
The Role of Heat Shock Protein 70 Subfamily in the Hyperplastic Prostate: From Molecular Mechanisms to Therapeutic Opportunities


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Benign Prostate Hyperplasia
3. Members of HSP70 Family
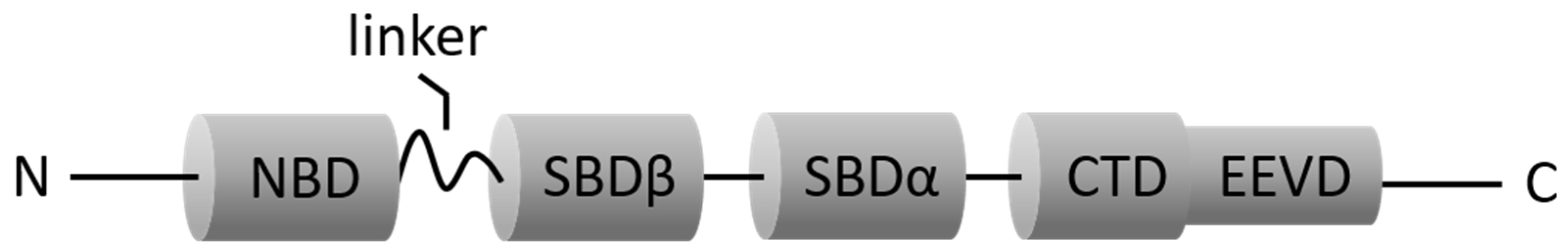
3.1. HSP70 (HSC70 and Inducible HSP70)
3.2. GRP78 (HSPA5)
3.3. Mortalin (GRP75, HSPA9)
4. The Association of HSP70s with BPH
4.1. HSP70s and Cell Survival, Proliferation and Apoptosis
4.2. HSP70s and Oxidative Stress
4.3. HSP70s and EMT Process
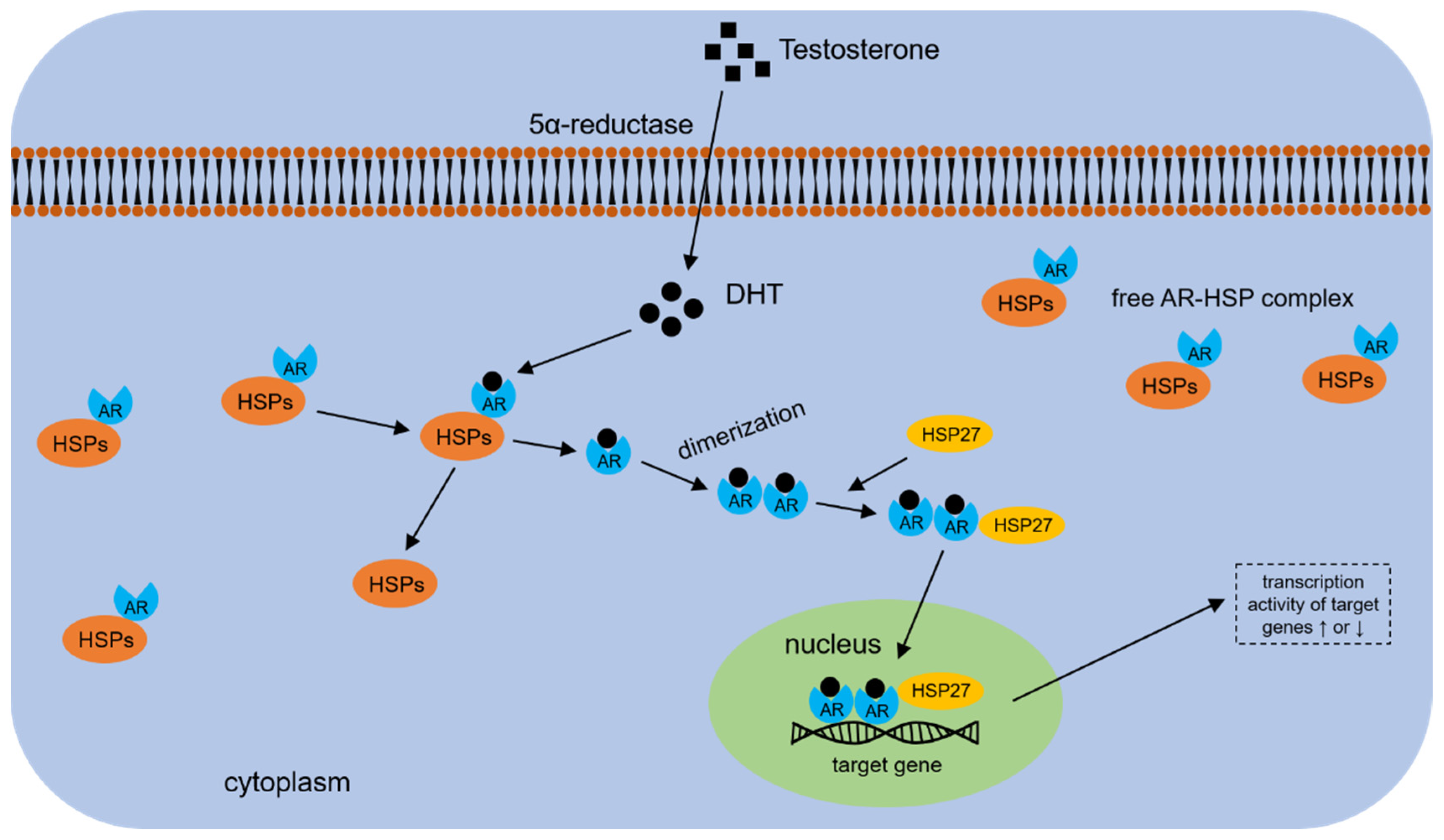
4.4. HSP70s and AR
5. HSP70s as Potential Therapeutic Targets for BPH
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lokeshwar, S.D.; Harper, B.T.; Webb, E.; Jordan, A.; Dykes, T.A.; Neal, D.J.; Terris, M.K.; Klaassen, Z. Epidemiology and treatment modalities for the management of benign prostatic hyperplasia. Transl. Androl. Urol. 2019, 8, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Launer, B.M.; McVary, K.T.; Ricke, W.A.; Lloyd, G.L. The rising worldwide impact of benign prostatic hyperplasia. BJU Int. 2021, 127, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.K. Benign Prostatic Hyperplasia and Male Lower Urinary Tract Symptoms: Epidemiology and Risk Factors. Curr. Bladder. Dysfunct. Rep. 2010, 5, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, G.; de la Taille, A.; Descazeaud, A. Epidemiology of benign prostatic hyperplasia. Prog. Urol. 2018, 28, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.J.; Coffey, D.S.; Walsh, P.C.; Ewing, L.L. The development of human benign prostatic hyperplasia with age. J. Urol. 1984, 132, 474–479. [Google Scholar] [CrossRef]
- Irwin, D.E.; Milsom, I.; Hunskaar, S.; Reilly, K.; Kopp, Z.; Herschorn, S.; Coyne, K.; Kelleher, C.; Hampel, C.; Artibani, W.; et al. Population-based survey of urinary incontinence, overactive bladder, and other lower urinary tract symptoms in five countries: Results of the EPIC study. Eur. Urol. 2006, 50, 1306–1315. [Google Scholar] [CrossRef]
- Parsons, J.K.; Bergstrom, J.; Silberstein, J.; Barrett-Connor, E. Prevalence and characteristics of lower urinary tract symptoms in men aged > or = 80 years. Urology 2008, 72, 318–321. [Google Scholar] [CrossRef] [Green Version]
- McConnell, J.D. Medical management of benign prostatic hyperplasia with androgen suppression. Prostate Suppl. 1990, 3, 49–59. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Brossner, C.; Reiner, A.; Cheng, G.; Sugiyama, N.; Warner, M.; Gustafsson, J.A. A role for epithelial-mesenchymal transition in the etiology of benign prostatic hyperplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 2859–2863. [Google Scholar] [CrossRef] [Green Version]
- Broster, S.A.; Kyprianou, N. Epithelial-mesenchymal transition in prostatic disease. Future. Oncol. 2015, 11, 3197–3206. [Google Scholar] [CrossRef]
- Aydin, A.; Arsova-Sarafinovska, Z.; Sayal, A.; Eken, A.; Erdem, O.; Erten, K.; Ozgok, Y.; Dimovski, A. Oxidative stress and antioxidant status in non-metastatic prostate cancer and benign prostatic hyperplasia. Clin. Biochem. 2006, 39, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Aryal, M.; Pandeya, A.; Bas, B.K.; Lamsal, M.; Majhi, S.; Pandit, R.; Agrawal, C.S.; Gautam, N.; Baral, N. Oxidative stress in patients with benign prostate hyperplasia. JNMA J. Nepal. Med. Assoc. 2007, 46, 103–106. [Google Scholar] [PubMed]
