial infarction | ||
| HC7 | 81 | F | Myocardial infarction | ||
| HC8 | 84 | F | Pulmonary embolism | ||
| HC9 | 85 | F | Cardiovascular failure | ||
| Case | AD | ||||
| Age | Gender | Duration of the Disease (years) | Cause of Death | NINCDS-ADRDA Diagnosis | |
| AD1 | 73 | F | 4 | Bronchopneumonia | Definitive AD |
| AD2 | 73 | M | 7 | Bronchopneumonia | Definitive AD |
| AD3 | 77 | M | 3.5 | Bronchopneumonia | Definitive AD |
| AD4 | 80 | F | 5 | Bronchopneumonia | Definitive AD |
| AD5 | 80 | F | 6 | Myocardial infarction | Definitive AD |
| AD6 | 81 | M | 3 | Bronchopneumonia | Definitive AD |
| AD7 | 83 | F | 5.5 | Cardiovascular failure | Probable AD |
| AD8 | 84 | F | 3.5 | Cardiovascular failure | Definitive AD |
| AD9 | 84 | F | 3.5 | Cardiovascular failure | Definitive AD |
| AD10 | 88 | F | 4.5 | Carcinoma | Probable AD |
| AD11 | 91 | F | 4 | Bronchopneumonia | Probable AD |
| Case | hCA3 Counts | Area | CA2/3 Counts | Area | CA1 Counts | Area | SUB Counts | Area | Total Counts | Σ Area | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | A | G | C | mm2 | N | A | G | C | mm2 | N | A | G | C | mm2 | N | A | G | C | mm2 | N | A | G | C | mm2 | |
| HC1 | 4 | 12 | 0 | 12 | 5981 | 23 | 26 | 0 | 1 | 3449 | 113 | 140 | 0 | 84 | 8421 | 0 | 0 | 0 | 0 | 0 | 140 | 178 | 0 | 97 | 17,852 |
| HC2 | 86 | 92 | 32 | 108 | 7837 | 0 | 0 | 1 | 23 | 0 | 16 | 17 | 3 | 51 | 10,702 | 63 | 53 | 4 | 55 | 14,885 | 165 | 162 | 40 | 237 | 33,424 |
| HC3 | 16 | 19 | 32 | 9 | 7738 | 38 | 24 | 47 | 11 | 11,277 | 30 | 50 | 50 | 9 | 17,299 | 67 | 32 | 22 | 55 | 12,825 | 151 | 125 | 151 | 84 | 49,139 |
| HC4 | 52 | 38 | 10 | 13 | 3953 | 188 | 139 | 24 | 75 | 2302 | 159 | 100 | 4 | 35 | 10,149 | 279 | 121 | 2 | 44 | 10,065 | 678 | 398 | 40 | 167 | 26,468 |
| HC5 | 54 | 95 | 81 | 98 | 7362 | 183 | 200 | 185 | 340 | 6829 | 113 | 109 | 252 | 230 | 22,223 | 57 | 155 | 43 | 55 | 23,678 | 407 | 559 | 561 | 723 | 60,092 |
| HC6 | 35 | 17 | 12 | 7 | 4201 | 45 | 20 | 9 | 9 | 1803 | 28 | 18 | 3 | 14 | 13,001 | 65 | 173 | 48 | 70 | 10,486 | 173 | 228 | 72 | 100 | 29,490 |
| HC7 | 6 | 149 | 17 | 43 | 4486 | 74 | 159 | 30 | 70 | 1596 | 44 | 131 | 11 | 22 | 12,506 | 96 | 160 | 41 | 53 | 9121 | 220 | 599 | 99 | 188 | 28,108 |
