
Glucocorticoid and PD-1 Cross-Talk: Does the Immune System Become Confused?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
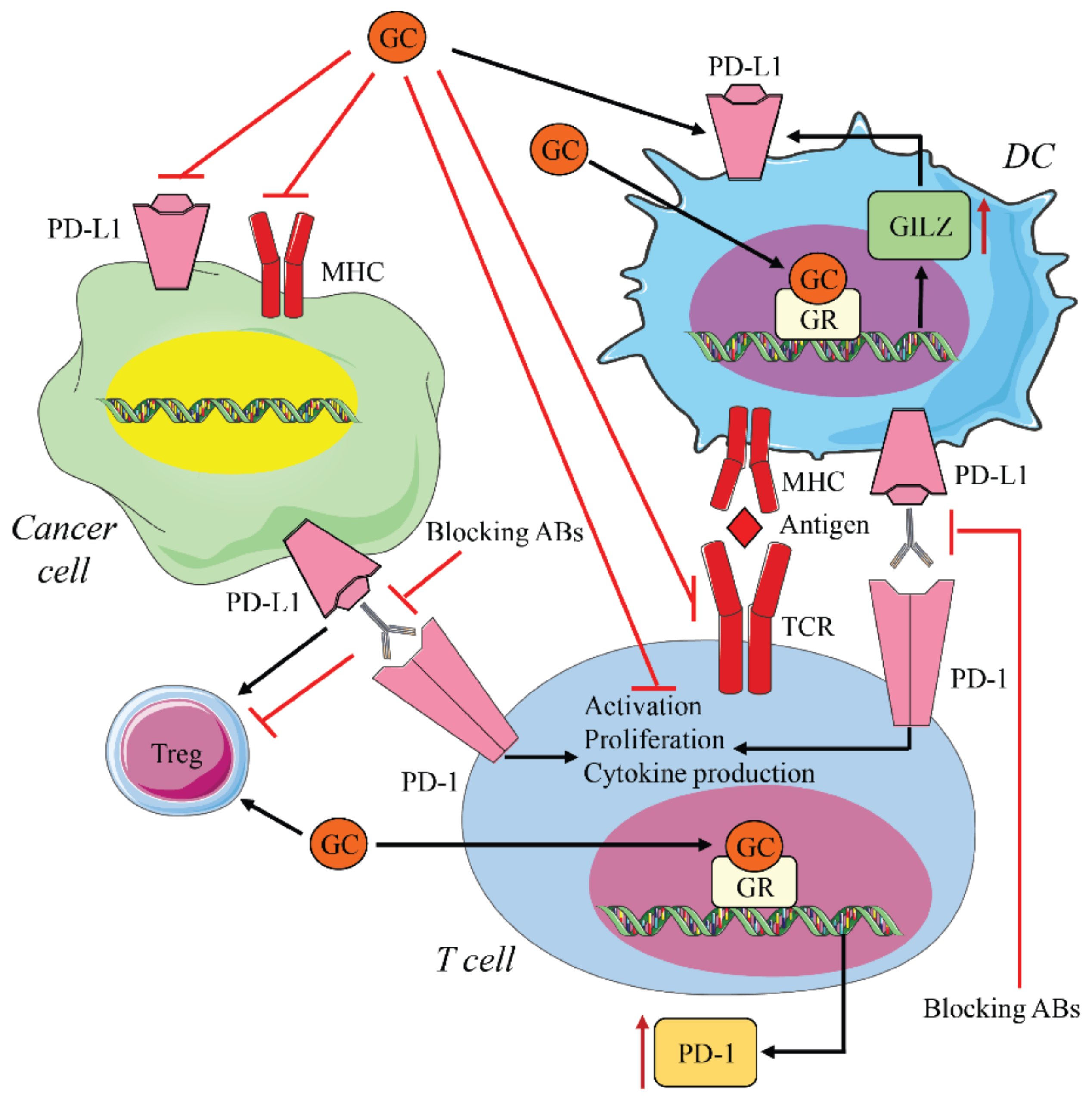
:1. Introduction
2. PD-1/PD-L1 in Health and Cancer
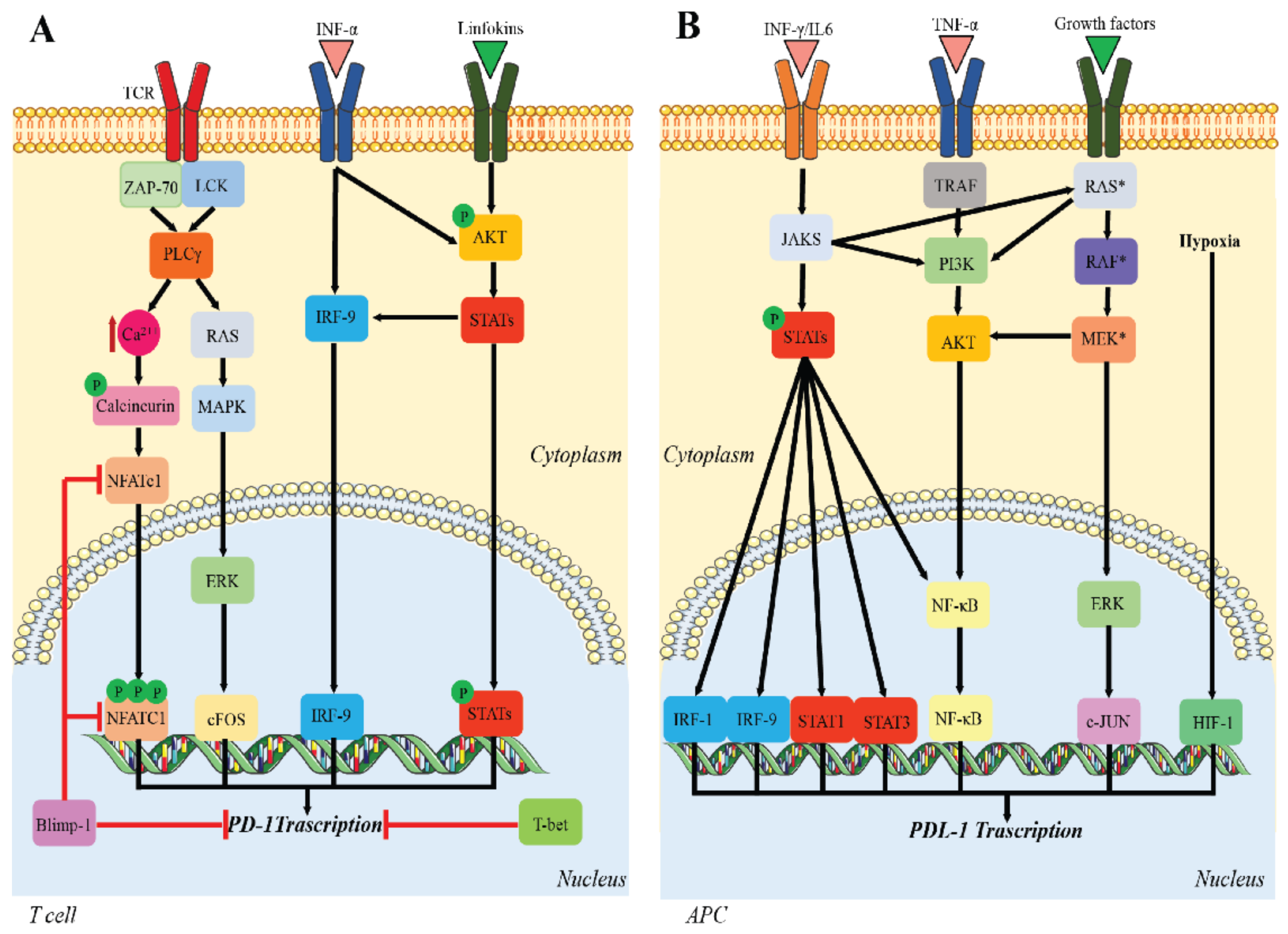
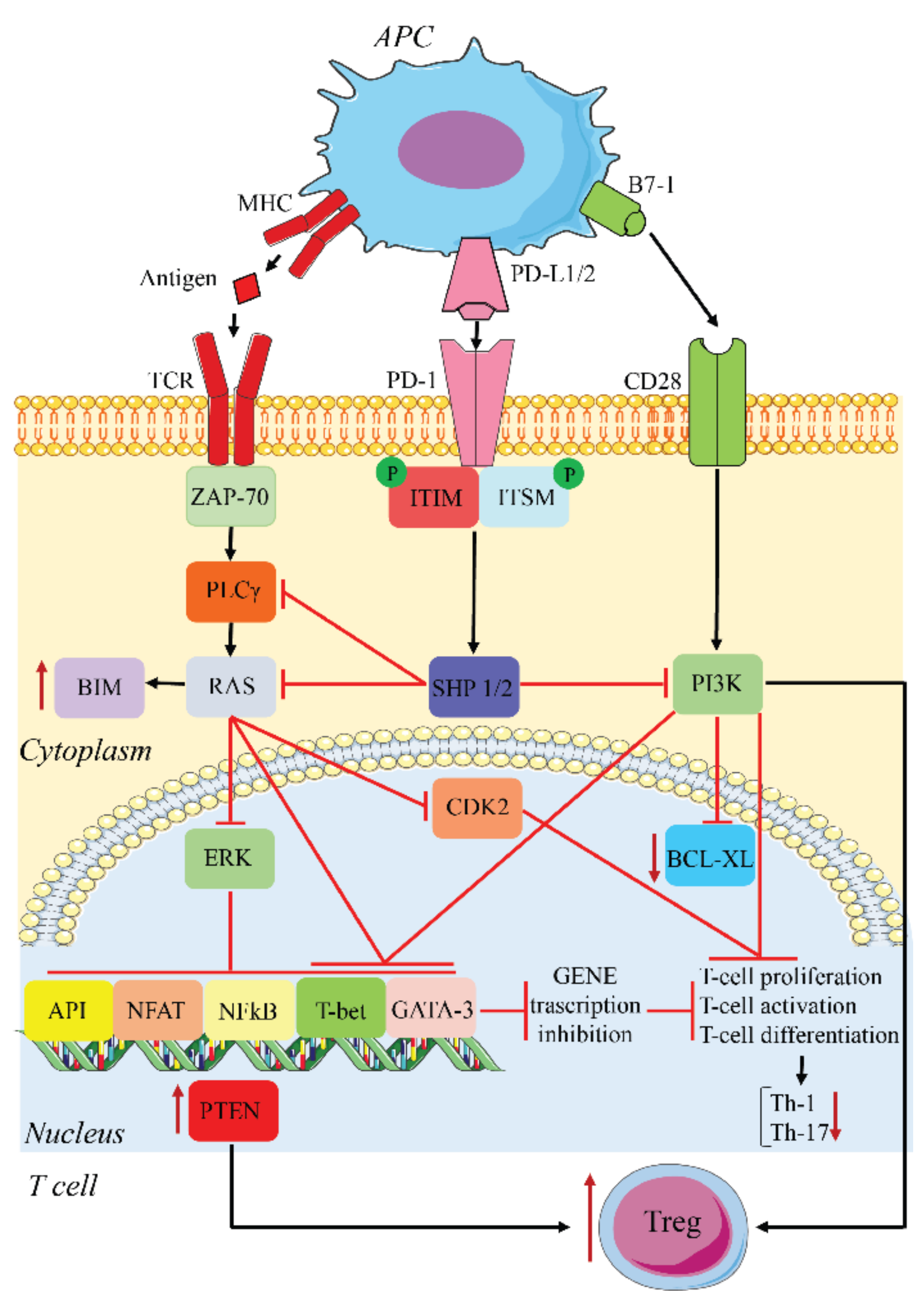
2.1. PD-1/PD-L1 Function
2.2. PD-1/PD-L1 and Cancer
3. Glucocorticoids in Health and Cancer
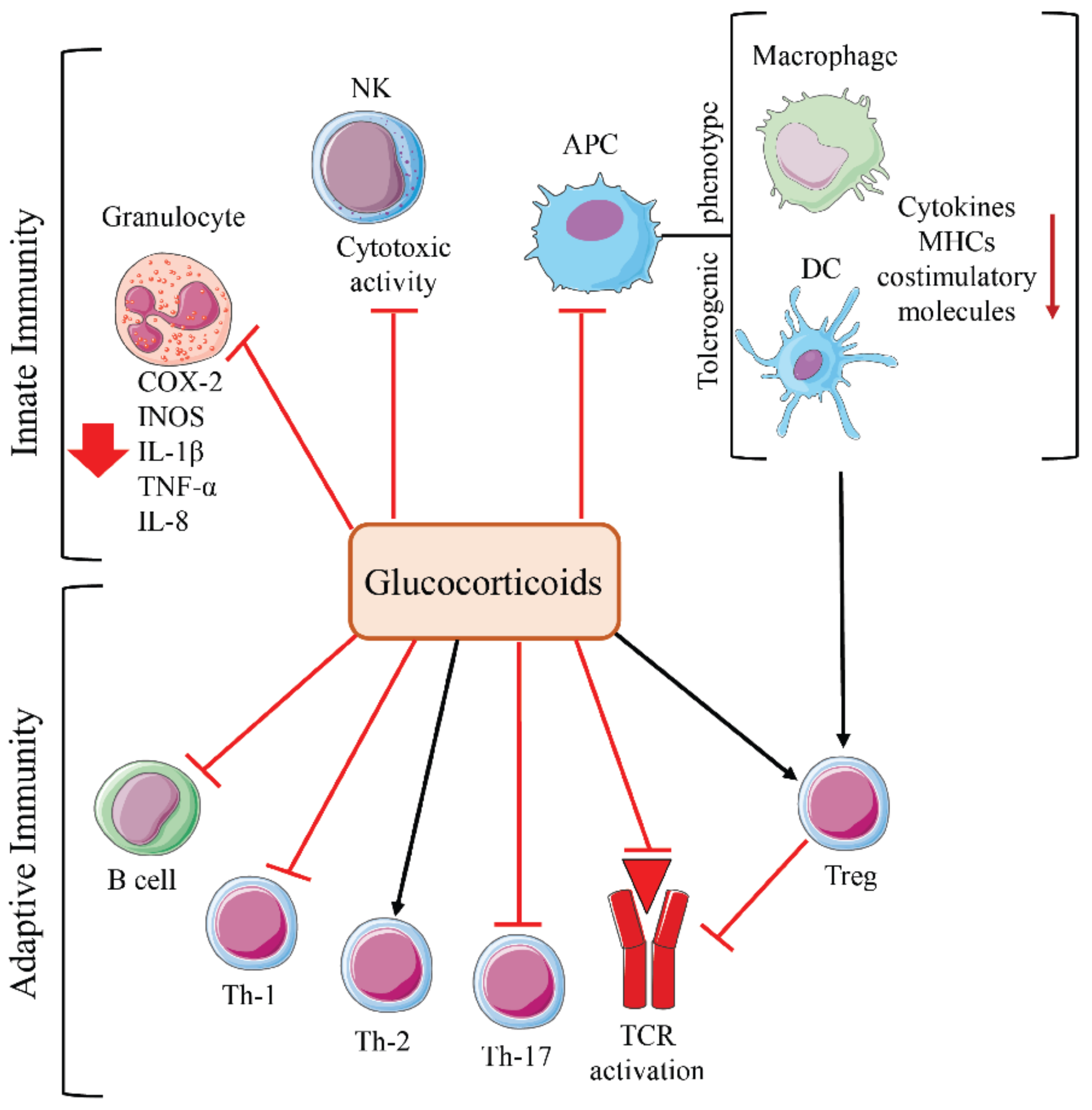
3.1. Immune Functions of Glucocorticoids
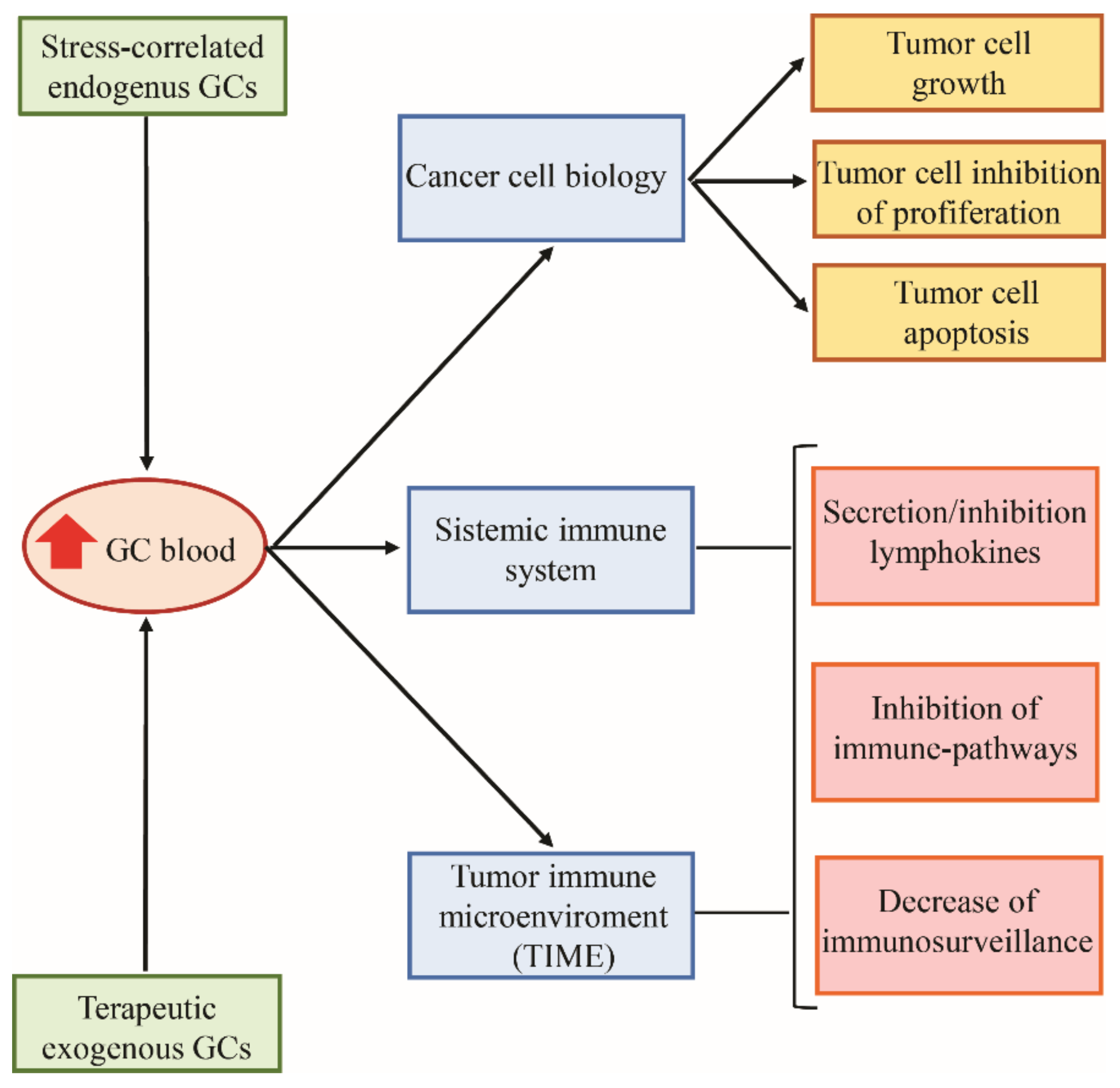
3.2. Glucocorticoids and Cancer
4. GC and PD-1/PD-L1 Pathways in Health and Cancer
4.1. Intersection of GC and PD-1/PD-L1 Pathways
4.2. Cross-Talk between GC and PD-1 Pathways in Tumor Immune Evasion
4.2.1. Role of Exogenous GCs in PD-1/PDL-1 Blockage
4.2.2. Role of Endogenous GCs in PD-1/PDL-1 Blockage
4.2.3. GCs and Resistance to PD-1/PDL-1 Blockage
Effect on Macrophage and DC
Effect on Tregs
Effect on Activated Lymphocytes
5. Conclusions: Does the Immune System Become Confused?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Gorska, M.; Politynska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Shin, K.J.; Lee, Y.J.; Yang, Y.R.; Park, S.; Suh, P.G.; Follo, M.Y.; Cocco, L.; Ryu, S.H. Molecular Mechanisms Underlying Psychological Stress and Cancer. Curr. Pharm. Des. 2016, 22, 2389–2402. [Google Scholar] [CrossRef]
- Ayroldi, E.; Cannarile, L.; Adorisio, S.; Delfino, D.V.; Riccardi, C. Role of Endogenous Glucocorticoids in Cancer in the Elderly. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, E.; Cravello, L.; Muzzoni, B.; Casarotti, D.; Paltro, M.; Solerte, S.B.; Fioravanti, M.; Cuzzoni, G.; Pontiggia, B.; Magri, F. Age-related changes of the hypothalamic-pituitary-adrenal axis: Pathophysiological correlates. Eur. J. Endocrinol. 2001, 144, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Pufall, M.A. Glucocorticoids and Cancer. Adv. Exp. Med. Biol. 2015, 872, 315–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.R. Anti-inflammatory functions of glucocorticoid-induced genes. Mol. Cell. Endocrinol. 2007, 275, 79–97. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; De Smedt, T.; Sornasse, T.; Tielemans, F.; Chentoufi, A.A.; Muraille, E.; Van Mechelen, M.; Urbain, J.; Leo, O. Glucocorticoids down-regulate dendritic cell function in vitro and in vivo. Eur. J. Immunol. 1995, 25, 2818–2824. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, R.; Luksik, A.S.; Garzon-Muvdi, T.; Hung, A.L.; Kim, E.S.; Wu, A.; Xia, Y.; Belcaid, Z.; Gorelick, N.; Choi, J.; et al. Contrasting impact of corticosteroids on anti-PD-1 immunotherapy efficacy for tumor histologies located within or outside the central nervous system. Oncoimmunology 2018, 7, e1500108. [Google Scholar] [CrossRef] [Green Version]
- Eigentler, T.K.; Hassel, J.C.; Berking, C.; Aberle, J.; Bachmann, O.; Grunwald, V.; Kahler, K.C.; Loquai, C.; Reinmuth, N.; Steins, M.; et al. Diagnosis, monitoring and management of immune-related adverse drug reactions of anti-PD-1 antibody therapy. Cancer Treat. Rev. 2016, 45, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bally, A.P.R.; Austin, J.W.; Boss, J.M. Genetic and Epigenetic Regulation of PD-1 Expression. J. Immunol. 2016, 196, 2431–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The Common gamma-Chain Cytokines IL-2, IL-7, IL-15, and IL-21 Induce the Expression of Programmed Death-1 and Its Ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Luong, G.; Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2017, 7, e1364828. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Lee, S.W.; Seo, S.K.; Choi, I.W.; Choi, I.; Lee, S.W. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-alpha-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim. Biophys. Acta 2008, 1779, 811–819. [Google Scholar] [CrossRef]