- Altinok, S.; Sanchez-Hodge, R.; Stewart, M.; Smith, K.; Schisler, J.C. With or without You: Co-Chaperones Mediate Health and Disease by Modifying Chaperone Function and Protein Triage. Cells 2021, 10, 3121. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Iturbe, B.; Lanaspa, M.A.; Johnson, R.J. The role of autoimmune reactivity induced by heat shock protein 70 in the pathogenesis of essential hypertension. Br. J. Pharmacol. 2019, 176, 1829–1838. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, W.; Lubkowski, M.; Lubkowska, A. Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 897. [Google Scholar] [CrossRef]
- Kabakov, A.E.; Gabai, V.L. HSP70s in Breast Cancer: Promoters of Tumorigenesis and Potential Targets/Tools for Therapy. Cells 2021, 10, 3446. [Google Scholar] [CrossRef]
- Fu, X.; Liu, J.; Liu, D.; Zhou, Y.; Guo, Y.; Wang, Z.; Yang, S.; He, W.; Chen, P.; Wang, X.; et al. Glucose-regulated protein 78 modulates cell growth, epithelial-mesenchymal transition, and oxidative stress in the hyperplastic prostate. Cell Death Dis. 2022, 13, 78. [Google Scholar] [CrossRef]
- Jones, E.L.; Zhao, M.J.; Stevenson, M.A.; Calderwood, S.K. The 70 kilodalton heat shock protein is an inhibitor of apoptosis in prostate cancer. Int. J. Hyperth. 2004, 20, 835–849. [Google Scholar] [CrossRef]
- Wang, X.J.; Ni, X.Q.; Zhao, S.; Zhao, R.Z.; Wang, X.H.; Xia, S.J.; Sun, X.W.; Zhuo, J. ROS-NLRP3 signaling pathway induces sterile inflammation after thulium laser resection of the prostate. J. Cell. Physiol. 2022, 237, 1923–1935. [Google Scholar] [CrossRef]
- Schuster, G.A.; Schuster, T.G. The relative amount of epithelium, muscle, connective tissue and lumen in prostatic hyperplasia as a function of the mass of tissue resected. J. Urol. 1999, 161, 1168–1173. [Google Scholar] [CrossRef]
- McNeal, J.E. Origin and evolution of benign prostatic enlargement. Investig. Urol. 1978, 15, 340–345. [Google Scholar]
- Strand, D.W.; Costa, D.N.; Francis, F.; Ricke, W.A.; Roehrborn, C.G. Targeting phenotypic heterogeneity in benign prostatic hyperplasia. Differentiation 2017, 96, 49–61. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; Koff, A.; Drobnjak, M.; Capodieci, P.; Osman, I.; Millard, S.S.; Gaudin, P.B.; Fazzari, M.; Zhang, Z.F.; Massague, J.; et al. Distinct altered patterns of p27KIP1 gene expression in benign prostatic hyperplasia and prostatic carcinoma. J. Natl. Cancer Inst. 1998, 90, 1284–1291. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Liu, J.; Li, Y.; Liu, H.; Hassan, H.M.; He, W.; Li, M.; Zhou, Y.; Fu, X.; Zhan, J.; et al. Upregulated bone morphogenetic protein 5 enhances proliferation and epithelial-mesenchymal transition process in benign prostatic hyperplasia via BMP/Smad signaling pathway. Prostate 2021, 81, 1435–1449. [Google Scholar] [CrossRef]
- Liu, J.; Yin, J.; Chen, P.; Liu, D.; He, W.; Li, Y.; Li, M.; Fu, X.; Zeng, G.; Guo, Y.; et al. Smoothened inhibition leads to decreased cell proliferation and suppressed tissue fibrosis in the development of benign prostatic hyperplasia. Cell Death Discov. 2021, 7, 115. [Google Scholar] [CrossRef]
- Kyprianou, N.; Tu, H.; Jacobs, S.C. Apoptotic versus proliferative activities in human benign prostatic hyperplasia. Hum. Pathol. 1996, 27, 668–675. [Google Scholar] [CrossRef]
