
Effects of High Dietary Carbohydrate and Lipid Intake on the Lifespan of C. elegans

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Impacts of Carbohydrate Metabolism on Lifespan
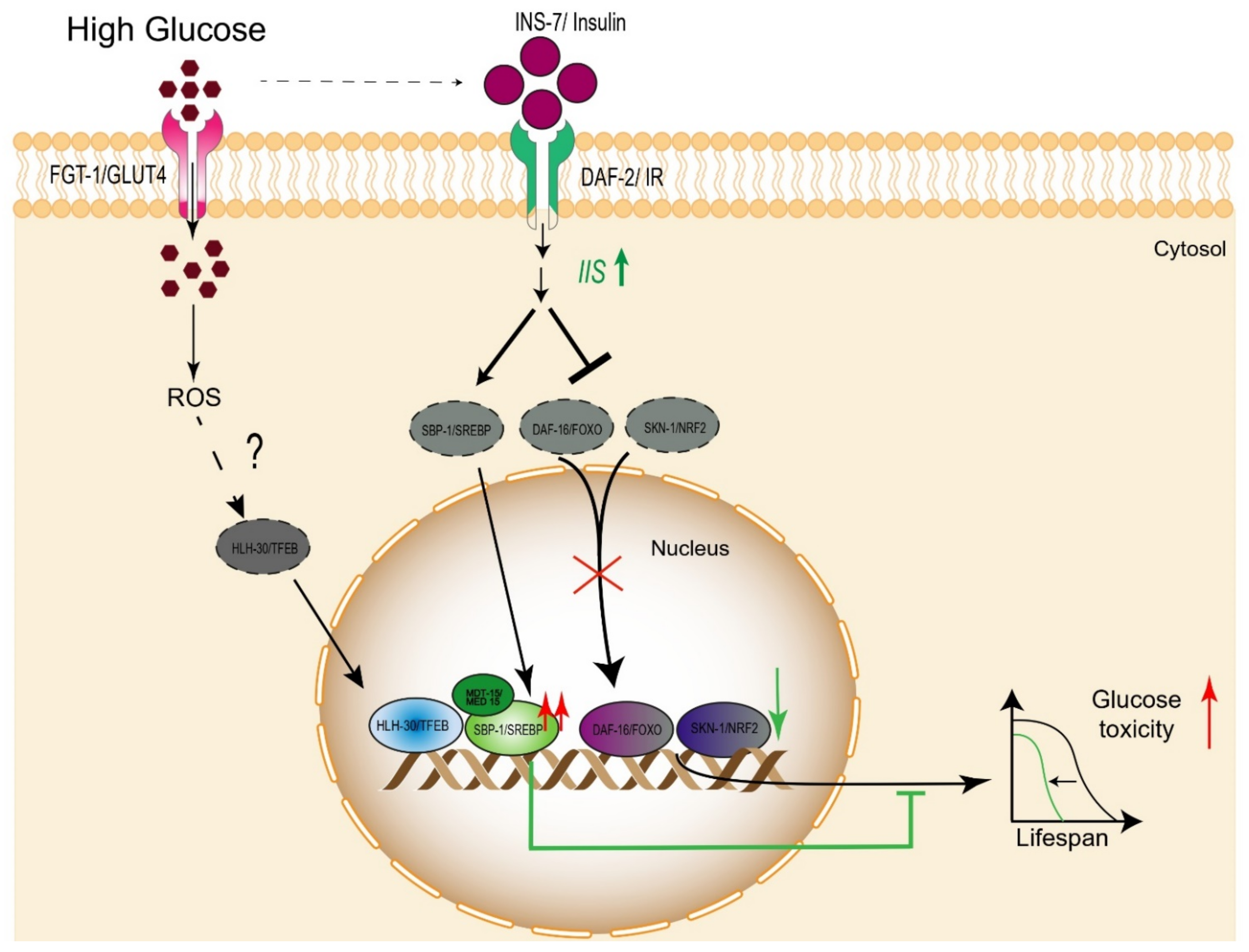
2.1. Glucose Breakdown
2.2. Lifespan-Related Pathways Affected by High Glucose
2.2.1. Daf-16/FOXO

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C. elegans Gene | Human Homologue | Gene Description | Effect on Lifespan | References | |
|---|---|---|---|---|---|
| Gain of Function | Loss of Function | ||||
| daf-2 | INSR | Insulin receptor | ND | Increase a,b | [55,56] |
| fgt-1 | SLC2A4 | Solute Carrier Family 2 Member 4 | ND | Increase a | [27] |
| daf-16 | FOXO | Forkhead Box O1 | Increase a | Decrease a | [53,57] |
| skn-1 | Nrf2 | Nuclear factor, erythroid 2-like 2 | Increase a,b | Decrease a | [58,59,60,61] |
| hlh-30 | TFEB | Transcription factor EB | Increase a | Decrease a | [62] |
| sbp-1 | SREBP | Sterol Regulatory Element Binding Transcription Factor | Increase a,b | Decrease a | [63] |
| mdt-15 | MED15 | Mediator Complex Subunit 15 | Increase a | ND | [64] |
| nhr-49 | PPARα | Peroxisome Proliferator Activated Receptor Alpha | Increase a | Decrease a | [65] |
| nhr-80 | HNF4A | Hepatocyte Nuclear Factor 4 Alpha | Increase a | Decrease a | [66,67] |
| xbp-1 | XBP1 | X-Box-Binding Protein 1 | Increase a | Decrease a | [68] |
2.2.2. SKN-1/Nrf
2.2.3. HLH-30/TFEB
2.2.4. SBP-1/SREBP
2.3. Fructose
2.4. Trehalose
3. Effects of Lipids on Lifespan
3.1. PUFAs
3.2. MUFAS
4. Pharmacologic Strategies That Extend the Organismal Lifespan
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.D.; Garber, A.J.; Farmer, J.A. Role of insulin signaling in maintaining energy homeostasis. Endocr. Pract. 2008, 14, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Organismal carbohydrate and lipid homeostasis. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Minehira, K.; Vega, N.; Vidal, H.; Acheson, K.; Tappy, L. Effect of carbohydrate overfeeding on whole body macronutrient metabolism and expression of lipogenic enzymes in adipose tissue of lean and overweight humans. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 1291–1298. [Google Scholar] [CrossRef] [Green Version]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef] [PubMed]
- Parry, S.A.; Hodson, L. Influence of dietary macronutrients on liver fat accumulation and metabolism. J. Investig. Med. 2017, 65, 1102–1115. [Google Scholar] [CrossRef]
- Markaki, M.; Tavernarakis, N. Modeling human diseases in Caenorhabditis elegans. Biotechnol. J. 2010, 5, 1261–1276. [Google Scholar] [CrossRef]
- Bouyanfif, A.; Jayarathne, S.; Koboziev, I.; Moustaid-Moussa, N. The Nematode Caenorhabditis elegans as a Model Organism to Study Metabolic Effects of omega-3 Polyunsaturated Fatty Acids in Obesity. Adv. Nutr. 2019, 10, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, J.; Ichikawa, K.; Shoura, M.J.; Artiles, K.L.; Gabdank, I.; Wahba, L.; Smith, C.L.; Edgley, M.L.; Rougvie, A.E.; Fire, A.Z.; et al. Recompleting the Caenorhabditis elegans genome. Genome Res. 2019, 29, 1009–1022. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, F.; Zhou, T.; Wang, G.; Li, Z. Caenorhabditis elegans as a Useful Model for Studying Aging Mutations. Front. Endocrinol. 2020, 11, 554994. [Google Scholar] [CrossRef]
- Hashmi, S.; Wang, Y.; Parhar, R.S.; Collison, K.S.; Conca, W.; Al-Mohanna, F.; Gaugler, R. A C. elegans model to study human metabolic regulation. Nutr. Metab. 2013, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Corsi, A.K.; Wightman, B.; Chalfie, M. A Transparent Window into Biology: A Primer on Caenorhabditis elegans. Genetics 2015, 200, 387–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.L.; Ristow, M. Lipid and Carbohydrate Metabolism in Caenorhabditis elegans. Genetics 2017, 207, 413–446. [Google Scholar] [PubMed]
