
Reverting TP53 Mutation in Breast Cancer Cells: Prime Editing Workflow and Technical Considerations

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
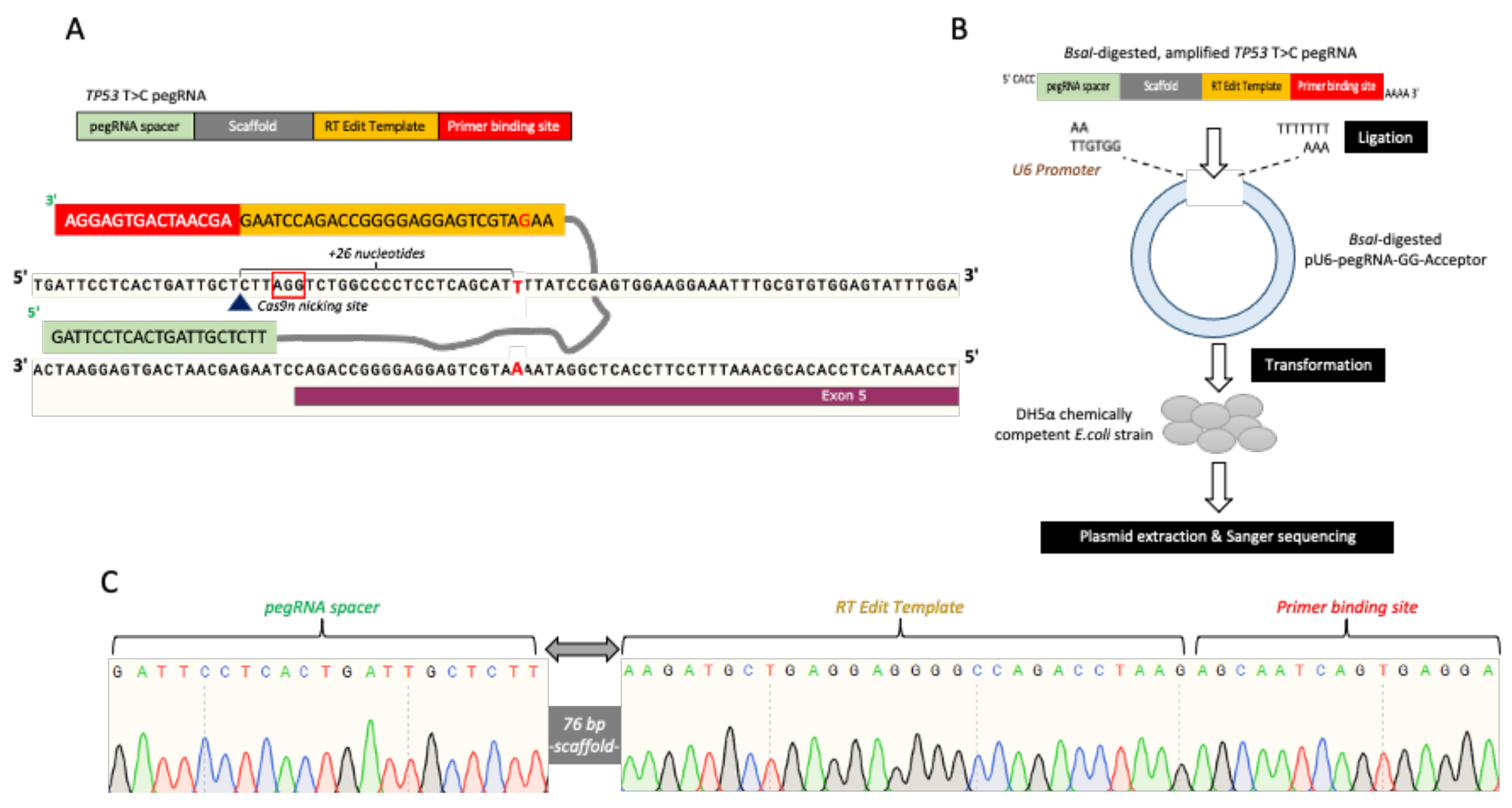
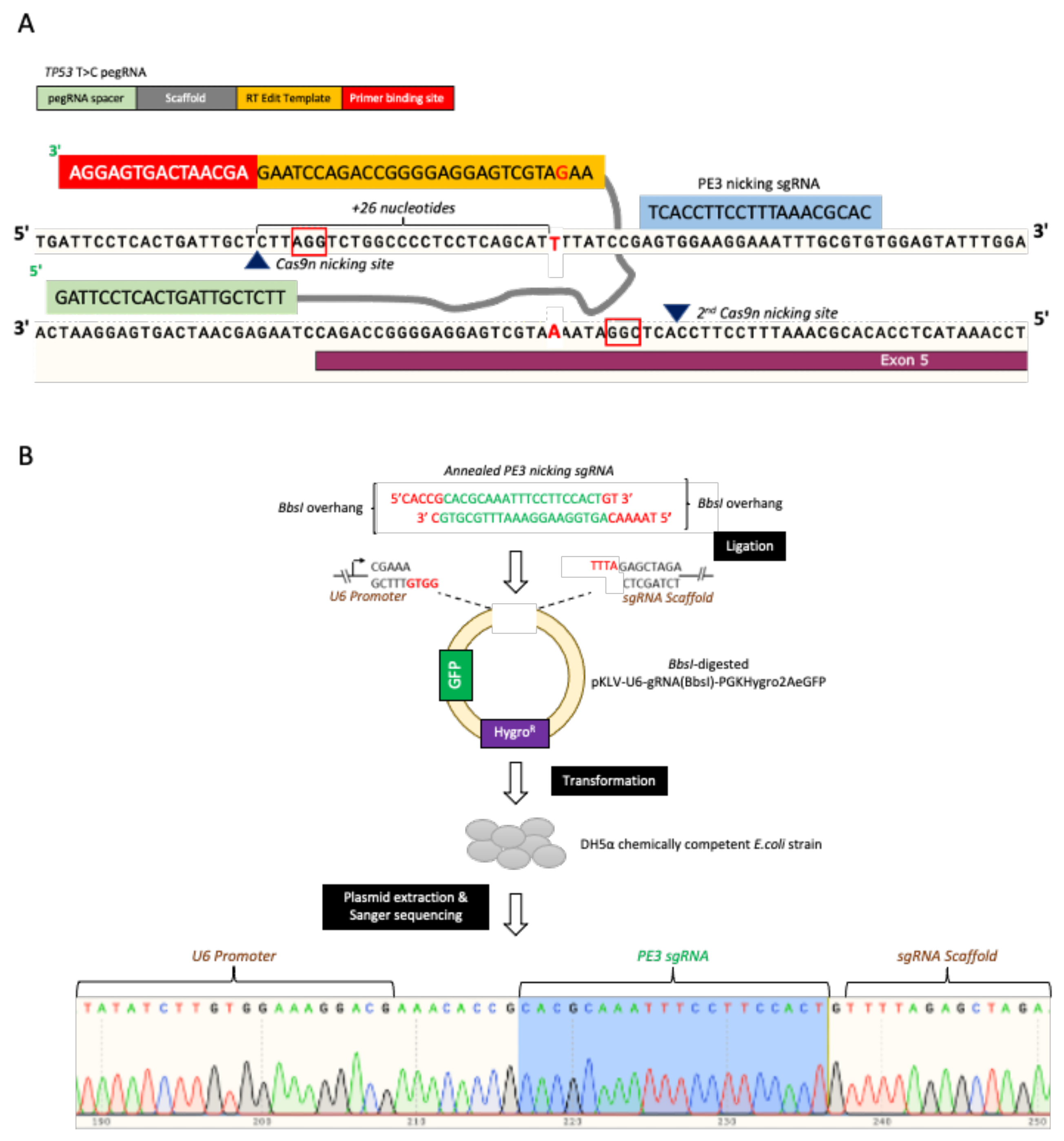
2.2. pegRNAs and Nicking sgRNA Plasmid Construction (PE3)
2.3. Plasmids, Genomic DNA and Proteins Extraction
2.4. Lentiviral Particles Production and Transduction
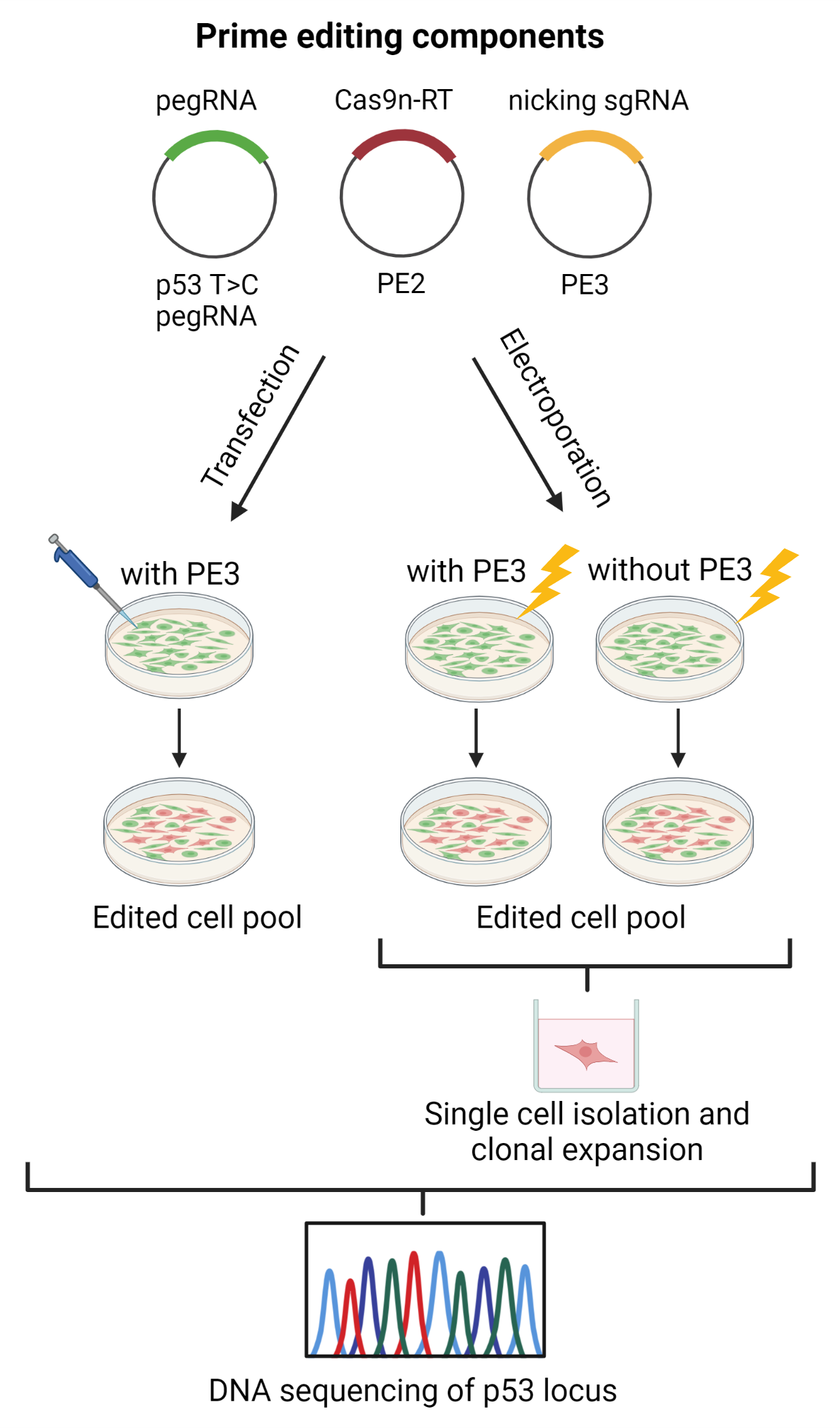
2.5. TP53 Prime Editing: T47D Cells Transfection
2.6. TP53/HEK3 Prime Editing: T47D and HEK293T Cell Electroporation
2.7. TP53 Genotyping Using Sanger Sequencing
2.8. Amplicon Targeted Sequencing
2.9. Western Blotting
3. Results
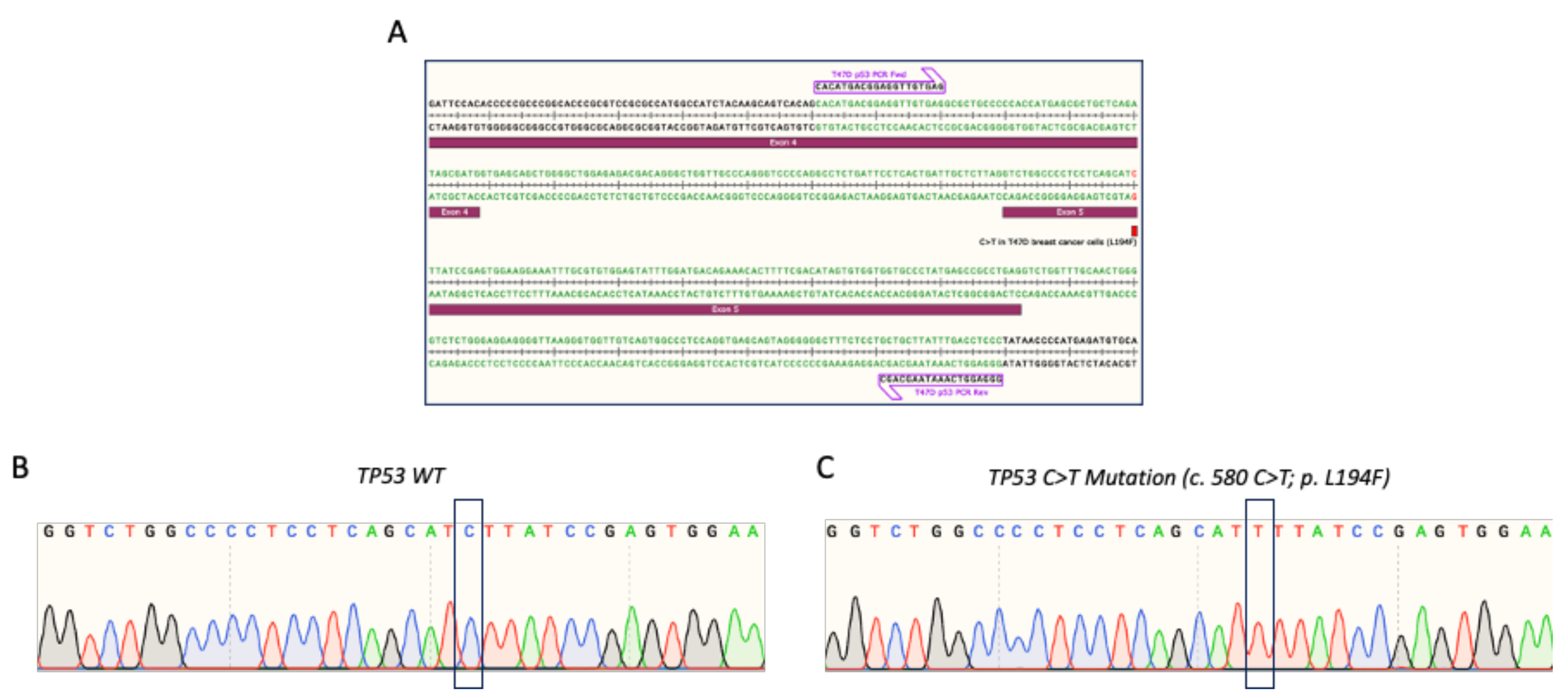
3.1. Verification of TP53 Mutation in T47D Breast Cancer Cells
3.2. pegRNAs and PE3 Plasmids Construction
3.3. Assessing the Prime Editing Components Efficiency
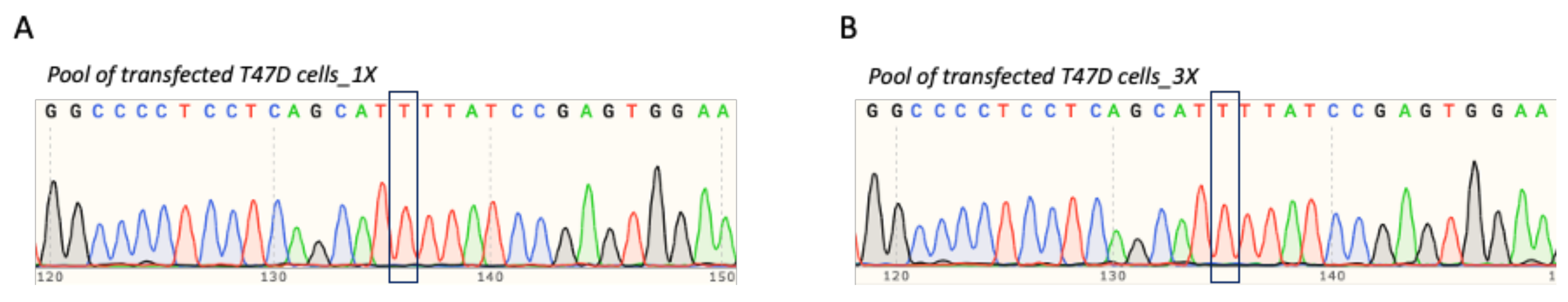
3.4. T47D Cell Transfection and Assessment of TP53 Prime Editing Efficiency
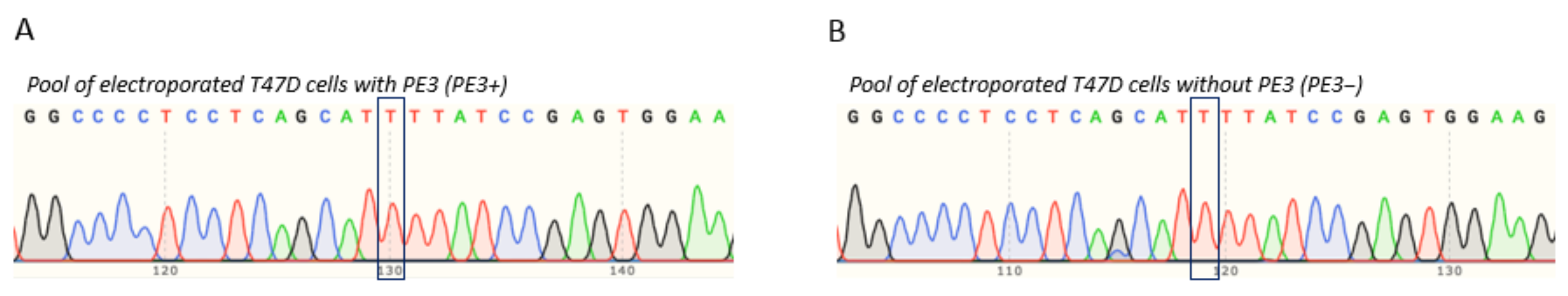
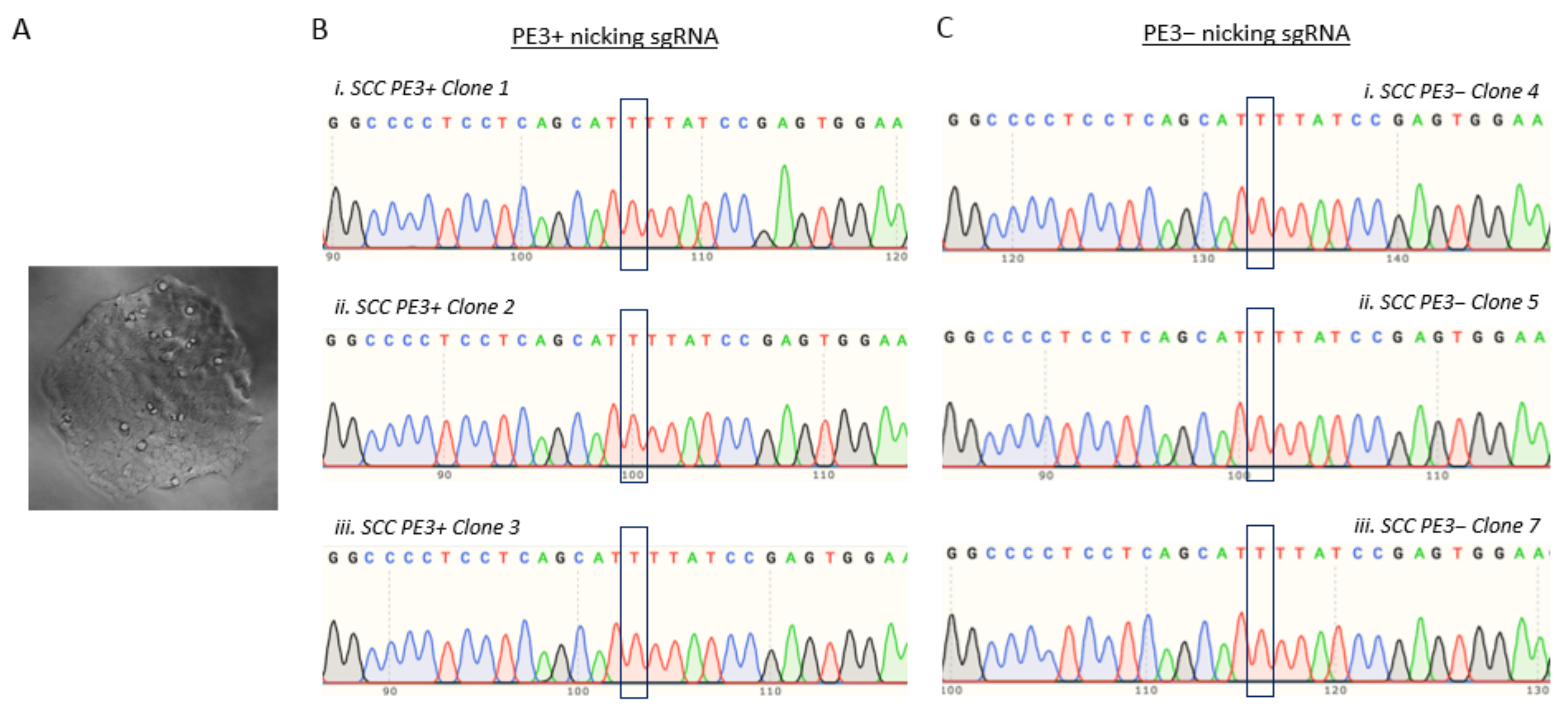
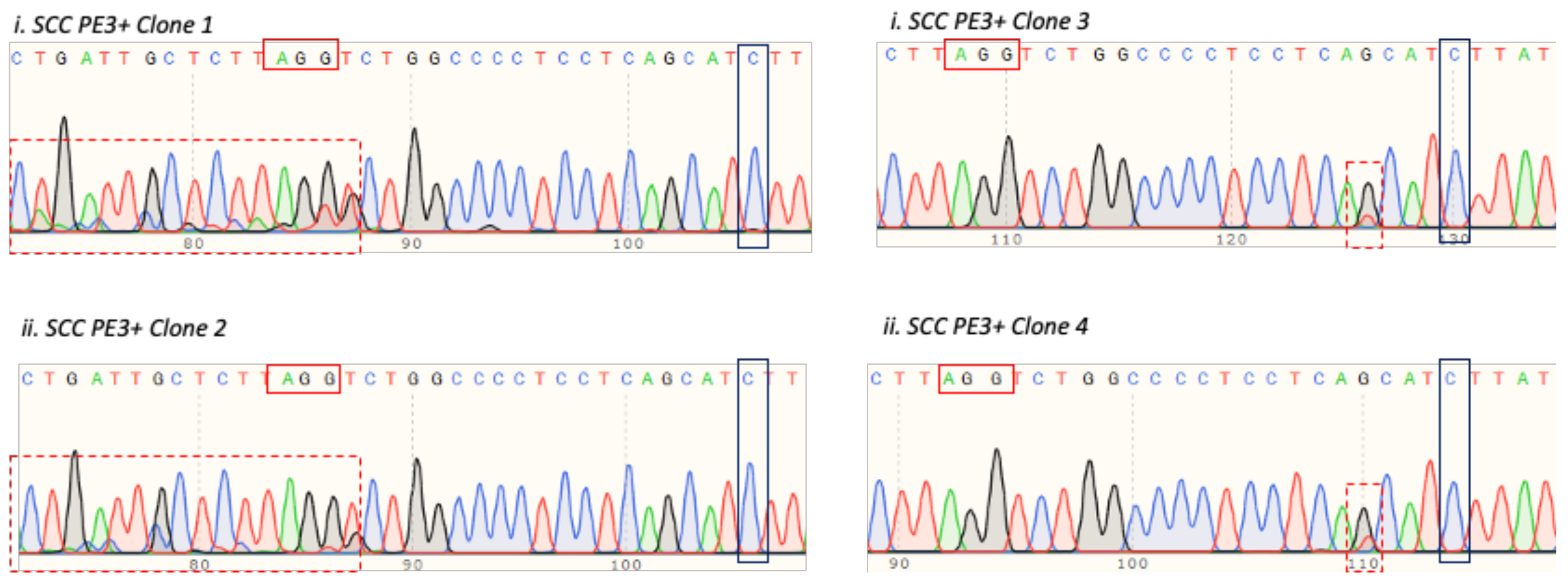
3.5. T47D Cell Electroporation and Assessment of the TP53 Prime Editing Efficiency