- Jiang, M.Y.; Han, Z.D.; Li, W.; Yue, F.; Ye, J.; Li, B.; Cai, Z.; Lu, J.M.; Dong, W.; Jiang, X.; et al. Mitochondrion-associated protein peroxiredoxin 3 promotes benign prostatic hyperplasia through autophagy suppression and pyroptosis activation. Oncotarget 2017, 8, 80295–80302. [Google Scholar] [CrossRef] [Green Version]
- Minciullo, P.L.; Inferrera, A.; Navarra, M.; Calapai, G.; Magno, C.; Gangemi, S. Oxidative stress in benign prostatic hyperplasia: A systematic review. Urol. Int. 2015, 94, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Udensi, U.K.; Tchounwou, P.B. Oxidative stress in prostate hyperplasia and carcinogenesis. J. Exp. Clin. Cancer Res. 2016, 35, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltaci, S.; Orhan, D.; Gogus, C.; Turkolmez, K.; Tulunay, O.; Gogus, O. Inducible nitric oxide synthase expression in benign prostatic hyperplasia, low- and high-grade prostatic intraepithelial neoplasia and prostatic carcinoma. BJU Int. 2001, 88, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Gradini, R.; Realacci, M.; Ginepri, A.; Naso, G.; Santangelo, C.; Cela, O.; Sale, P.; Berardi, A.; Petrangeli, E.; Gallucci, M.; et al. Nitric oxide synthases in normal and benign hyperplastic human prostate: Immunohistochemistry and molecular biology. J. Pathol. 1999, 189, 224–229. [Google Scholar] [CrossRef]
- Pace, G.; di Massimo, C.; de Amicis, D.; Corbacelli, C.; di Renzo, L.; Vicentini, C.; Miano, L.; Tozzi, C.M. Oxidative stress in benign prostatic hyperplasia and prostate cancer. Urol. Int. 2010, 85, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Zastawny, T.H.; Foksinski, M.; Barecki, A.; Dizdaroglu, M. DNA base modifications and antioxidant enzyme activities in human benign prostatic hyperplasia. Free. Radic. Biol. Med. 1995, 18, 807–813. [Google Scholar] [CrossRef]
- Fraga, C.H.; True, L.D.; Kirk, D. Enhanced expression of the mesenchymal marker, vimentin, in hyperplastic versus normal human prostatic epithelium. J. Urol. 1998, 159, 270–274. [Google Scholar] [CrossRef]
- Barrack, E.R.; Bujnovszky, P.; Walsh, P.C. Subcellular distribution of androgen receptors in human normal, benign hyperplastic, and malignant prostatic tissues: Characterization of nuclear salt-resistant receptors. Cancer Res. 1983, 43, 1107–1116. [Google Scholar]
- Rennie, P.; Bruchovsky, N.; Goldenberg, S. Relationship of androgen receptors to the growth and regression of the prostate. Am. J. Clin. Oncol. 1988, 11, S13–S17. [Google Scholar] [CrossRef]
- Bierhoff, E.; Vogel, J.; Benz, M.; Giefer, T.; Wernert, N.; Pfeifer, U. Stromal nodules in benign prostatic hyperplasia. Eur. Urol. 1996, 29, 345–354. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, C.; Lin, C.C.; Niu, Y.; Lai, K.P.; Chang, H.C.; Yeh, S.D.; Chang, C.; Yeh, S. Altered prostate epithelial development and IGF-1 signal in mice lacking the androgen receptor in stromal smooth muscle cells. Prostate 2011, 71, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Yeh, C.R.; Niu, Y.; Chang, H.C.; Tsai, Y.C.; Moses, H.L.; Shyr, C.R.; Chang, C.; Yeh, S. Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate 2012, 72, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.P.; Yamashita, S.; Vitkus, S.; Shyr, C.R.; Yeh, S.; Chang, C. Suppressed prostate epithelial development with impaired branching morphogenesis in mice lacking stromal fibromuscular androgen receptor. Mol. Endocrinol. 