- Schlotterer, A.; Kukudov, G.; Bozorgmehr, F.; Hutter, H.; Du, X.; Oikonomou, D.; Ibrahim, Y.; Pfisterer, F.; Rabbani, N.; Thornalley, P.; et al. C. elegans as model for the study of high glucose- mediated life span reduction. Diabetes 2009, 58, 2450–2456. [Google Scholar] [CrossRef] [Green Version]
- Jequier, E. Carbohydrates as a source of energy. Am. J. Clin. Nutr. 1994, 59 (Suppl. 3), 682S–685S. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Models Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Chawla, S.; Tessarolo Silva, F.; Amaral Medeiros, S.; Mekary, R.A.; Radenkovic, D. The Effect of Low-Fat and Low-Carbohydrate Diets on Weight Loss and Lipid Levels: A Systematic Review and Meta-Analysis. Nutrients 2020, 12, 3774. [Google Scholar] [CrossRef]
- Ingram, D.K.; Roth, G.S. Glycolytic inhibition: An effective strategy for developing calorie restriction mimetics. Geroscience 2021, 43, 1159–1169. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Glucose intolerance and aging. Diabetes Care 1981, 4, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Onken, B.; Kalinava, N.; Driscoll, M. Gluconeogenesis and PEPCK are critical components of healthy aging and dietary restriction life extension. PLoS Genet. 2020, 16, e1008982. [Google Scholar] [CrossRef] [PubMed]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [Green Version]
- Zorzano, A.; Palacin, M.; Guma, A. Mechanisms regulating GLUT4 glucose transporter expression and glucose transport in skeletal muscle. Acta Physiol. 2005, 183, 43–58. [Google Scholar] [CrossRef]
- Kitaoka, S.; Morielli, A.D.; Zhao, F.Q. FGT-1-mediated glucose uptake is defective in insulin/IGF-like signaling mutants in Caenorhabditis elegans. FEBS Open Bio. 2016, 6, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaoka, S.; Morielli, A.D.; Zhao, F.Q. FGT-1 is a mammalian GLUT2-like facilitative glucose transporter in Caenorhabditis elegans whose malfunction induces fat accumulation in intestinal cells. PLoS ONE 2013, 8, e68475. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Williams, B.G.; Koumanov, F.; Wolstenholme, A.J.; Holman, G.D. FGT-1 is the major glucose transporter in C. elegans and is central to aging pathways. Biochem. J. 2013, 456, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, J.M.; Benite-Ribeiro, S.A.; Queiroz, G.; Duarte, J.A. The effect of age on glucose uptake and GLUT1 and GLUT4 expression in rat skeletal muscle. Cell Biochem. Funct. 2012, 30, 191–197. [Google Scholar] [CrossRef]
- Meneilly, G.S.; Elahi, D. Metabolic alterations in middle-aged and elderly lean patients with type 2 diabetes. Diabetes Care 2005, 28, 1498–1499. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.C.; Park, M.C.; Keam, B.; Choi, J.M.; Cho, Y.; Hyun, S.; Park, S.C.; Lee, J. DDS, 4,4’-diaminodiphenylsulfone, extends organismic lifespan. Proc. Natl. Acad. Sci. USA 2010, 107, 19326–19331. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Kingsley, S.; Walker, G.; Mondoux, M.A.; Tissenbaum, H.A. Metabolic shift from glycogen to trehalose promotes lifespan and healthspan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2018, 115, E2791–E2800. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Lee, R.Y.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003, 33, 40–48. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipkiss, A.R. Does chronic glycolysis accelerate aging? Could this explain how dietary restriction works? Ann. N. Y. Acad. Sci. 2006, 1067, 361–368. [Google Scholar] [CrossRef]
- Sami, W.; Ansari, T.; Butt, N.S.; Hamid, M.R.A. Effect of diet on type 2 diabetes mellitus: A review. Int. J. Health Sci. 2017, 11, 65–71. [Google Scholar]
- Everitt, A.V.; Hilmer, S.N.; Brand-Miller, J.C.; Jamieson, H.A.; Truswell, A.S.; Sharma, A.P.; Mason, R.S.; Morris, B.J.; Le Couteur, D.G. Dietary approaches that delay age-related diseases. Clin. Interv. Aging 2006, 1, 11–31. [Google Scholar] [CrossRef]