- Bally, A.P.; Lu, P.; Tang, Y.; Austin, J.W.; Scharer, C.D.; Ahmed, R.; Boss, J.M. NF-kappaB regulates PD-1 expression in macrophages. J. Immunol. 2015, 194, 4545–4554. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Bai, C.; Gao, W.; Strom, T.B.; Rothstein, T.L. Suppression of expression and function of negative immune regulator PD-1 by certain pattern recognition and cytokine receptor signals associated with immune system danger. Int. Immunol. 2004, 16, 1181–1188. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Klein, S.R.; Freeman, G.J.; Sharpe, A.H. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity 2016, 44, 955–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Shi, L.; Zhang, W.; Han, J.; Zhang, S.; Fu, Z.; Cai, J. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget 2018, 9, 5208–5215. [Google Scholar] [CrossRef] [Green Version]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, T.; Taniwaki, M.; Ishida, Y.; Kawaichi, M.; Honjo, T. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics 1994, 23, 704–706. [Google Scholar] [CrossRef]
- Nielsen, C.; Ohm-Laursen, L.; Barington, T.; Husby, S.; Lillevang, S.T. Alternative splice variants of the human PD-1 gene. Cell Immunol. 2005, 235, 109–116. [Google Scholar] [CrossRef]
- Machiraju, D.; Wiecken, M.; Lang, N.; Hulsmeyer, I.; Roth, J.; Schank, T.E.; Eurich, R.; Halama, N.; Enk, A.; Hassel, J.C. Soluble immune checkpoints and T-cell subsets in blood as biomarkers for resistance to immunotherapy in melanoma patients. Oncoimmunology 2021, 10, 1926762. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol. 2008, 181, 4832–4839. [Google Scholar] [CrossRef]
- Lu, P.; Youngblood, B.A.; Austin, J.W.; Mohammed, A.U.; Butler, R.; Ahmed, R.; Boss, J.M. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J. Exp. Med. 2014, 211, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Kao, C.; Oestreich, K.J.; Paley, M.A.; Crawford, A.; Angelosanto, J.M.; Ali, M.A.A.; Intlekofer, A.M.; Boss, J.M.; Reiner, S.L.; Weinmann, A.S.; et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8(+) T cell responses during chronic infection. Nat. Immunol. 2011, 12, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.W.; Schwartz, J.C.D.; Guo, X.L.; Bhatia, S.; Cao, E.H.; Lorenz, M.; Cammer, M.; Chen, L.P.; Zhang, Z.Y.; Edidin, M.A.; et al. Structural and functional analysis of the costimulatory receptor programmed death-1 (vol 20, pg 337, 2004). Immunity 2004, 20, 651. [Google Scholar] [CrossRef] [Green Version]
- Munari, E.; Mariotti, F.R.; Quatrini, L.; Bertoglio, P.; Tumino, N.; Vacca, P.; Eccher, A.; Ciompi, F.; Brunelli, M.; Martignoni, G.; et al. PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Philips, E.A.; Techova, A.S.; Mor, A.; Kong, X.P. Structural, functional, and evolutionary differences between PD-L1 and PD-L2. J. Immunol. 2018, 200. [Google Scholar]
- Fabrizio, F.P.; Trombetta, D.; Rossi, A.; Sparaneo, A.; Castellana, S.; Muscarella, L.A. Gene code CD274/PD-L1: From molecular basis toward cancer immunotherapy. Ther. Adv. Med. Oncol. 2018, 10, 1758835918815598. [Google Scholar] [CrossRef] [Green Version]