2012, 26, 52–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lin, W.J.; Izumi, K.; Jiang, Q.; Lai, K.P.; Xu, D.; Fang, L.Y.; Lu, T.; Li, L.; Xia, S.; et al. Increased infiltrated macrophages in benign prostatic hyperplasia (BPH): Role of stromal androgen receptor in macrophage-induced prostate stromal cell proliferation. J. Biol. Chem. 2012, 287, 18376–18385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Jha, R.; Melamed, J.; Shapiro, E.; Hayward, S.W.; Lee, P. Stromal androgen receptor in prostate development and cancer. Am. J. Pathol. 2014, 184, 2598–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, C.; Boufaied, N.; Mills, I.G.; Franco, O.E.; Hayward, S.W.; Thomson, A.A. Genome-wide analysis of AR binding and comparison with transcript expression in primary human fetal prostate fibroblasts and cancer associated fibroblasts. Mol. Cell. Endocrinol. 2018, 471, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Lin, W.J.; Izumi, K.; Wang, X.; Xu, D.; Fang, L.Y.; Li, L.; Jiang, Q.; Jin, J.; Chang, C. Targeting androgen receptor to suppress macrophage-induced EMT and benign prostatic hyperplasia (BPH) development. Mol. Endocrinol. 2012, 26, 1707–1715. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Daugaard, M.; Rohde, M.; Jaattela, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [Green Version]
- Havalova, H.; Ondrovicova, G.; Keresztesova, B.; Bauer, J.A.; Pevala, V.; Kutejova, E.; Kunova, N. Mitochondrial HSP70 Chaperone System-The Influence of Post-Translational Modifications and Involvement in Human Diseases. Int. J. Mol. Sci. 2021, 22, 8077. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Dworniczak, B.; Mirault, M.E. Structure and expression of a human gene coding for a 71 kd heat shock ‘ognate’ protein. Nucleic Acids Res. 1987, 15, 5181–5197. [Google Scholar] [CrossRef] [Green Version]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C. Molecular chaperones and protein quality control. Protein Pept. Lett. 2011, 18, 100. [Google Scholar] [CrossRef]
- Andrade-Tomaz, M.; de Souza, I.; Rocha, C.; Gomes, L.R. The Role of Chaperone-Mediated Autophagy in Cell Cycle Control and Its Implications in Cancer. Cells 2020, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Kennedy, A.L.; Isingizwe, Z.R.; Javadian, P.; Benbrook, D.M. Similarities and Differences of Hsp70, hsc70, Grp78 and Mortalin as Cancer Biomarkers and Drug Targets. Cells 2021, 10, 2996. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [Green Version]
- Cesa, L.C.; Shao, H.; Srinivasan, S.R.; Tse, E.; Jain, C.; Zuiderweg, E.; Southworth, D.R.; Mapp, A.K.; Gestwicki, J.E. X-linked inhibitor of apoptosis protein (XIAP) is a client of heat shock protein 70 (Hsp70) and a biomarker of its inhibition. J. Biol. Chem. 2018, 293, 2370–2380. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Fernandez, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J.M. Hsp70—A master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.V.; Clarke, P.A.; Workman, P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 2008, 14, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Ni, M.; Lee, A.S. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007, 581, 3641–3651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, D.M.; Long, A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol. 2012, 34, 286–297. [Google Scholar]