3.6. Introducing c.580 C > T Mutation in TP53 Gene in HEK293T Cells
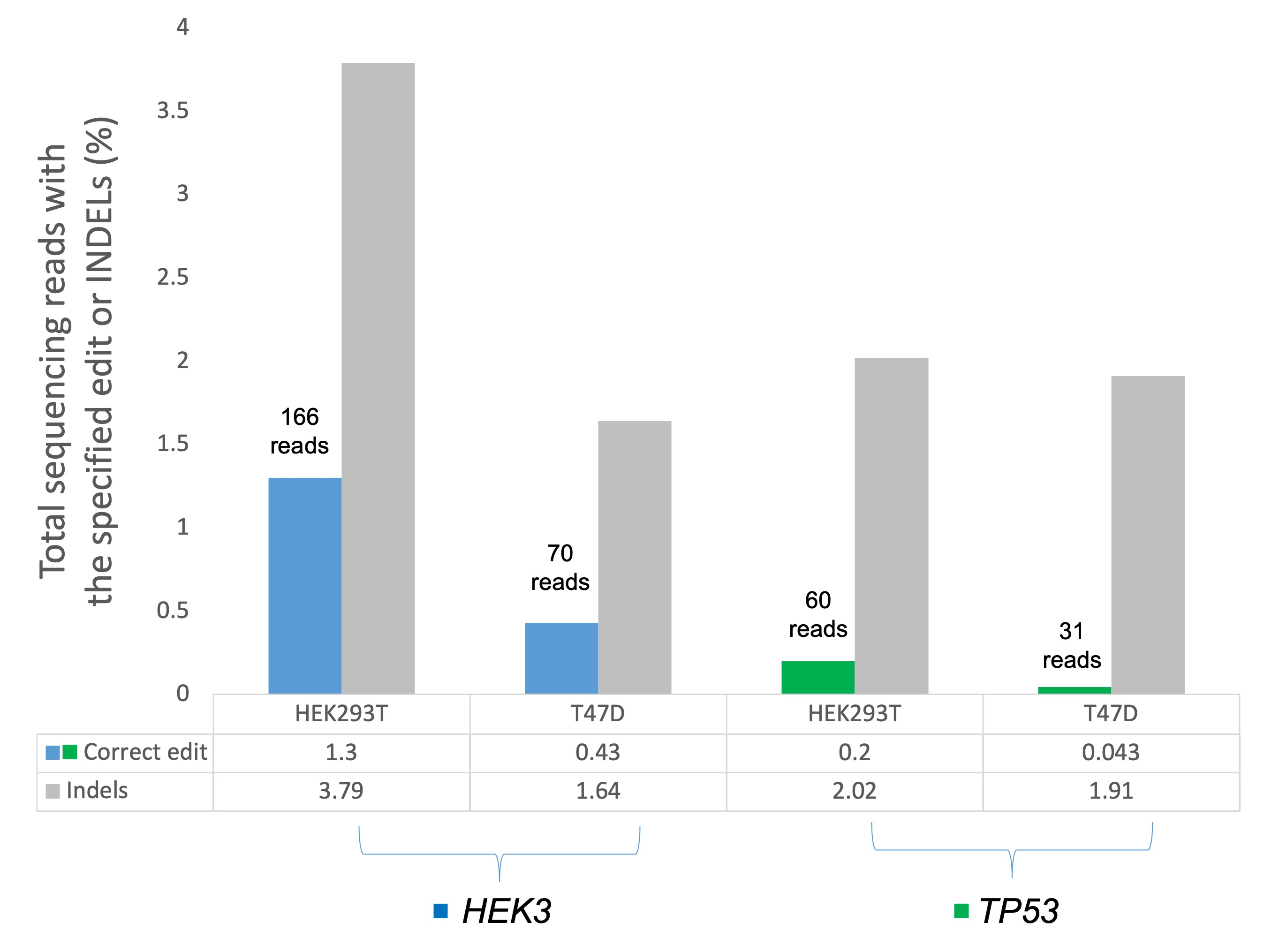
3.7. Assessing TP53 and HEK3 Primer Editing Using Amplicon Target Sequencing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primer 2019, 5, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef] [PubMed]
- Peart, O. Metastatic Breast Cancer. Radiol. Technol. 2017, 88, 519M–539M. [Google Scholar]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Zardavas, D.; Irrthum, A.; Swanton, C.; Piccart, M. Clinical management of breast cancer heterogeneity. Nat. Rev. Clin. Oncol. 2015, 12, 381–394. [Google Scholar] [CrossRef]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.; Lee, C.D.; Guye, M.L.; Van Sciver, R.E.; Lee, M.P.; Lafever, A.C.; Pang, A.; Tang-Tan, A.M.; Winston, J.S.; Samli, B.; et al. Unmet Clinical Need: Developing Prognostic Biomarkers and Precision Medicine to Forecast Early Tumor Relapse, Detect Chemo-Resistance and Improve Overall Survival in High-Risk Breast Cancer. Ann. Breast Cancer Ther. 2020, 4, 48–57. [Google Scholar]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [Green Version]