| HC8 | 16 | 41 | 4 | 13 | 3445 | 32 | 149 | 8 | 78 | 1388 | 19 | 119 | 14 | 20 | 6647 | 20 | 96 | 0 | 15 | 15,423 | 87 | 405 | 26 | 126 | 26,903 |
| HC9 | 73 | 83 | 7 | 13 | 5412 | 67 | 36 | 0 | 5 | 3916 | 69 | 26 | 0 | 16 | 14,360 | 34 | 42 | 0 | 24 | 14,239 | 243 | 187 | 7 | 58 | 37,926 |
| AD1 | 58 | 109 | 15 | 114 | 6247 | 73 | 129 | 51 | 102 | 2162 | 28 | 39 | 14 | 124 | 6441 | 24 | 99 | 13 | 146 | 6131 | 183 | 376 | 93 | 486 | 20,980 |
| AD2 | 60 | 278 | 48 | 90 | 3897 | 127 | 124 | 58 | 80 | 1180 | 104 | 36 | 10 | 141 | 4680 | 284 | 300 | 36 | 129 | 9659 | 575 | 738 | 152 | 440 | 19,416 |
| AD3 | 7 | 123 | 26 | 33 | 4018 | 96 | 81 | 45 | 41 | 2619 | 16 | 32 | 6 | 70 | 11,055 | 44 | 60 | 13 | 10 | 11,419 | 163 | 296 | 90 | 154 | 29,111 |
| AD4 | 88 | 74 | 23 | 44 | 12,189 | 200 | 113 | 8 | 50 | 2671 | 180 | 27 | 2 | 171 | 7269 | 179 | 119 | 0 | 170 | 15,626 | 647 | 333 | 33 | 435 | 37,754 |
| AD5 | 38 | 54 | 10 | 23 | 7351 | 152 | 131 | 36 | 25 | 1571 | 79 | 100 | 38 | 207 | 7029 | 98 | 170 | 11 | 130 | 10,083 | 367 | 455 | 95 | 385 | 26,034 |
| AD6 | 84 | 156 | 21 | 39 | 8899 | 148 | 121 | 7 | 39 | 2588 | 100 | 14 | 0 | 79 | 5571 | 132 | 142 | 9 | 40 | 7837 | 464 | 433 | 37 | 197 | 24,894 |
| AD7 | 38 | 227 | 44 | 60 | 4881 | 141 | 278 | 60 | 69 | 2720 | 65 | 82 | 15 | 226 | 7956 | 257 | 146 | 8 | 97 | 7240 | 501 | 733 | 127 | 452 | 22,797 |
| AD8 | 45 | 67 | 22 | 74 | 8738 | 151 | 78 | 17 | 29 | 3386 | 105 | 38 | 29 | 439 | 8296 | 67 | 29 | 12 | 210 | 6921 | 368 | 212 | 80 | 752 | 27,342 |
| AD9 | 4 | 45 | 11 | 29 | 3622 | 19 | 88 | 44 | 35 | 1995 | 48 | 19 | 16 | 275 | 6168 | 6 | 74 | 14 | 180 | 7741 | 77 | 226 | 85 | 519 | 19,527 |
| AD10 | 20 | 17 | 12 | 22 | 4356 | 187 | 84 | 27 | 61 | 4201 | 66 | 54 | 24 | 215 | 21,832 | 109 | 45 | 4 | 245 | 6501 | 382 | 200 | 67 | 543 | 36,890 |
| AD11 | 32 | 140 | 23 | 63 | 7476 | 113 | 172 | 8 | 67 | 2562 | 87 | 52 | 5 | 25 | 6547 | 32 | 117 | 0 | 2 | 3276 | 264 | 481 | 36 | 157 | 19,862 |
| Case | NLRP1 (Cells/mm2) | ASC (Cells/mm2) | GSDMD (Cells/mm2) | CASP-6 (Cells/mm2) |
|---|---|---|---|---|
| HC1 | 0.784 | 1.000 | 0.000 | 0.543 |
| HC2 | 0.494 | 0.480 | 0.120 | 0.709 |
| HC3 | 0.307 | 0.250 | 0.307 | 0.171 |
| HC4 | 2.562 | 1.500 | 0.151 | 0.631 |
| HC5 | 0.677 | 0.930 | 0.934 | 1.203 |
| HC6 | 0.587 | 0.770 | 0.244 | 0.339 |
| HC7 | 0.783 | 2.130 | 0.352 | 0.669 |