- Elfiky, A.A.; Baghdady, A.M.; Ali, S.A.; Ahmed, M.I. GRP78 targeting: Hitting two birds with a stone. Life Sci. 2020, 260, 118317. [Google Scholar] [CrossRef]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- Munro, S.; Pelham, H.R. An Hsp70-like protein in the ER: Identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 1986, 46, 291–300. [Google Scholar] [CrossRef]
- Wiersma, V.R.; Michalak, M.; Abdullah, T.M.; Bremer, E.; Eggleton, P. Mechanisms of Translocation of ER Chaperones to the Cell Surface and Immunomodulatory Roles in Cancer and Autoimmunity. Front. Oncol. 2015, 5, 7. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Zhang, Y.; Tseng, C.C.; Stanciauskas, R.; Pinaud, F.; Lee, A.S. Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J. Biol. Chem. 2015, 290, 8049–8064. [Google Scholar] [CrossRef] [Green Version]
- Misra, U.K.; Gonzalez-Gronow, M.; Gawdi, G.; Hart, J.P.; Johnson, C.E.; Pizzo, S.V. The role of Grp 78 in alpha 2-macroglobulin-induced signal transduction. Evidence from RNA interference that the low density lipoprotein receptor-related protein is associated with, but not necessary for, GRP78-mediated signal transduction. J. Biol. Chem. 2002, 277, 42082–42087. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gronow, M.; Kaczowka, S.J.; Payne, S.; Wang, F.; Gawdi, G.; Pizzo, S.V. Plasminogen structural domains exhibit different functions when associated with cell surface GRP78 or the voltage-dependent anion channel. J. Biol. Chem. 2007, 282, 32811–32820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tseng, C.C.; Tsai, Y.L.; Fu, X.; Schiff, R.; Lee, A.S. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS ONE 2013, 8, e80071. [Google Scholar] [CrossRef]
- Misra, U.K.; Pizzo, S.V. Ligation of cell surface GRP78 with antibody directed against the COOH-terminal domain of GRP78 suppresses Ras/MAPK and PI 3-kinase/AKT signaling while promoting caspase activation in human prostate cancer cells. Cancer Biol. Ther. 2010, 9, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlseid, J.N.; Lill, R.; Green, J.M.; Xu, X.; Qiu, Y.; Pierce, S.K. PBP74, a new member of the mammalian 70-kDa heat shock protein family, is a mitochondrial protein. Mol. Biol. Cell 1994, 5, 1265–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Londono, C.; Osorio, C.; Gama, V.; Alzate, O. Mortalin, apoptosis, and neurodegeneration. Biomolecules 2012, 2, 143–164. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [Green Version]
- Bohnert, M.; Pfanner, N.; van der Laan, M. A dynamic machinery for import of mitochondrial precursor proteins. FEBS Lett. 2007, 581, 2802–2810. [Google Scholar] [CrossRef] [Green Version]
- Dolezal, P.; Likic, V.; Tachezy, J.; Lithgow, T. Evolution of the molecular machines for protein import into mitochondria. Science 2006, 313, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Mokranjac, D.; Neupert, W. Thirty years of protein translocation into mitochondria: Unexpectedly complex and still puzzling. Biochim. Biophys. Acta 2009, 1793, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperone machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef] [Green Version]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; de Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Ryu, J.; Kaul, Z.; Yoon, A.R.; Liu, Y.; Yaguchi, T.; Na, Y.; Ahn, H.M.; Gao, R.; Choi, I.K.; Yun, C.O.; et al. Identification and functional characterization of nuclear mortalin in human carcinogenesis. J. Biol. Chem. 2014, 289, 24832–24844. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, R.; Takano, S.; Robert, M.; Yoshida, A.; Nomura, H.; Reddel, R.R.; Mitsui, Y.; Kaul, S.C. Inactivation of tumor suppressor p53 by mot-2, a hsp70 family member. J. Biol. Chem. 1998, 273, 29586–29591. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, R.; Taira, K.; Kaul, S.C. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: What, when, and where? Cell Stress Chaperones 2002, 7, 309–316. [Google Scholar] [CrossRef]