- Zhu, G.; Yin, F.; Wang, L.; Wei, W.; Jiang, L.; Qin, J. Modeling type 2 diabetes-like hyperglycemia in C. elegans on a microdevice. Integr. Biol. 2016, 8, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Jeong, D.E.; Son, H.G.; Yamaoka, Y.; Kim, H.; Seo, K.; Khan, A.A.; Roh, T.Y.; Moon, D.W.; Lee, Y.; et al. SREBP and MDT-15 protect C. elegans from glucose-induced accelerated aging by preventing accumulation of saturated fat. Genes Dev. 2015, 29, 2490–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaza, G.; Khan, A.K.; Rashid, R.; Muneer, S.; Hasan, S.M.F.; Chen, J. FOXO Transcriptional Factors and Long-Term Living. Oxid. Med. Cell. Longev. 2017, 2017, 3494289. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.J.; Cao, H.; Lu, J.; Wu, C.; Hu, F.Y.; Guo, J.; Zhao, L.; Yang, F.; Zhang, Y.X.; et al. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum. Mol. Genet. 2009, 18, 4897–4904. [Google Scholar] [CrossRef] [Green Version]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [Green Version]
- Anselmi, C.V.; Malovini, A.; Roncarati, R.; Novelli, V.; Villa, F.; Condorelli, G.; Bellazzi, R.; Puca, A.A. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009, 12, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, I.; Komatsu, T.; Hayashi, N.; Kim, S.E.; Kawata, T.; Park, S.; Hayashi, H.; Yamaza, H.; Chiba, T.; Mori, R. The life-extending effect of dietary restriction requires Foxo3 in mice. Aging Cell 2015, 14, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Ma, J.; Gao, X.; Tian, P.; Li, W.; Zhang, L. High glucose induced oxidative stress and apoptosis in cardiac microvascular endothelial cells are regulated by FoxO3a. PLoS ONE 2013, 8, e79739. [Google Scholar] [CrossRef]
- O-Sullivan, I.S.; Zhang, W.; Wasserman, D.H.; Liew, C.W.; Liu, J.; Paik, J.; DePinho, R.A.; Stolz, D.B.; Kahn, C.R.; Schwartz, M.W.; et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat. Commun. 2015, 6, 7079. [Google Scholar] [CrossRef] [Green Version]
- Raju, I.; Kannan, K.; Abraham, E.C. FoxO3a Serves as a Biomarker of Oxidative Stress in Human Lens Epithelial Cells under Conditions of Hyperglycemia. PLoS ONE 2013, 8, e67126. [Google Scholar] [CrossRef] [Green Version]
- Zecic, A.; Braeckman, B.P. DAF-16/FoxO in Caenorhabditis elegans and Its Role in Metabolic Remodeling. Cells 2020, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Yen, K.; Narasimhan, S.D.; Tissenbaum, H.A. DAF-16/Forkhead box O transcription factor: Many paths to a single Fork(head) in the road. Antioxid. Redox. Signal. 2011, 14, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesp, K.; Smant, G.; Kammenga, J.E. Caenorhabditis elegans DAF-16/FOXO transcription factor and its mammalian homologs associate with age-related disease. Exp. Gerontol. 2015, 72, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, T.; Iino, Y.; Tomioka, M. DAF-16/FOXO promotes taste avoidance learning independently of axonal insulin-like signaling. PLoS Genet. 2019, 15, e1008297. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Murphy, C.T.; Kenyon, C. Glucose shortens the life span of C. elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab. 2009, 10, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.K.; Park, S.K. Phosphatidylserine modulates response to oxidative stress through hormesis and increases lifespan via DAF-16 in Caenorhabditis elegans. Biogerontology 2020, 21, 231–244. [Google Scholar] [CrossRef]
- Hibshman, J.D.; Doan, A.E.; Moore, B.T.; Kaplan, R.E.; Hung, A.; Webster, A.K.; Bhatt, D.P.; Chitrakar, R.; Hirschey, M.D.; Baugh, L.R. daf-16/FoxO promotes gluconeogenesis and trehalose synthesis during starvation to support survival. Elife 2017, 6, 1–29. [Google Scholar] [CrossRef]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.D.; Tissenbaum, H.A.; Liu, Y.; Ruvkun, G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 1997, 277, 942–946. [Google Scholar] [CrossRef]