- Zerdes, I.; Matikas, A.; Bergh, J.; Rassidakis, G.Z.; Foukakis, T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: Biology and clinical correlations. Oncogene 2018, 37, 4639–4661. [Google Scholar] [CrossRef] [Green Version]
- Eppihimer, M.J.; Gunn, J.; Freeman, G.J.; Greenfield, E.A.; Chernova, T.; Erickson, J.; Leonard, J.P. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 2002, 9, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Loke, P.; Allison, J.P. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5336–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef]
- Kondo, A.; Yamashita, T.; Tamura, H.; Zhao, W.; Tsuji, T.; Shimizu, M.; Shinya, E.; Takahashi, H.; Tamada, K.; Chen, L.; et al. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood 2010, 116, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, L.; Huang, F.; Zhang, Q.; Liu, S.; Ma, L.; You, Z. Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 2017, 184, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.Q.; Boussiotis, V.A. Selective Effects of PD-1 on Akt and Ras Pathways Regulate Molecular Components of the Cell Cycle and Inhibit T Cell Proliferation. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaiopoulos, A.G.; Kostakis, I.D.; Koutsilieris, M.; Papavassiliou, A.G. Colorectal cancer stem cells. Stem Cells 2012, 30, 363–371. [Google Scholar] [CrossRef]
- Maccalli, C.; Rasul, K.I.; Elawad, M.; Ferrone, S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin. Cancer Biol. 2018, 53, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharm. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kusnadi, A.; Lee, E.J.; Kim, W.W.; Cho, B.C.; Lee, I.J.; Seong, J.; Ha, S.J. Tumor-infiltrating regulatory T cells delineated by upregulation of PD-1 and inhibitory receptors. Cell. Immunol. 2012, 278, 76–83. [Google Scholar] [CrossRef]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Sari, D.; Boussiotis, V.A. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 2012, 11, 4305–4309. [Google Scholar] [CrossRef] [Green Version]
- Arasanz, H.; Gato-Canas, M.; Zuazo, M.; Ibanez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945. [Google Scholar] [CrossRef] [Green Version]
- Almozyan, S.; Colak, D.; Mansour, F.; Alaiya, A.; Al-Harazi, O.; Qattan, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int. J. Cancer 2017, 141, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, N.; Mokhtarzadeh, A.; Baghbanzadeh, A.; Hajiasgharzadeh, K.; Shahgoli, V.K.; Hemmat, N.; Safarzadeh, E.; Baradaran, B. Immune checkpoints in tumor microenvironment and their relevance to the development of cancer stem cells. Life Sci. 2020, 256, 118005. [Google Scholar] [CrossRef] [PubMed]
- Karwacz, K.; Bricogne, C.; MacDonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 2011, 3, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.; Busselaar, J.; Bosma, D.M.T.; Ossendorp, F. Mechanism of action of PD-1 receptor/ligand targeted cancer immunotherapy. Eur. J. Immunol. 2021. [Google Scholar] [CrossRef]
- Luo, X.Y.; Wu, K.M.; He, X.X. Advances in drug development for hepatocellular carcinoma: Clinical trials and potential therapeutic targets. J. Exp. Clin. Cancer Res. 2021, 40, 172. [Google Scholar] [CrossRef]
- Bailly, C.; Thuru, X.; Quesnel, B. Soluble Programmed Death Ligand-1 (sPD-L1): A Pool of Circulating Proteins Implicated in Health and Diseases. Cancers 2021, 13. [Google Scholar] [CrossRef]
- Chen, J.; Song, Y.; Miao, F.; Chen, G.; Zhu, Y.; Wu, N.; Pang, L.; Chen, Z.; Chen, X. PDL1-positive exosomes suppress antitumor immunity by inducing tumor-specific CD8(+) T cell exhaustion during metastasis. Cancer Sci. 2021. [Google Scholar] [CrossRef]