- Bouvard, V.; Zaitchouk, T.; Vacher, M.; Duthu, A.; Canivet, M.; Choisy-Rossi, C.; Nieruchalski, M.; May, E. Tissue and cell-specific expression of the p53-target genes: Bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene 2000, 19, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Kaul, S.C.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274. [Google Scholar] [CrossRef]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brittingham, A.; Morrison, C.J.; McMaster, W.R.; McGwire, B.S.; Chang, K.P.; Mosser, D.M. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J. Immunol. 1995, 155, 3102–3111. [Google Scholar] [CrossRef]
- Pilzer, D.; Saar, M.; Koya, K.; Fishelson, Z. Mortalin inhibitors sensitize K562 leukemia cells to complement-dependent cytotoxicity. Int. J. Cancer. 2010, 126, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Saar, R.M.; Moskovich, O.; Iosefson, O.; Fishelson, Z. Mortalin/GRP75 binds to complement C9 and plays a role in resistance to complement-dependent cytotoxicity. J. Biol. Chem. 2014, 289, 15014–15022. [Google Scholar] [CrossRef] [Green Version]
- Mazkereth, N.; Rocca, F.; Schubert, J.R.; Geisler, C.; Hillman, Y.; Egner, A.; Fishelson, Z. Complement triggers relocation of Mortalin/GRP75 from mitochondria to the plasma membrane. Immunobiology 2016, 221, 1395–1406. [Google Scholar] [CrossRef]
- Brunnert, D.; Langer, C.; Zimmermann, L.; Bargou, R.C.; Burchardt, M.; Chatterjee, M.; Stope, M.B. The heat shock protein 70 inhibitor VER155008 suppresses the expression of HSP27, HOP and HSP90beta and the androgen receptor, induces apoptosis, and attenuates prostate cancer cell growth. J. Cell. Biochem. 2020, 121, 407–417. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, Y.; Xia, X.; Shao, Z.; Huang, C.; He, J.; Jiang, L.; Tang, D.; Liu, J.; Huang, H. Targeting GRP78-dependent AR-V7 protein degradation overcomes castration-resistance in prostate cancer therapy. Theranostics 2020, 10, 3366–3381. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. GRP78 Regulates Apoptosis, Cell Survival and Proliferation in 5-Fluorouracil-resistant SNUC5 Colon Cancer Cells. Anticancer Res. 2017, 37, 4943–4951. [Google Scholar]
- Liu, R.; Li, X.; Gao, W.; Zhou, Y.; Wey, S.; Mitra, S.K.; Krasnoperov, V.; Dong, D.; Liu, S.; Li, D.; et al. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clin. Cancer Res. 2013, 19, 6802–6811. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1alpha to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar]