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183, 1436–1456.e31. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [Green Version]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Olivier, M.; Langer, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bi, I.; et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Silwal-Pandit, L.; Vollan, H.K.M.; Chin, S.-F.; Rueda, O.M.; McKinney, S.; Osako, T.; Quigley, D.A.; Kristensen, V.N.; Aparicio, S.; Børresen-Dale, A.-L.; et al. TP53 Mutation Spectrum in Breast Cancer Is Subtype Specific and Has Distinct Prognostic Relevance. Clin. Cancer Res. 2014, 20, 3569–3580. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Miller, L.D.; Smeds, J.; George, J.; Vega, V.B.; Vergara, L.; Ploner, A.; Pawitan, Y.; Hall, P.; Klaar, S.; Liu, E.T.; et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. USA 2005, 102, 13550–13555. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Melino, G. Context is everything: Extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov. 2020, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E.A. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S. The Heroes of CRISPR. Cell 2016, 164, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.A. CRISPR-Cas9: A fascinating journey from bacterial immune system to human gene editing. Prog. Mol. Biol. Transl. Sci. 2021, 178, 63–83. [Google Scholar] [PubMed]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Brocken, D.J.W.; Tark-Dame, M.; Dame, R.T. dCas9: A Versatile Tool for Epigenome Editing. Curr. Issues Mol. Biol. 2018, 26, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR—Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Scholefield, J.; Harrison, P.T. Prime editing—An update on the field. Gene Ther. 2021, 28, 396–401. [Google Scholar] [CrossRef]
- Jiang, Y.-Y.; Chai, Y.-P.; Lu, M.-H.; Han, X.-L.; Lin, Q.; Zhang, Y.; Zhang, Q.; Zhou, Y.; Wang, X.-C.; Gao, C.; et al. Prime editing efficiently generates W542L and S621I double mutations in two ALS genes in maize. Genome Biol. 2020, 21, 257. [Google Scholar] [CrossRef]
- Sürün, D.; Schneider, A.; Mircetic, J.; Neumann, K.; Lansing, F.; Paszkowski-Rogacz, M.; Hänchen, V.; Lee-Kirsch, M.A.; Buchholz, F. Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors. Genes 2020, 11, 511. [Google Scholar] [CrossRef]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.M.; van Boxtel, R.; van der Doef, H.P.J.; van der Laan, L.J.W.; Verstegen, M.M.A.; van Hasselt, P.M.; et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020, 11, 5352. [Google Scholar] [CrossRef]
- Gao, P.; Lyu, Q.; Ghanam, A.R.; Lazzarotto, C.R.; Newby, G.A.; Zhang, W.; Choi, M.; Slivano, O.J.; Holden, K.; Walker, J.A.; et al. Prime editing in mice reveals the essentiality of a single base in driving tissue-specific gene expression. Genome Biol. 2021, 22, 83. [Google Scholar] [CrossRef]
- Kim, H.K.; Yu, G.; Park, J.; Min, S.; Lee, S.; Yoon, S.; Kim, H.H. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 2021, 39, 198–206. [Google Scholar] [CrossRef]
- Rousseau, J.; Mbakam, C.H.; Guyon, A.; Tremblay, G.; Begin, F.G.; Tremblay, J.P. Specific mutations in genes responsible for Alzheimer and for Duchenne Muscular Dystrophy introduced by Base editing and PRIME editing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Geurts, M.H.; de Poel, E.; Pleguezuelos-Manzano, C.; Oka, R.; Carrillo, L.; Andersson-Rolf, A.; Boretto, M.; Brunsveld, J.E.; van Boxtel, R.; Beekman, J.M.; et al. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Syafruddin, S.E.; Rodrigues, P.; Vojtasova, E.; Patel, S.A.; Zaini, M.N.; Burge, J.; Warren, A.Y.; Stewart, G.D.; Eisen, T.; Bihary, D.; et al. A KLF6-driven transcriptional network links lipid homeostasis and tumour growth in renal carcinoma. Nat. Commun. 2019, 10, 1152. [Google Scholar] [CrossRef] [Green Version]
- Koike-Yusa, H.; Li, Y.; Tan, E.-P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267–273. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Geurts, M.H.; de Poel, E.; Amatngalim, G.D.; Oka, R.; Meijers, F.M.; Kruisselbrink, E.; van Mourik, P.; Berkers, G.; de Winter-de Groot, K.M.; Michel, S.; et al. CRISPR-Based Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell 2020, 26, 503–510.e7. [Google Scholar] [CrossRef]
- Schene, I.F.; Joore, I.P.; Baijens, J.H.L.; Stevelink, R.; Kok, G.; Shehata, S.; Ilcken, E.F.; Nieuwenhuis, E.C.M.; Bolhuis, D.P.; van Rees, R.C.M.; et al. Mutation-specific reporter for optimization and enrichment of prime editing. Nat. Commun. 2022, 13, 1028. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zong, Y.; Xue, C.; Wang, S.; Jin, S.; Zhu, Z.; Wang, Y.; Anzalone, A.V.; Raguram, A.; Doman, J.L.; et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 2020, 38, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Jeong, T.Y.; Shin, S.K.; Yoon, D.E.; Lim, S.