- Yin, Z.; Yu, M.; Ma, T.; Zhang, C.; Huang, S.; Karimzadeh, M.R.; Momtazi-Borojeni, A.A.; Chen, S. Mechanisms underlying low-clinical responses to PD-1/PD-L1 blocking antibodies in immunotherapy of cancer: A key role of exosomal PD-L1. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Biddie, S.C.; Conway-Campbell, B.L.; Lightman, S.L. Dynamic regulation of glucocorticoid signalling in health and disease. Rheumatology 2012, 51, 403–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New insights into the anti-inflammatory mechanisms of glucocorticoids: An emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology 2013, 154, 993–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. 1998, 94, 557–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef]
- Van Laethem, F.; Baus, E.; Smyth, L.A.; Andris, F.; Bex, F.; Urbain, J.; Kioussis, D.; Leo, O. Glucocorticoids attenuate T cell receptor signaling. J. Exp. Med. 2001, 193, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Cannarile, L.; Delfino, D.V.; Adorisio, S.; Riccardi, C.; Ayroldi, E. Implicating the Role of GILZ in Glucocorticoid Modulation of T-Cell Activation. Front. Immunol. 2019, 10, 1823. [Google Scholar] [CrossRef] [PubMed]
- Ayroldi, E.; Cannarile, L.; Delfino, D.V.; Riccardi, C. A dual role for glucocorticoid-induced leucine zipper in glucocorticoid function: Tumor growth promotion or suppression? Cell Death Dis. 2018, 9, 463. [Google Scholar] [CrossRef]
- Mayayo-Peralta, I.; Zwart, W.; Prekovic, S. Duality of glucocorticoid action in cancer: Tumor-suppressor or oncogene? Endocr.-Relat. Cancer 2021, 28, R157–R171. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Chrousos, G.P.; Binder, E.B.; Zannas, A.S. Life stress, glucocorticoid signaling, and the aging epigenome: Implications for aging-related diseases. Neurosci. Biobehav. Rev. 2017, 74, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Azher, S.; Azami, O.; Amato, C.; McCullough, M.; Celentano, A.; Cirillo, N. The Non-Conventional Effects of Glucocorticoids in Cancer. J. Cell. Physiol. 2016, 231, 2368–2373. [Google Scholar] [CrossRef]
- Herr, I.; Pfitzenmaier, J. Glucocorticoid use in prostate cancer and other solid tumours: Implications for effectiveness of cytotoxic treatment and metastases. Lancet Oncol. 2006, 7, 425–430. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Porta, C.; Sica, A.; Allavena, P. Inflammation and cancer: Breast cancer as a prototype. Breast 2007, 16 (Suppl. 2), S27–S33. [Google Scholar] [CrossRef]
- Landwehr, L.S.; Altieri, B.; Schreiner, J.; Sbiera, I.; Weigand, I.; Kroiss, M.; Fassnacht, M.; Sbiera, S. Interplay between glucocorticoids and tumor-infiltrating lymphocytes on the prognosis of adrenocortical carcinoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Cirillo, N.; Morgan, D.J.; Pedicillo, M.C.; Celentano, A.; Lo Muzio, L.; McCullough, M.J.; Prime, S.S. Characterisation of the cancer-associated glucocorticoid system: Key role of 11beta-hydroxysteroid dehydrogenase type 2. Br. J. Cancer 2017, 117, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidler, D.; Renzulli, P.; Schnoz, C.; Berger, B.; Schneider-Jakob, S.; Fluck, C.; Inderbitzin, D.; Corazza, N.; Candinas, D.; Brunner, T. Colon cancer cells produce immunoregulatory glucocorticoids. Oncogene 2011, 30, 2411–2419. [Google Scholar] [CrossRef] [Green Version]
- Cain, D.W.; Cidlowski, J.A. Specificity and sensitivity of glucocorticoid signaling in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef] [Green Version]