- Gao, Y.; Han, C.; Huang, H.; Xin, Y.; Xu, Y.; Luo, L.; Yin, Z. Heat shock protein 70 together with its co-chaperone CHIP inhibits TNF-alpha induced apoptosis by promoting proteasomal degradation of apoptosis signal-regulating kinase1. Apoptosis 2010, 15, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jaattela, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Sigua, C.; Bali, P.; George, P.; Fiskus, W.; Scuto, A.; Annavarapu, S.; Mouttaki, A.; Sondarva, G.; Wei, S.; et al. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood 2005, 105, 1246–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabai, V.L.; Mabuchi, K.; Mosser, D.D.; Sherman, M.Y. Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol. Cell. Biol. 2002, 22, 3415–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaglom, J.A.; Ekhterae, D.; Gabai, V.L.; Sherman, M.Y. Regulation of necrosis of H9c2 myogenic cells upon transient energy deprivation. Rapid deenergization of mitochondria precedes necrosis and is controlled by reactive oxygen species, stress kinase JNK, HSP72 and ARC. J. Biol. Chem. 2003, 278, 50483–50496. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Mun, S.; Harada, A.; Ohkawa, Y.; Inagaki, A.; Sano, S.; Takahashi, K.; Izumi, Y.; Osada-Oka, M.; Wanibuchi, H.; et al. Hsc70 contributes to cancer cell survival by preventing Rab1A degradation under stress conditions. PLoS ONE 2014, 9, e96785. [Google Scholar]
- Ouyang, Y.B.; Xu, L.J.; Sun, Y.J.; Giffard, R.G. Overexpression of inducible heat shock protein 70 and its mutants in astrocytes is associated with maintenance of mitochondrial physiology during glucose deprivation stress. Cell Stress Chaperones 2006, 11, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Murtuza, B.; Sammut, I.A.; Latif, N.; Jayakumar, J.; Smolenski, R.T.; Kaneda, Y.; Sawa, Y.; Matsuda, H.; Yacoub, M.H. Heat shock protein 72 enhances manganese superoxide dismutase activity during myocardial ischemia-reperfusion injury, associated with mitochondrial protection and apoptosis reduction. Circulation 2002, 106, I270–I276. [Google Scholar] [CrossRef]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.S.; Li, L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007, 5, e172. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Duan, M.; Ouyang, Y.; Emery, J.F.; Stoy, C.; Giffard, R.G. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J. Cereb. Blood Flow Metab. 2008, 28, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, W.; Song, X.D.; Zuo, J. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol. Cell. Biochem. 2005, 268, 45–51. [Google Scholar] [CrossRef]
- Cultrara, C.N.; Kozuch, S.D.; Ramasundaram, P.; Heller, C.J.; Shah, S.; Beck, A.E.; Sabatino, D.; Zilberberg, J. GRP78 modulates cell adhesion markers in prostate Cancer and multiple myeloma cell lines. BMC Cancer 2018, 18, 1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldscholte, J.; Berrevoets, C.A.; Brinkmann, A.O.; Grootegoed, J.A.; Mulder, E. Anti-androgens and the mutated androgen receptor of LNCaP cells: Differential effects on binding affinity, heat-shock protein interaction, and transcription activation. Biochemistry 1992, 31, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar] [PubMed]
- Cano, L.Q.; Lavery, D.N.; Bevan, C.L. Mini-review: Foldosome regulation of androgen receptor action in prostate cancer. Mol. Cell. Endocrinol. 2013, 369, 52–62. [Google Scholar] [CrossRef]
- Dong, J.; Wu, Z.; Wang, D.; Pascal, L.E.; Nelson, J.B.; Wipf, P.; Wang, Z. Hsp70 Binds to the Androgen Receptor N-terminal Domain and Modulates the Receptor Function in Prostate Cancer Cells. Mol. Cancer Ther. 2019, 18, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.K.; Andriola, I.F.; Kampinga, H.H.; Merry, D.E. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2002, 11, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Moses, M.A.; Kim, Y.S.; Rivera-Marquez, G.M.; Oshima, N.; Watson, M.J.; Beebe, K.E.; Wells, C.; Lee, S.; Zuehlke, A.D.; Shao, H.; et al. Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Tan, Z.; Wortman, M.; Lu, S.; Dong, Z. Regulation of heat shock protein 70-1 expression by androgen receptor and its signaling in human prostate cancer cells. Int. J. Oncol. 2010, 36, 459–467. [Google Scholar]