- Bansal, A.; Kwon, E.S.; Conte, D., Jr.; Liu, H.; Gilchrist, M.J.; MacNeil, L.T.; Tissenbaum, H.A. Transcriptional regulation of Caenorhabditis elegans FOXO/DAF-16 modulates lifespan. Longev. Healthspan 2014, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.N.; Wason, E.; Edrey, Y.H.; Kristan, D.M.; Nevo, E.; Buffenstein, R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc. Natl. Acad. Sci. USA 2015, 112, 3722–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Dai, Y.; Tang, H.; Pang, S. SKN-1 Is a Negative Regulator of DAF-16 and Somatic Stress Resistance in Caenorhabditis elegans. G3 2020, 10, 1707–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Arriola, E.; Cardenas-Rodriguez, N.; Coballase-Urrutia, E.; Pedraza-Chaverri, J.; Carmona-Aparicio, L.; Ortega-Cuellar, D. Caenorhabditis elegans: A useful model for studying metabolic disorders in which oxidative stress is a contributing factor. Oxid. Med. Cell. Longev. 2014, 2014, 705253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Chen, Y.; Chenzhao, C.; Fu, S.; Xu, Q.; Zhao, J. Glucose negatively affects Nrf2/SKN-1-mediated innate immunity in C. elegans. Aging 2018, 10, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, E.J.; Ruvkun, G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 2013, 15, 668–676. [Google Scholar] [CrossRef]
- Kobayashi, M.; Fujii, N.; Narita, T.; Higami, Y. SREBP-1c-Dependent Metabolic Remodeling of White Adipose Tissue by Caloric Restriction. Int. J. Mol. Sci. 2018, 19, 3335. [Google Scholar] [CrossRef] [Green Version]
- Taubert, S.; Hansen, M.; Van Gilst, M.R.; Cooper, S.B.; Yamamoto, K.R. The Mediator subunit MDT-15 confers metabolic adaptation to ingested material. PLoS Genet. 2008, 4, e1000021. [Google Scholar] [CrossRef] [Green Version]
- Argmann, C.; Dobrin, R.; Heikkinen, S.; Auburtin, A.; Pouilly, L.; Cock, T.A.; Koutnikova, H.; Zhu, J.; Schadt, E.E.; Auwerx, J. Ppargamma2 is a key driver of longevity in the mouse. PLoS Genet. 2009, 5, e1000752. [Google Scholar] [CrossRef]
- Qi, W.; Gutierrez, G.E.; Gao, X.; Dixon, H.; McDonough, J.A.; Marini, A.M.; Fisher, A.L. The omega-3 fatty acid alpha-linolenic acid extends Caenorhabditis elegans lifespan via NHR-49/PPARalpha and oxidation to oxylipins. Aging Cell 2017, 16, 1125–1135. [Google Scholar] [CrossRef] [Green Version]
- Goudeau, J.; Bellemin, S.; Toselli-Mollereau, E.; Shamalnasab, M.; Chen, Y.; Aguilaniu, H. Fatty acid desaturation links germ cell loss to longevity through NHR-80/HNF4 in C. elegans. PLoS Biol. 2011, 9, e1000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imanikia, S.; Sheng, M.; Castro, C.; Griffin, J.L.; Taylor, R.C. XBP-1 Remodels Lipid Metabolism to Extend Longevity. Cell Rep. 2019, 28, 581–589.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox. Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef] [PubMed]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137–4142. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med. 2015, 88, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wang, F.; Zhang, L.; Cao, Y.; Liu, W.; Hao, J.; Liu, Q.; Duan, H. Modulation of Nrf2 expression alters high glucose-induced oxidative stress and antioxidant gene expression in mouse mesangial cells. Cell. Signal. 2011, 23, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Nhan, J.D.; Turner, C.D.; Anderson, S.M.; Yen, C.A.; Dalton, H.M.; Cheesman, H.K.; Ruter, D.L.; Uma Naresh, N.; Haynes, C.M.; Soukas, A.A.; et al. Redirection of SKN-1 abates the negative metabolic outcomes of a perceived pathogen infection. Proc. Natl. Acad. Sci. USA 2019, 116, 22322–22330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, S.; Lynn, D.A.; Lo, J.Y.; Paek, J.; Curran, S.P. SKN-1 and Nrf2 couples proline catabolism with lipid metabolism during nutrient deprivation. Nat. Commun. 2014, 5, 5048. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Robida-Stubbs, S.; Tullet, J.M.; Rual, J.F.; Vidal, M.; Blackwell, T.K. RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet. 2010, 6, e1001048. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.H.L.; Vong, C.T.; Kwan, Y.W.; Lee, S.M.; Hoi, M.P.M. Lysosomal Ca(2+) Signaling Regulates High Glucose-Mediated Interleukin-1beta Secretion via Transcription Factor EB in Human Monocytic Cells. Front. Immunol. 2017, 8, 1161. [Google Scholar] [CrossRef] [Green Version]