- Vettorazzi, S.; Nalbantoglu, D.; Gebhardt, J.C.M.; Tuckermann, J. A guide to changing paradigms of glucocorticoid receptor function—A model system for genome regulation and physiology. FEBS J. 2021. [Google Scholar] [CrossRef]
- Matthews, L.C.; Berry, A.A.; Morgan, D.J.; Poolman, T.M.; Bauer, K.; Kramer, F.; Spiller, D.G.; Richardson, R.V.; Chapman, K.E.; Farrow, S.N.; et al. Glucocorticoid receptor regulates accurate chromosome segregation and is associated with malignancy. Proc. Natl. Acad. Sci. USA 2015, 112, 5479–5484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veneris, J.T.; Darcy, K.M.; Mhawech-Fauceglia, P.; Tian, C.; Lengyel, E.; Lastra, R.R.; Pejovic, T.; Conzen, S.D.; Fleming, G.F. High glucocorticoid receptor expression predicts short progression-free survival in ovarian cancer. Gynecol. Oncol. 2017, 146, 153–160. [Google Scholar] [CrossRef]
- Tangen, I.L.; Veneris, J.T.; Halle, M.K.; Werner, H.M.; Trovik, J.; Akslen, L.A.; Salvesen, H.B.; Conzen, S.D.; Fleming, G.F.; Krakstad, C. Expression of glucocorticoid receptor is associated with aggressive primary endometrial cancer and increases from primary to metastatic lesions. Gynecol. Oncol. 2017, 147, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, L.M.; Tredan, O.; Hussein, N.; Badran, B.; Le Romancer, M.; Poulard, C. Glucocorticoid Receptor: A Multifaceted Actor in Breast Cancer. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Vilasco, M.; Communal, L.; Mourra, N.; Courtin, A.; Forgez, P.; Gompel, A. Glucocorticoid receptor and breast cancer. Breast Cancer Res. Treat. 2011, 130, 1–10. [Google Scholar] [CrossRef]
- Yemelyanov, A.; Czwornog, J.; Chebotaev, D.; Karseladze, A.; Kulevitch, E.; Yang, X.; Budunova, I. Tumor suppressor activity of glucocorticoid receptor in the prostate. Oncogene 2007, 26, 1885–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.S.; Lien, H.C.; Yeh, P.Y.; Kuo, S.H.; Chang, W.C.; Kuo, M.L.; Cheng, A.L. Glucocorticoid receptor expression in advanced non-small cell lung cancer: Clinicopathological correlation and in vitro effect of glucocorticoid on cell growth and chemosensitivity. Lung Cancer 2006, 53, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Yao, Q.Y.; Xue, J.S.; Tian, X.Y.; An, Q.M.; Cui, L.X.; Xu, C.; Su, H.; Yang, L.; Feng, Y.Y.; et al. Dexamethasone inhibits pancreatic tumor growth in preclinical models: Involvement of activating glucocorticoid receptor. Toxicol. Appl. Pharm. 2020, 401, 115118. [Google Scholar] [CrossRef]
- Mikosz, C.A.; Brickley, D.R.; Sharkey, M.S.; Moran, T.W.; Conzen, S.D. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J. Biol. Chem. 2001, 276, 16649–16654. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Maruhashi, T.; Sugiura, D.; Shimizu, K.; Okazaki, I.M.; Okazaki, T. Glucocorticoids potentiate the inhibitory capacity of programmed cell death 1 by up-regulating its expression on T cells. J. Biol. Chem. 2019, 294, 19896–19906. [Google Scholar] [CrossRef]
- Quatrini, L.; Vacca, P.; Tumino, N.; Besi, F.; Di Pace, A.L.; Scordamaglia, F.; Martini, S.; Munari, E.; Mingari, M.C.; Ugolini, S.; et al. Glucocorticoids and the cytokines IL-12, IL-15, and IL-18 present in the tumor microenvironment induce PD-1 expression on human natural killer cells. J. Allergy Clin. Immunol. 2021, 147, 349–360. [Google Scholar] [CrossRef]
- Xing, K.; Gu, B.; Zhang, P.; Wu, X. Dexamethasone enhances programmed cell death 1 (PD-1) expression during T cell activation: An insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunol. 2015, 16, 39. [Google Scholar] [CrossRef] [Green Version]