| HC8 | 0.323 | 1.510 | 0.097 | 0.468 |
| HC9 | 0.641 | 0.490 | 0.019 | 0.153 |
| AD1 | 0.872 | 1.792 | 0.443 | 2.316 |
| AD2 | 2.961 | 3.801 | 0.783 | 2.266 |
| AD3 | 0.560 | 1.017 | 0.309 | 0.529 |
| AD4 | 1.714 | 0.882 | 0.087 | 1.152 |
| AD5 | 1.410 | 1.748 | 0.365 | 1.479 |
| AD6 | 1.864 | 1.740 | 0.149 | 0.791 |
| AD7 | 2.198 | 3.215 | 0.557 | 1.983 |
| AD8 | 1.346 | 0.775 | 0.293 | 2.750 |
| AD9 | 0.394 | 1.157 | 0.435 | 2.658 |
| AD10 | 1.036 | 0.542 | 0.182 | 1.472 |
| AD11 | 1.329 | 2.422 | 0.181 | 0.790 |
| Case | hCA3 | CA2/3 | CA1 | SUB | Σ | ||||
|---|---|---|---|---|---|---|---|---|---|
| NN (106) | NFTs (106) | NN (106) | NFTs (106) | NN (106) | NFTs (106) | NN (106) | NFTs (106) | NFTs (106) | |
| HC1 | 0.84 | 0 | 1.2 | 0 | 7.18 | 0 | 2.61 | 0 | 0 |
| HC2 | 1.29 | 0 | 1.41 | 0 | 8.41 | 0 | 4.34 | 0 | 0 |
| HC3 | 1.57 | 0 | 1.94 | 0 | 2.62 | 0 | 2 | 0 | 0 |
| HC4 | 1.27 | 0 | 2.9 | 0 | 5.35 | 0 | 1.47 | 0 | 0 |
| HC5 | 1.4 | 0 | 2.48 | 0 | 8.28 | 0 | 4.85 | 0.05 | 0.05 |
| HC6 | 0.83 | 0 | 1.68 | 0 | 2.43 | 0 | 1.79 | 0.03 | 0.03 |
| HC7 | 0.87 | 0 | 1.61 | 0 | 3.42 | 0 | 2.29 | 0 | 0 |
| HC8 | 1.35 | 0 | 1.45 | 0 | 7.89 | 0.01 | 4.89 | 0 | 0 |
| HC9 | 1.29 | 0 | 2.16 | 0 | 2.26 | 0 | 1.59 | 0 | 0 |
| AD1 | 1 | 0.08 | 0.98 | 0.22 | 4.93 | 3.45 | 0.93 | 0.71 | 4.45 |
| AD2 | 1.63 | 0.08 | 1.45 | 0.51 | 5.48 | 4.02 | 3.47 | 0.54 | 5.14 |
| AD3 | 0.57 | 0.13 | 1.64 | 0 | 5 | 0.81 | 1.42 | 0.25 | 1.19 |
| AD4 | 0.93 | 0.16 | 1.82 | 0.11 | 10.9 | 1.95 | 2.78 | 0.43 | 2.66 |
| AD5 | 0.3 | 0 | 0.92 | 0.18 | 4.38 | 3.15 | 2.4 | 0.52 | 3.85 |
| AD6 | 1.8 | 0.3 | 0.78 | 0.2 | 4.59 | 2.88 | 3.16 | 1.19 | 4.57 |
| AD7 | 0.37 | 0 | 1 | 0 | 3.09 | 1.99 | 1.25 | 0.13 | 2.13 |
| AD8 | 1.67 | 0.07 | 1.39 | 0.11 | 7.65 | 2.42 | 3.81 | 0.55 | 3.15 |
| AD9 | 0.17 | 0.08 | 1.26 | 0 | 2.06 | 1.42 | 1.01 | 0.3 | 1.8 |
| AD10 | 0.23 | 0 | 0.84 | 0.19 | 3.27 | 1.88 | 1.79 | 0.68 | 2.74 |
| AD11 | 0.53 | 0.08 | 0.96 | 0.37 | 1.14 | 0.55 | 0.84 | 0.27 | 1.27 |
| Case | hCA3 Counts | CA2/3 Counts | CA1 Counts | SUB Counts | Total Counts per Sample | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | H | C | I | H | C | I | H | C | I | H | C | I | H | C | |
| HC1 | 3 | 3 | 3 | 2 | 3 | 2 | 0 | 2 | 2 | 1 | 2 | 2 | 6 | 10 | 9 |
| HC2 | 3 | 2 | 2 | 3 | 2 | 3 | 2 | 3 | 1 | 3 | 2 | 1 | 11 | 9 | 7 |