- Peiro, C.; Lorenzo, O.; Carraro, R.; Sanchez-Ferrer, C.F. IL-1beta Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus. Front. Pharmacol. 2017, 8, 363. [Google Scholar] [CrossRef] [Green Version]
- La Spina, M.; Contreras, P.S.; Rissone, A.; Meena, N.K.; Jeong, E.; Martina, J.A. MiT/TFE Family of Transcription Factors: An Evolutionary Perspective. Front. Cell Dev. Biol. 2020, 8, 609683. [Google Scholar] [CrossRef]
- Ferrari, L.; Martens, S. Out of Phase: How IPMK Inhibits TFEB. Dev. Cell 2020, 55, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Visvikis, O.; Ihuegbu, N.; Labed, S.A.; Luhachack, L.G.; Alves, A.F.; Wollenberg, A.C.; Stuart, L.M.; Stormo, G.D.; Irazoqui, J.E. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 2014, 40, 896–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Diab, H.I.; Brady, O.A.; Puertollano, R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016, 35, 479–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Akayama, S.; Yoshimori, T. Autophagy-independent function of lipidated LC3 essential for TFEB activation during the lysosomal damage responses. Autophagy 2021, 17, 581–583. [Google Scholar] [CrossRef]
- Sharma, M.; Pandey, R.; Saluja, D. ROS is the major player in regulating altered autophagy and lifespan in sin-3 mutants of C. elegans. Autophagy 2018, 14, 1239–1255. [Google Scholar] [CrossRef] [Green Version]
- Franco-Juarez, B.; Mejia-Martinez, F.; Moreno-Arriola, E.; Hernandez-Vazquez, A.; Gomez-Manzo, S.; Marcial-Quino, J.; Arreguin-Espinosa, R.; Velazquez-Arellano, A.; Ortega-Cuellar, D. A high glucose diet induces autophagy in a HLH-30/TFEB-dependent manner and impairs the normal lifespan of C. elegans. Aging 2018, 10, 2657–2667. [Google Scholar] [CrossRef]
- Mellor, K.M.; Bell, J.R.; Young, M.J.; Ritchie, R.H.; Delbridge, L.M. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J. Mol. Cell. Cardiol. 2011, 50, 1035–1043. [Google Scholar] [CrossRef]
- Zeng, W.; Xiao, T.; Cai, A.; Cai, W.; Liu, H.; Liu, J.; Li, J.; Tan, M.; Xie, L.; Liu, Y.; et al. Inhibiting ROS-TFEB-Dependent Autophagy Enhances Salidroside-Induced Apoptosis in Human Chondrosarcoma Cells. Cell. Physiol. Biochem. 2017, 43, 1487–1502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeisser, K.; Parker, J.A. Pleiotropic Effects of mTOR and Autophagy During Development and Aging. Front. Cell Dev. Biol. 2019, 7, 192. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, T.K.; Sewell, A.K.; Wu, Z.; Han, M. TOR Signaling in Caenorhabditis elegans Development, Metabolism, and Aging. Genetics 2019, 213, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolio, R.; Napoletano, F.; Mano, M.; Maurer-Stroh, S.; Fantuz, M.; Zannini, A.; Bicciato, S.; Sorrentino, G.; Del Sal, G. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 2019, 10, 1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Du, W.; Liu, Y.; Cheng, M.; Wang, X.; Zhang, C.; Lv, X.; Li, F.; Zhao, S.; Hao, J. Prolonged high-glucose exposure decreased SREBP-1/FASN/ACC in Schwann cells of diabetic mice via blocking PI3K/Akt pathway. J. Cell. Biochem. 2019, 120, 5777–5789. [Google Scholar] [CrossRef]
- Nomura, T.; Horikawa, M.; Shimamura, S.; Hashimoto, T.; Sakamoto, K. Fat accumulation in Caenorhabditis elegans is mediated by SREBP homolog SBP-1. Genes Nutr. 2010, 5, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Schroeder, E.A.; Silva-Garcia, C.G.; Hebestreit, K.; Mair, W.B.; Brunet, A. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 2017, 544, 185–190. [Google Scholar] [CrossRef]