- Biron, C.A. IMMUNOPATHOLOGY Glucocorticoids and NK cell PD-1. Nat. Immunol. 2018, 19, 908. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, A.R. Commentary: Endogenous glucocorticoids control host resistance to viral infection through tissue-specific regulation of PD-1 expression on NK cells. Cell. Mol. Immunol. 2019, 16, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Koop, A.; Meier, F.; Hassel, J.C.; Terheyden, P.; Zimmer, L.; Heinzerling, L.; Ugurel, S.; Pfohler, C.; Gesierich, A.; et al. Programmed cell death protein-1 (PD-1) inhibitor therapy in patients with advanced melanoma and preexisting autoimmunity or ipilimumab-triggered autoimmunity. Eur. J. Cancer 2017, 75, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Zam, W.; Ali, L. Immune checkpoint inhibitors in the treatment of cancer. Curr. Clin. Pharm. 2021. [Google Scholar] [CrossRef]
- Aston, W.J.; Hope, D.E.; Cook, A.M.; Boon, L.; Dick, I.; Nowak, A.K.; Lake, R.A.; Lesterhuis, W.J. Dexamethasone differentially depletes tumour and peripheral blood lymphocytes and can impact the efficacy of chemotherapy/checkpoint blockade combination treatment. Oncoimmunology 2019, 8, e1641390. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.S.; Barroso-Sousa, R.; Tolaney, S.M.; Hodi, F.S.; Kaiser, U.B.; Min, L. Endocrine Toxicity of Cancer Immunotherapy Targeting Immune Checkpoints. Endocr. Rev. 2019, 40, 17–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuswanto, W.F.; MacFarlane, L.A.; Gedmintas, L.; Mulloy, A.; Choueiri, T.K.; Bermas, B.L. Rheumatologic symptoms in oncologic patients on PD-1 inhibitors. Semin. Arthritis Rheum. 2018, 47, 907–910. [Google Scholar] [CrossRef]
- Gaucher, L.; Adda, L.; Sejourne, A.; Joachim, C.; Chaby, G.; Poulet, C.; Liabeuf, S.; Gras-Champel, V.; Masmoudi, K.; Moreira, A.; et al. Impact of the corticosteroid indication and administration route on overall survival and the tumor response after immune checkpoint inhibitor initiation. Ther. Adv. Med. Oncol. 2021, 13. [Google Scholar] [CrossRef]
- Goleva, E.; Lyubchenko, T.; Kraehenbuehl, L.; Lacouture, M.E.; Leung, D.Y.M.; Kern, J.A. Our current understanding of checkpoint inhibitor therapy in cancer immunotherapy. Ann. Allergy Asthma Immunol. 2021, 126, 630–638. [Google Scholar] [CrossRef]
- Johnson, D.B.; Jakubovic, B.D.; Sibaud, V.; Sise, M.E. Balancing Cancer Immunotherapy Efficacy and Toxicity. J. Allergy Clin. Immunol. Pr. 2020, 8, 2898–2906. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Madi, A.; Zhang, H.; Klapholz, M.; Escobar, G.; Dulberg, S.; Christian, E.; Ferreira, M.; Dixon, K.O.; Fell, G.; et al. Endogenous Glucocorticoid Signaling Regulates CD8(+) T Cell Differentiation and Development of Dysfunction in the Tumor Microenvironment. Immunity 2020, 53, 658–671.e6. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, Y.; Shi, T.; Wang, W.; Shao, D.; Zheng, X.; Sun, M.; He, K.; Chen, L. Depression Promotes Hepatocellular Carcinoma Progression through a Glucocorticoids Mediated Up-Regulation of PD-1 Expression in Tumor infiltrating NK Cells. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Cui, S.; Lin, H.; Cui, Y.; Wen, W.; Cui, X.; Shen, C.; Mo, H.; Yang, L.; Bai, S.; Shi, Y.; et al. Depression promotes lung carcinoma progression by regulating the tumor microenvironment in tumor-bearing models of C57BL/6J mice. Neurosci. Lett. 2021, 754, 135851. [Google Scholar] [CrossRef]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef]