-Y.; Kim, S.P.; Choi, J.; Lee, H.; Hong, J.-I.; Ahn, J.; et al. Targeted mutagenesis in mouse cells and embryos using an enhanced prime editor. Genome Biol. 2021, 22, 170. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Liang, S.-Q.; Zheng, C.; Mintzer, E.; Zhao, Y.G.; Ponnienselvan, K.; Mir, A.; Sontheimer, E.J.; Gao, G.; Flotte, T.R.; et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021, 12, 2121. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.Y.; Grünewald, J.; Szalay, R.; Shih, J.; Anzalone, A.V.; Lam, K.C.; Shen, M.W.; Petri, K.; Liu, D.R.; Joung, J.K.; et al. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 2021, 12, 1034. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.D.; Chen, J.S.; Shen, J.; Chen, S. A web tool for the design of prime-editing guide RNAs. Nat. Biomed. Eng. 2021, 5, 190–194. [Google Scholar] [CrossRef]
- Hwang, G.-H.; Jeong, Y.K.; Habib, O.; Hong, S.-A.; Lim, K.; Kim, J.-S.; Bae, S. PE-Designer and PE-Analyzer: Web-based design and analysis tools for CRISPR prime editing. Nucleic Acids Res. 2021, 49, W499–W504. [Google Scholar] [CrossRef]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E.; et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abuhamad, A.Y.; Mohamad Zamberi, N.N.; Sheen, L.; Naes, S.M.; Mohd Yusuf, S.N.H.; Ahmad Tajudin, A.; Mohtar, M.A.; Amir Hamzah, A.S.; Syafruddin, S.E. Reverting TP53 Mutation in Breast Cancer Cells: Prime Editing Workflow and Technical Considerations. Cells 2022, 11, 1612. https://doi.org/10.3390/cells11101612
Abuhamad AY, Mohamad Zamberi NN, Sheen L, Naes SM, Mohd Yusuf SNH, Ahmad Tajudin A, Mohtar MA, Amir Hamzah AS, Syafruddin SE. Reverting TP53 Mutation in Breast Cancer Cells: Prime Editing Workflow and Technical Considerations. Cells. 2022; 11(10):1612. https://doi.org/10.3390/cells11101612
Chicago/Turabian StyleAbuhamad, Asmaa Y., Nurul Nadia Mohamad Zamberi, Ling Sheen, Safaa M. Naes, Siti Nur Hasanah Mohd Yusuf, Asilah Ahmad Tajudin, M. Aiman Mohtar, Amir Syahir Amir Hamzah, and Saiful Effendi Syafruddin. 2022. "Reverting TP53 Mutation in Breast Cancer Cells: Prime Editing Workflow and Technical Considerations" Cells 11, no. 10: 1612. https://doi.org/10.3390/cells11101612