- Mejia-Martinez, F.; Franco-Juarez, B.; Moreno-Arriola, E.; Hernandez-Vazquez, A.; Martinez-Avila, M.; Gomez-Manzo, S.; Marcial-Quino, J.; Carvajal, K.; Velazquez-Arellano, A.; Ortega-Cuellar, D. The MXL-3/SBP-1 Axis Is Responsible for Glucose-Dependent Fat Accumulation in C. elegans. Genes 2017, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Kwon, S.; Ham, S.; Lee, D.; Park, H.H.; Yamaoka, Y.; Jeong, D.E.; Artan, M.; Altintas, O.; Park, S.; et al. Caenorhabditis elegans Lipin 1 moderates the lifespan-shortening effects of dietary glucose by maintaining omega-6 polyunsaturated fatty acids. Aging Cell 2020, 19, e13150. [Google Scholar] [CrossRef]
- Shi, Y.N.; Liu, Y.J.; Xie, Z.; Zhang, W.J. Fructose and metabolic diseases: Too much to be good. Chin. Med. J. 2021, 134, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Loza-Medrano, S.S.; Baiza-Gutman, L.A.; Ibanez-Hernandez, M.A.; Cruz-Lopez, M.; Diaz-Flores, M. Molecular alterations induced by fructose and its impact on metabolic diseases. Rev. Med. Inst. Mex. Seguro Soc. 2019, 56, 491–504. [Google Scholar] [PubMed]
- Moreira, P.I. High-sugar diets, type 2 diabetes and Alzheimer’s disease. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 440–445. [Google Scholar] [CrossRef]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef] [Green Version]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and localization of the principal fructose-metabolizing enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappy, L.; Rosset, R. Health outcomes of a high fructose intake: The importance of physical activity. J. Physiol. 2019, 597, 3561–3571. [Google Scholar] [CrossRef] [Green Version]
- Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef] [Green Version]
- Thirunavukkarasu, V.; Anitha Nandhini, A.T.; Anuradha, C.V. Lipoic acid attenuates hypertension and improves insulin sensitivity, kallikrein activity and nitrite levels in high fructose-fed rats. J. Comp. Physiol. B 2004, 174, 587–592. [Google Scholar] [CrossRef]
- Di Luccia, B.; Crescenzo, R.; Mazzoli, A.; Cigliano, L.; Venditti, P.; Walser, J.C.; Widmer, A.; Baccigalupi, L.; Ricca, E.; Iossa, S. Rescue of Fructose-Induced Metabolic Syndrome by Antibiotics or Faecal Transplantation in a Rat Model of Obesity. PLoS ONE 2015, 10, e0134893. [Google Scholar]
- Ruff, J.S.; Suchy, A.K.; Hugentobler, S.A.; Sosa, M.M.; Schwartz, B.L.; Morrison, L.C.; Gieng, S.H.; Shigenaga, M.K.; Potts, W.K. Human-relevant levels of added sugar consumption increase female mortality and lower male fitness in mice. Nat. Commun. 2013, 4, 2245. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Gao, C.; Wang, M.; Tran, P.; Mai, N.; Finley, J.W.; Heymsfield, S.B.; Greenway, F.L.; Li, Z.; Heber, D.; et al. Lower Doses of Fructose Extend Lifespan in Caenorhabditis elegans. J. Diet. Suppl. 2017, 14, 264–277. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, L.; Zhang, L.; Wang, W.; Wei, S.; Wang, J.; Che, H.; Zhang, Y. Effects of excess sugars and lipids on the growth and development of Caenorhabditis elegans. Genes Nutr. 2020, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Lodha, D.; Rajasekaran, S.; Jayavelu, T.; Subramaniam, J.R. Detrimental effects of fructose on mitochondria in mouse motor neurons and on C. elegans healthspan. Nutr. Neurosci. 2020, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Elbein, A.D.; Pan, Y.T.; Pastuszak, I.; Carroll, D. New insights on trehalose: A multifunctional molecule. Glycobiology 2003, 13, 17R–27R. [Google Scholar] [CrossRef]
- Burke, M. Carbohydrate Intolerance and Disaccharidase Measurement-A Mini-Review. Clin. Biochem. Rev. 2019, 40, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Yaribeygi, A.; Sathyapalan, T.; Sahebkar, A. Molecular mechanisms of trehalose in modulating glucose homeostasis in diabetes. Diabetes Metab. Syndr. 2019, 13, 2214–2218. [Google Scholar] [CrossRef]
- Avonce, N.; Mendoza-Vargas, A.; Morett, E.; Iturriaga, G. Insights on the evolution of trehalose biosynthesis. BMC Evol. Biol. 2006, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Pellerone, F.I.; Archer, S.K.; Behm, C.A.; Grant, W.N.; Lacey, M.J.; Somerville, A.C. Trehalose metabolism genes in Caenorhabditis elegans and filarial nematodes. Int. J. Parasitol. 2003, 33, 1195–1206. [Google Scholar] [CrossRef]