- Ghahremanloo, A.; Soltani, A.; Modaresi, S.M.S.; Hashemy, S.I. Recent advances in the clinical development of immune checkpoint blockade therapy. Cell Oncol. 2019, 42, 609–626. [Google Scholar] [CrossRef]
- Rodriguez, F.; Redondo, M.; Ruiz-Cabello, F. Dexamethasone induces altered binding of regulatory factors to HLA class I enhancer sequence in MCF-7 breast tumour cell line. Cancer Immunol. Immunother. 1998, 46, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Navajas, J.M.; Fan, D.D.; Yang, S.; Yang, F.M.; Lozano-Ruiz, B.; Shen, L.; Lee, J. The Impact of Tregs on the Anticancer Immunity and the Efficacy of Immune Checkpoint Inhibitor Therapies. Front. Immunol. 2021, 12, 625783. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Young, A.; Jacquelot, N.; Yamazaki, T.; Enot, D.; Zitvogel, L.; Smyth, M.J. A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1. Cancer Res. 2015, 75, 3800–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci. Transl. Med. 2011, 3, 111ra120. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.L.; Kuchroo, J.R.; Sage, P.T.; Liang, D.; Francisco, L.M.; Buck, J.; Thaker, Y.R.; Zhang, Q.; McArdel, S.L.; Juneja, V.R.; et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Pauken, K.E.; Torchia, J.A.; Chaudhri, A.; Sharpe, A.H.; Freeman, G.J. Emerging concepts in PD-1 checkpoint biology. Semin. Immunol. 2021, 101480. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Xiong, Y.; Neskey, D.M.; Horton, J.D.; Paulos, C.M.; Knochelmann, H.M.; Armeson, K.E.; Young, M.R.I. Immunological effects of nivolumab immunotherapy in patients with oral cavity squamous cell carcinoma. BMC Cancer 2020, 20, 229. [Google Scholar] [CrossRef]
- Kim, K.H.; Hur, J.Y.; Cho, J.; Ku, B.M.; Koh, J.; Koh, J.Y.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; et al. Immune-related adverse events are clustered into distinct subtypes by T-cell profiling before and early after anti-PD-1 treatment. Oncoimmunology 2020, 9, 1722023. [Google Scholar] [CrossRef] [Green Version]
- Koh, J.; Hur, J.Y.; Lee, K.Y.; Kim, M.S.; Heo, J.Y.; Ku, B.M.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; et al. Regulatory (FoxP3(+)) T cells and TGF-beta predict the response to anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci. Rep. 2020, 10, 18994. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Mehdizadeh, S.; Smith, J.; Young, A.; Mufazalov, I.A.; Mowery, C.T.; Daud, A.; Bluestone, J.A. Regulatory T cell control of systemic immunity and immunotherapy response in liver metastasis. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Lau, J.; Cheung, J.; Navarro, A.; Lianoglou, S.; Haley, B.; Totpal, K.; Sanders, L.; Koeppen, H.; Caplazi, P.; McBride, J.; et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat. Commun. 2017, 8, 14572. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.C.; Li, C.W.; Xia, W.; Hsu, J.M.; Lee, H.H.; Cha, J.H.; Wang, H.L.; Yang, W.H.; Yen, E.Y.; Chang, W.C.; et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J. Clin. Investig. 2019, 129, 3324–3338. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.M.; Sodre, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Zhou, Z.; Song, S.; Li, J.; Ji, J.; Yan, R.; Wang, J.; Cai, W.; Hu, W.; Zang, L.; et al. Dexamethasone suppresses immune evasion by inducing GR/STAT3 mediated downregulation of PD-L1 and IDO1 pathways. Oncogene 2021. [Google Scholar] [CrossRef] [PubMed]