- Tan, S.S.; Ahmad, I.; Bennett, H.L.; Singh, L.; Nixon, C.; Seywright, M.; Barnetson, R.J.; Edwards, J.; Leung, H.Y. GRP78 up-regulation is associated with androgen receptor status, Hsp70-Hsp90 client proteins and castrate-resistant prostate cancer. J. Pathol. 2011, 223, 81–87. [Google Scholar] [CrossRef]
- Hebert-Schuster, M.; Rotta, B.E.; Kirkpatrick, B.; Guibourdenche, J.; Cohen, M. The Interplay between Glucose-Regulated Protein 78 (GRP78) and Steroids in the Reproductive System. Int. J. Mol. Sci. 2018, 19, 1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.C.; Fu, H.C.; Hsiao, B.L.; Sobue, G.; Adachi, H.; Huang, F.J.; Hsuuw, Y.D.; Wei, K.T.; Chang, C.; Huang, K.E.; et al. Androgen receptor inclusions acquire GRP78/BiP to ameliorate androgen-induced protein misfolding stress in embryonic stem cells. Cell Death Dis. 2013, 4, e607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyrd-Hansen, M.; Nylandsted, J.; Jaattela, M. Heat shock protein 70 promotes cancer cell viability by safeguarding lysosomal integrity. Cell Cycle 2004, 3, 1484–1485. [Google Scholar] [CrossRef]
- Rodina, A.; Vilenchik, M.; Moulick, K.; Aguirre, J.; Kim, J.; Chiang, A.; Litz, J.; Clement, C.C.; Kang, Y.; She, Y.; et al. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat. Chem. Biol. 2007, 3, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, M.; Andrulis, M.; Stuhmer, T.; Muller, E.; Hofmann, C.; Steinbrunn, T.; Heimberger, T.; Schraud, H.; Kressmann, S.; Einsele, H.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E.; et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. USA 2011, 108, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Krause, S.W.; Gastpar, R.; Andreesen, R.; Gross, C.; Ullrich, H.; Thonigs, G.; Pfister, K.; Multhoff, G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: A clinical phase i trial. Clin. Cancer Res. 2004, 10, 3699–3707. [Google Scholar] [CrossRef] [Green Version]
- Trimble, C.L.; Peng, S.; Kos, F.; Gravitt, P.; Viscidi, R.; Sugar, E.; Pardoll, D.; Wu, T.C. A phase I trial of a human papillomavirus DNA vaccine for HPV16+ cervical intraepithelial neoplasia 2/3. Clin. Cancer Res. 2009, 15, 361–367. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, X.; Liu, H.; Liu, J.; DiSanto, M.E.; Zhang, X. The Role of Heat Shock Protein 70 Subfamily in the Hyperplastic Prostate: From Molecular Mechanisms to Therapeutic Opportunities. Cells 2022, 11, 2052. https://doi.org/10.3390/cells11132052
Fu X, Liu H, Liu J, DiSanto ME, Zhang X. The Role of Heat Shock Protein 70 Subfamily in the Hyperplastic Prostate: From Molecular Mechanisms to Therapeutic Opportunities. Cells. 2022; 11(13):2052. https://doi.org/10.3390/cells11132052
Chicago/Turabian StyleFu, Xun, Huan Liu, Jiang Liu, Michael E. DiSanto, and Xinhua Zhang. 2022. "The Role of Heat Shock Protein 70 Subfamily in the Hyperplastic Prostate: From Molecular Mechanisms to Therapeutic Opportunities" Cells 11, no. 13: 2052. https://doi.org/10.3390/cells11132052