- Kormish, J.D.; McGhee, J.D. The C. elegans lethal gut-obstructed gob-1 gene is trehalose-6-phosphate phosphatase. Dev. Biol. 2005, 287, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Rasulova, M.; Zecic, A.; Monje Moreno, J.M.; Vandemeulebroucke, L.; Dhondt, I.; Braeckman, B.P. Elevated Trehalose Levels in C. elegans daf-2 Mutants Increase Stress Resistance, Not Lifespan. Metabolites 2021, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Tanaka, M.; Honda, S. Trehalose extends longevity in the nematode Caenorhabditis elegans. Aging Cell 2010, 9, 558–569. [Google Scholar] [CrossRef]
- Mutlu, A.S.; Duffy, J.; Wang, M.C. Lipid metabolism and lipid signals in aging and longevity. Dev. Cell 2021, 56, 1394–1407. [Google Scholar] [CrossRef] [PubMed]
- Skowronska-Krawczyk, D.; Budin, I. Aging membranes: Unexplored functions for lipids in the lifespan of the central nervous system. Exp. Gerontol. 2020, 131, 110817. [Google Scholar] [CrossRef]
- Ishikawa, M.; Maekawa, K.; Saito, K.; Senoo, Y.; Urata, M.; Murayama, M.; Tajima, Y.; Kumagai, Y.; Saito, Y. Plasma and serum lipidomics of healthy white adults shows characteristic profiles by subjects’ gender and age. PLoS ONE 2014, 9, e91806. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.W.; Buffenstein, R.; Hulbert, A.J. Membrane phospholipid composition may contribute to exceptional longevity of the naked mole-rat (Heterocephalus glaber): A comparative study using shotgun lipidomics. Exp. Gerontol. 2007, 42, 1053–1062. [Google Scholar] [CrossRef]
- Eyster, K.M. The membrane and lipids as integral participants in signal transduction: Lipid signal transduction for the non-lipid biochemist. Adv. Physiol. Educ. 2007, 31, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, K.W. Advances in Understanding of the Role of Lipid Metabolism in Aging. Cells 2021, 10, 880. [Google Scholar] [CrossRef] [PubMed]
- Weiser, M.J.; Butt, C.M.; Mohajeri, M.H. Docosahexaenoic Acid and Cognition throughout the Lifespan. Nutrients 2016, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Shan, K.; Chen, H.; Chen, Y.Q. n-3 Polyunsaturated Fatty Acids and their Role in Cancer Chemoprevention. Curr. Pharmacol. Rep. 2015, 1, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhamid, A.S.; Martin, N.; Bridges, C.; Brainard, J.S.; Wang, X.; Brown, T.J.; Hanson, S.; Jimoh, O.F.; Ajabnoor, S.M.; Deane, K.H.; et al. Polyunsaturated fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2018, 7, CD012345. [Google Scholar]
- Lee, J.M.; Lee, H.; Kang, S.; Park, W.J. Fatty Acid Desaturases, Polyunsaturated Fatty Acid Regulation, and Biotechnological Advances. Nutrients 2016, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Ren, H.; Yao, X.; Shi, Z.; Liang, F.; Kang, J.X.; Wan, J.B.; Pei, Z.; Su, K.P.; Su, H. Enriched Brain Omega-3 Polyunsaturated Fatty Acids Confer Neuroprotection against Microinfarction. EBioMedicine 2018, 32, 50–61. [Google Scholar] [CrossRef]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef]
- Rangel-Huerta, O.D.; Gil, A. Effect of omega-3 fatty acids on cognition: An updated systematic review of randomized clinical trials. Nutr. Rev. 2018, 76, 1–20. [Google Scholar] [CrossRef]
- Valencak, T.G.; Ruf, T. N-3 polyunsaturated fatty acids impair lifespan but have no role for metabolism. Aging Cell 2007, 6, 15–25. [Google Scholar] [CrossRef]