- Draxler, D.F.; Bain, C.R.; Taylor, R.; Wallace, S.; Gouldthorpe, O.; Corcoran, T.B.; Myles, P.S.; Bozaoglu, K.; Medcalf, R.L. Data on the modulatory effects of a single bolus dexamethasone on the surface marker expression of various leucocyte subsets. Data Brief 2020, 32, 106117. [Google Scholar] [CrossRef]
- Cohen, N.; Mouly, E.; Hamdi, H.; Maillot, M.C.; Pallardy, M.; Godot, V.; Capel, F.; Balian, A.; Naveau, S.; Galanaud, P.; et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood 2006, 107, 2037–2044. [Google Scholar] [CrossRef]
- Vetillard, M.; Schlecht-Louf, G. Glucocorticoid-Induced Leucine Zipper: Fine-Tuning of Dendritic Cells Function. Front. Immunol. 2018, 9, 1232. [Google Scholar] [CrossRef] [PubMed]
- Cathelin, D.; Met, O.; Svane, I.M. Silencing of the glucocorticoid-induced leucine zipper improves the immunogenicity of clinical-grade dendritic cells. Cytotherapy 2013, 15, 740–749. [Google Scholar] [CrossRef]
- Yang, H.; Xia, L.; Chen, J.; Zhang, S.; Martin, V.; Li, Q.; Lin, S.; Chen, J.; Calmette, J.; Lu, M.; et al. Stress-glucocorticoid-TSC22D3 axis compromises therapy-induced antitumor immunity. Nat. Med. 2019, 25, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.B.; Huang, Y.F.; Li, Y.; Han, G.C.; Li, Y.R.; Wang, D.J.; Du, G.P.; Yu, J.F.; Song, J. Experimental study of the mechanism of tolerance induction in dexamethasone-treated dendritic cells. Med. Sci. Monit. 2011, 17, Br125–Br131. [Google Scholar] [CrossRef] [Green Version]
- Tokita, D.; Mazariegos, G.V.; Zahorchak, A.F.; Chien, N.; Abe, M.; Raimondi, G.; Thomson, A.W. High PD-L1/CD86 ratio on plasmacytoid dendritic cells correlates with elevated T-regulatory cells in liver transplant tolerance. Transplantation 2008, 85, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Zha, H.R.; Jiang, Y.; Wang, X.; Shang, J.; Wang, N.; Yu, L.; Zhao, W.; Li, Z.H.; An, J.; Zhang, X.C.; et al. Non-canonical PD-1 signaling in cancer and its potential implications in clinic. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Horvat, T.Z.; Adel, N.G.; Dang, T.O.; Momtaz, P.; Postow, M.A.; Callahan, M.K.; Carvajal, R.D.; Dickson, M.A.; D’Angelo, S.P.; Woo, K.M.; et al. Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients With Melanoma Treated With Ipilimumab at Memorial Sloan Kettering Cancer Center. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3193–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinelli, D.; Giusti, R.; Mazzotta, M.; Filetti, M.; Krasniqi, E.; Pizzuti, L.; Landi, L.; Tomao, S.; Cappuzzo, F.; Ciliberto, G.; et al. Palliative- and non-palliative indications for glucocorticoids use in course of immune-checkpoint inhibition. Current evidence and future perspectives. Crit. Rev. Oncol. Hematol. 2021, 157, 103176. [Google Scholar] [CrossRef]
- Yang, R.; Yu, Y. Glucocorticoids are double-edged sword in the treatment of COVID-19 and cancers. Int. J. Biol. Sci. 2021, 17, 1530–1537. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adorisio, S.; Cannarile, L.; Delfino, D.V.; Ayroldi, E. Glucocorticoid and PD-1 Cross-Talk: Does the Immune System Become Confused? Cells 2021, 10, 2333. https://doi.org/10.3390/cells10092333
Adorisio S, Cannarile L, Delfino DV, Ayroldi E. Glucocorticoid and PD-1 Cross-Talk: Does the Immune System Become Confused? Cells. 2021; 10(9):2333. https://doi.org/10.3390/cells10092333
Chicago/Turabian StyleAdorisio, Sabrina, Lorenza Cannarile, Domenico V. Delfino, and Emira Ayroldi. 2021. "Glucocorticoid and PD-1 Cross-Talk: Does the Immune System Become Confused?" Cells 10, no. 9: 2333. https://doi.org/10.3390/cells10092333