| HC3 | 2 | 0 | 1 | 2 | 0 | 2 | 2 | 0 | 1 | 1 | 0 | 1 | 7 | 0 | 5 |
| HC4 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 2 | 3 | 0 | 2 | 9 | 0 | 6 |
| HC5 | 2 | 2 | 2 | 1 | 2 | 2 | 0 | 2 | 1 | 0 | 2 | 1 | 3 | 8 | 6 |
| HC6 | 2 | 2 | 2 | 1 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 7 | 9 | 8 |
| HC7 | 0 | 1 | 2 | 1 | 0 | 2 | 1 | 0 | 1 | 0 | 2 | 3 | 2 | 3 | 8 |
| HC8 | 3 | 0 | 2 | 2 | 0 | 2 | 2 | 0 | 1 | 2 | 1 | 2 | 9 | 1 | 7 |
| HC9 | 2 | 0 | 3 | 1 | 0 | 2 | 2 | 0 | 2 | 2 | 0 | 2 | 7 | 0 | 9 |
| AD1 | 0 | 0 | 3 | 0 | 2 | 2 | 0 | 1 | 4 | 0 | 1 | 3 | 0 | 4 | 12 |
| AD2 | 2 | 1 | 4 | 3 | 1 | 3 | 3 | 2 | 4 | 3 | 1 | 4 | 11 | 5 | 15 |
| AD3 | 0 | 1 | 3 | 2 | 0 | 3 | 1 | 2 | 4 | 0 | 2 | 3 | 3 | 5 | 13 |
| AD4 | 2 | 1 | 3 | 3 | 0 | n.a. | 2 | 3 | 4 | 3 | 4 | 4 | 10 | 8 | 11 |
| AD5 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 4 | 3 | 3 | 3 | 11 | 12 | 13 |
| AD6 | 2 | 2 | 4 | 3 | 0 | 3 | 3 | 3 | 4 | 2 | 2 | 3 | 10 | 7 | 14 |
| AD7 | 0 | 2 | 1 | 3 | 0 | 3 | 2 | 4 | 3 | 4 | 4 | 4 | 9 | 10 | 11 |
| AD8 | 0 | 4 | 4 | 1 | 3 | 3 | 1 | 4 | 4 | 0 | 4 | 4 | 2 | 15 | 15 |
| AD9 | 0 | 2 | 4 | 3 | 0 | 3 | 3 | 2 | 4 | 2 | 0 | 3 | 8 | 4 | 14 |
| AD10 | 3 | 0 | 2 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 2 | 4 | 12 | 7 | 12 |
| AD11 | 3 | 3 | 2 | 3 | 2 | 2 | 3 | 3 | 2 | 2 | 3 | 3 | 11 | 11 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Španić, E.; Langer Horvat, L.; Ilić, K.; Hof, P.R.; Šimić, G. NLRP1 Inflammasome Activation in the Hippocampal Formation in Alzheimer’s Disease: Correlation with Neuropathological Changes and Unbiasedly Estimated Neuronal Loss. Cells 2022, 11, 2223. https://doi.org/10.3390/cells11142223
Španić E, Langer Horvat L, Ilić K, Hof PR, Šimić G. NLRP1 Inflammasome Activation in the Hippocampal Formation in Alzheimer’s Disease: Correlation with Neuropathological Changes and Unbiasedly Estimated Neuronal Loss. Cells. 2022; 11(14):2223. https://doi.org/10.3390/cells11142223
Chicago/Turabian StyleŠpanić, Ena, Lea Langer Horvat, Katarina Ilić, Patrick R. Hof, and Goran Šimić. 2022. "NLRP1 Inflammasome Activation in the Hippocampal Formation in Alzheimer’s Disease: Correlation with Neuropathological Changes and Unbiasedly Estimated Neuronal Loss" Cells 11, no. 14: 2223. https://doi.org/10.3390/cells11142223