- Van Gilst, M.R.; Hadjivassiliou, H.; Yamamoto, K.R. A Caenorhabditis elegans nutrient response system partially dependent on nuclear receptor NHR-49. Proc. Natl. Acad. Sci. USA 2005, 102, 13496–13501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, E.J.; Kuballa, P.; Xavier, R.; Ruvkun, G. omega-6 Polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 2013, 27, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.L.; Browse, J. Dietary manipulation implicates lipid signaling in the regulation of germ cell maintenance in C. elegans. Dev. Biol. 2006, 292, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cinar, R.; Xiong, K.; Godlewski, G.; Jourdan, T.; Lin, Y.; Ntambi, J.M.; Kunos, G. Monounsaturated fatty acids generated via stearoyl CoA desaturase-1 are endogenous inhibitors of fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2013, 110, 18832–18837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, C.; Krumsiek, J.; Lehrbach, N.J.; Murfitt, S.A.; Miska, E.A.; Griffin, J.L. A study of Caenorhabditis elegans DAF-2 mutants by metabolomics and differential correlation networks. Mol. Biosyst. 2013, 9, 1632–1642. [Google Scholar] [CrossRef]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef] [Green Version]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Arriola, E.; El Hafidi, M.; Ortega-Cuellar, D.; Carvajal, K. AMP-Activated Protein Kinase Regulates Oxidative Metabolism in Caenorhabditis elegans through the NHR-49 and MDT-15 Transcriptional Regulators. PLoS ONE 2016, 11, e0148089. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhou, B.; Oshiro-Rapley, N.; Li, M.; Paulo, J.A.; Webster, C.M.; Mou, F.; Kacergis, M.C.; Talkowski, M.E.; Carr, C.E.; et al. An Ancient, Unified Mechanism for Metformin Growth Inhibition in C. elegans and Cancer. Cell 2016, 167, 1705–1718.e13. [Google Scholar] [CrossRef] [Green Version]
- Galiullina, L.F.; Scheidt, H.A.; Huster, D.; Aganov, A.; Klochkov, V. Interaction of statins with phospholipid bilayers studied by solid-state NMR spectroscopy. Biochim. Biophys. Acta Biomembr. 2019, 1861, 584–593. [Google Scholar] [CrossRef]
- Stone, N.J.; Turin, A.; Spitz, J.A.; Valle, C.W.; Kazmi, S. Statin therapy across the lifespan: Evidence in major age groups. Expert Rev. Cardiovasc. Ther. 2016, 14, 341–366. [Google Scholar] [CrossRef] [PubMed]
- Jahn, A.; Scherer, B.; Fritz, G.; Honnen, S. Statins Induce a DAF-16/Foxo-dependent Longevity Phenotype via JNK-1 through Mevalonate Depletion in C. elegans. Aging Dis. 2020, 11, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, T.M.; Weinkove, D.; Houthoofd, K.; Gems, D.; Partridge, L. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech. Ageing Dev. 2007, 128, 546–552. [Google Scholar] [CrossRef]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Leontieva, O.V.; Demidenko, Z.N.; Blagosklonny, M.V. Rapamycin reverses insulin resistance (IR) in high-glucose medium without causing IR in normoglycemic medium. Cell Death Dis. 2014, 5, e1214. [Google Scholar] [CrossRef] [Green Version]
- Makki, K.; Taront, S.; Molendi-Coste, O.; Bouchaert, E.; Neve, B.; Eury, E.; Lobbens, S.; Labalette, M.; Duez, H.; Staels, B.; et al. Beneficial metabolic effects of rapamycin are associated with enhanced regulatory cells in diet-induced obese mice. PLoS ONE 2014, 9, e92684. [Google Scholar] [CrossRef] [PubMed]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012, 15, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco-Juárez, B.; Gómez-Manzo, S.; Hernández-Ochoa, B.; Cárdenas-Rodríguez, N.; Arreguin-Espinosa, R.; Pérez de la Cruz, V.; Ortega-Cuellar, D. Effects of High Dietary Carbohydrate and Lipid Intake on the Lifespan of C. elegans. Cells 2021, 10, 2359. https://doi.org/10.3390/cells10092359
Franco-Juárez B, Gómez-Manzo S, Hernández-Ochoa B, Cárdenas-Rodríguez N, Arreguin-Espinosa R, Pérez de la Cruz V, Ortega-Cuellar D. Effects of High Dietary Carbohydrate and Lipid Intake on the Lifespan of C. elegans. Cells. 2021; 10(9):2359. https://doi.org/10.3390/cells10092359
Chicago/Turabian StyleFranco-Juárez, Berenice, Saúl Gómez-Manzo, Beatriz Hernández-Ochoa, Noemi Cárdenas-Rodríguez, Roberto Arreguin-Espinosa, Verónica Pérez de la Cruz, and Daniel Ortega-Cuellar. 2021. "Effects of High Dietary Carbohydrate and Lipid Intake on the Lifespan of C. elegans" Cells 10, no. 9: 2359. https://doi.org/10.3390/cells10092359