
Dissecting Highly Mutagenic Processing of Complex Clustered DNA Damage in Yeast Saccharomyces cerevisiae

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Oligonucleotides and Plasmids
2.2. Yeast Strains and Media
2.3. Yeast Transformation and Analysis of Transformants
2.4. Preparation of Yeast Cell Extract
2.5. Cleavage of the Lesion-Containing Duplexes by Yeast Cell Extract
2.6. Repair Synthesis Assay on MDS1 by Yeast Whole-Cell Extract
2.7. Statistical Analysis
3. Results
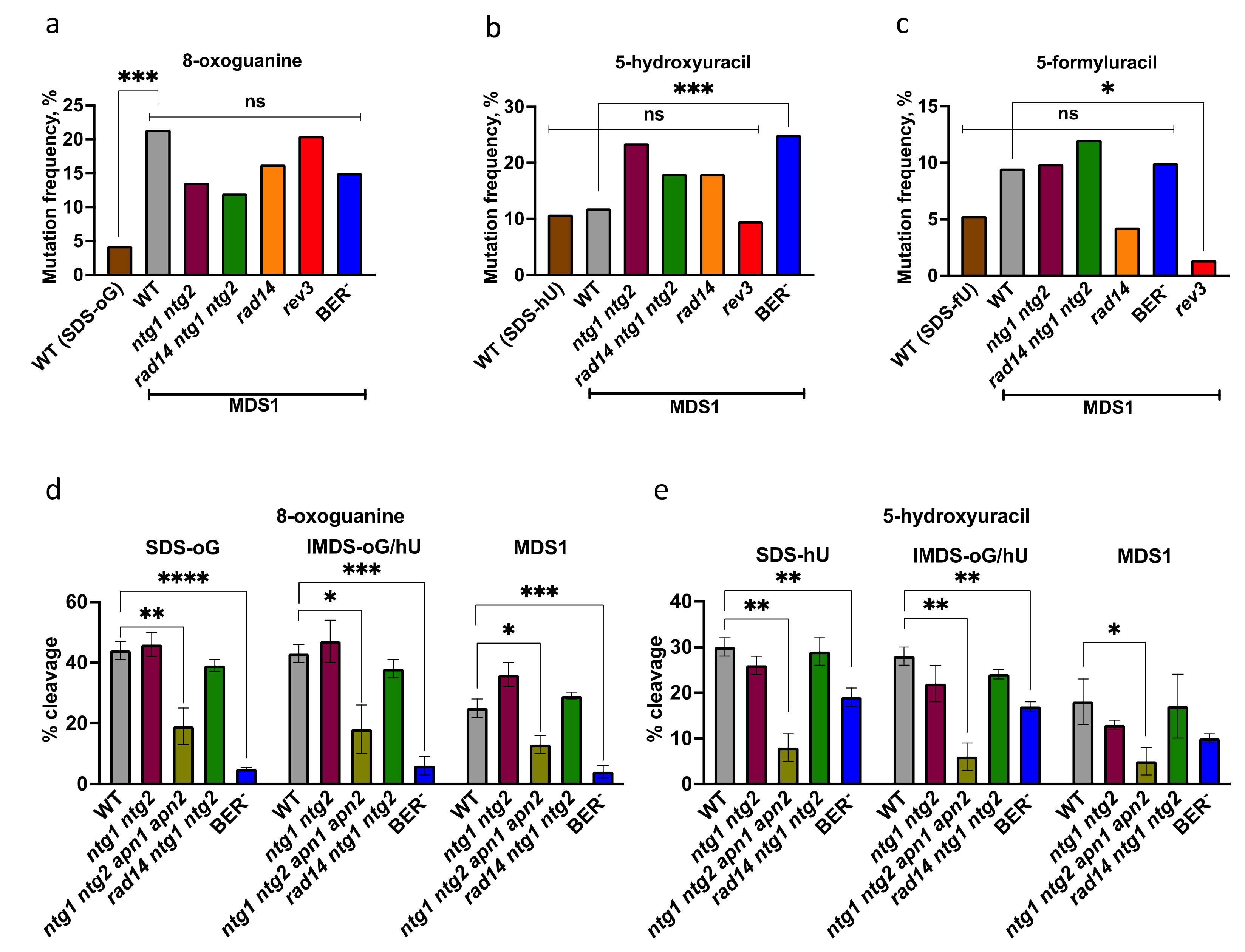
3.1. Elevated Mutagenesis Is Targeted at Damage within MDS
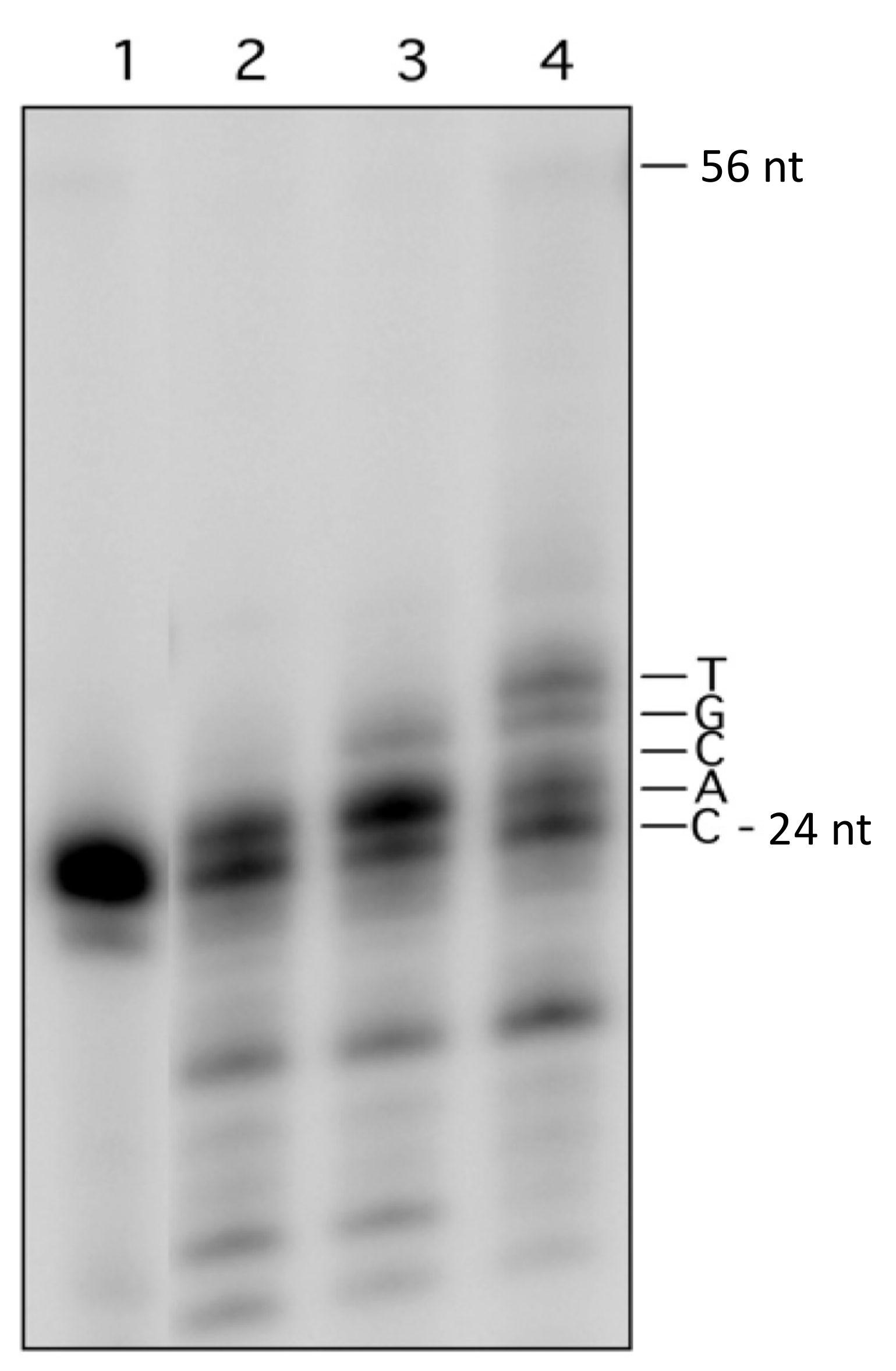
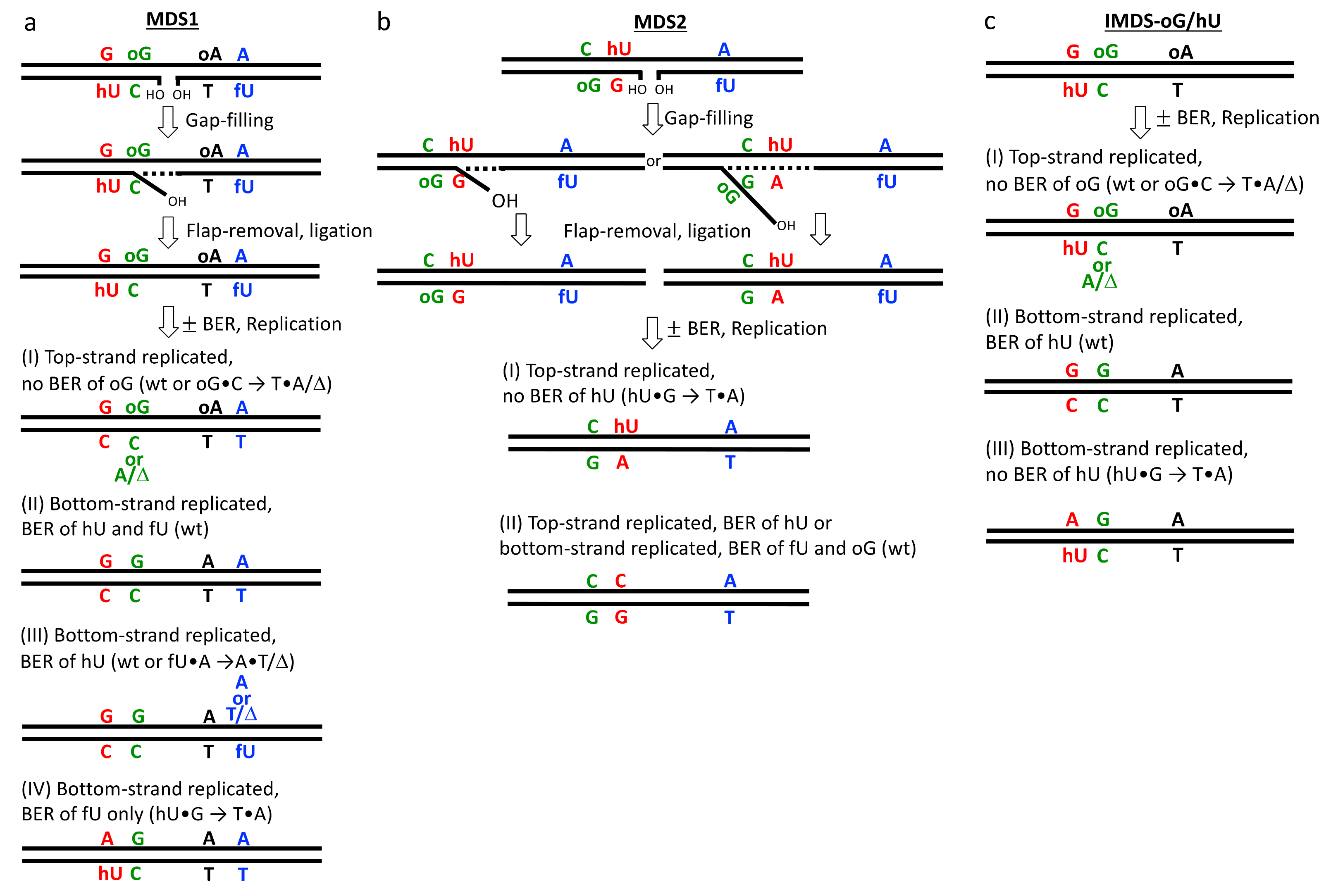
3.2. Processing of Lesions within MDS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, J.F. The complexity of DNA damage: Relevance to biological consequences. Int. J. Radiat. Biol. 1994, 66, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A. Radiation Track Structure: How the Spatial Distribution of Energy Deposition Drives Biological Response. Clin. Oncol. 2020, 32, 75–83. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Bennett, P.V.; Sidorkina, O.; Laval, J. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc. Natl. Acad. Sci. USA 2000, 97, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.M.; Bennett, P.V.; Sutherland, J.C.; Laval, J. Clustered DNA damages induced by x rays in human cells. Radiat. Res. 2002, 157, 611–616. [Google Scholar] [CrossRef]
- Gulston, M.; Fulford, J.; Jenner, T.; de Lara, C.; O’Neill, P. Clustered DNA damage induced by gamma radiation in human fibroblasts (HF19), hamster (V79-4) cells and plasmid DNA is revealed as Fpg and Nth sensitive sites. Nucleic. Acids Res. 2002, 30, 3464–3472. [Google Scholar] [CrossRef] [Green Version]
- Nikjoo, H.; O’Neill, P.; Wilson, W.E.; Goodhead, D.T. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat. Res. 2001, 156, 577–583. [Google Scholar] [CrossRef]
- Semenenko, V.A.; Stewart, R.D. A fast Monte Carlo algorithm to simulate the spectrum of DNA damages formed by ionizing radiation. Radiat. Res. 2004, 161, 451–457. [Google Scholar] [CrossRef]
- Watanabe, R.; Rahmanian, S.; Nikjoo, H. Spectrum of radiation-induced clustered non-DSB damage—A Monte Carlo track structure modeling and calculations. Radiat. Res. 2015, 183, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Nakano, T.; Tsuda, M.; Kanamoto, R.; Hirayama, R.; Uzawa, A.; Ide, H. Direct observation of damage clustering in irradiated DNA with atomic force microscopy. Nucleic Acids Res. 2020, 48, e18. [Google Scholar] [CrossRef]
- Akamatsu, K.; Shikazono, N.; Saito, T. Fluorescence anisotropy study of radiation-induced DNA damage clustering based on FRET. Anal. Bioanal. Chem. 2021, 4, 1185–1192. [Google Scholar] [CrossRef]
- Asaithamby, A.; Hu, B.R.; Chen, D.J. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8293–8298. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Gao, Y.; Liu, W.; Gao, T.; Zheng, Y.; Sanche, L. Clustered DNA Damage Induced by 2−20 eV Electrons and Transient Anions: General Mechanism and Correlation to Cell Death. J. Phys. Chem. Lett. 2019, 10, 2985–2990. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Collins, L.B.; Chen, T.H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Nikitaki, Z.; Pariset, E.; Sudar, D.; Costes, S.V.; Georgakilas, A.G. In Situ Detection of Complex DNA Damage Using Microscopy: A Rough Road Ahead. Cancers 2020, 12, 3288. [Google Scholar] [CrossRef] [PubMed]
- Lorat, Y.; Brunner, C.U.; Schanz, S.; Jakob, B.; Taucher-Scholz, G.; Rube, C.E. Nanoscale analysis of clustered DNA damage after high-LET irradiation by quantitative electron microscopy—The heavy burden to repair. DNA Repair 2015, 28, 93–106. [Google Scholar] [CrossRef]
- Lorat, Y.; Schanz, S.; Rübe, C.E. Ultrastructural Insights into the Biological Significance of Persisting DNA Damage Foci after Low Doses of Ionizing Radiation. Clin. Cancer Res. 2016, 22, 5300–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA damage concentrated in particle trajectories causes persistent large-scale rearrangements in chromatin architecture. Radiother Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Niimi, A.; Isono, M.; Yamauchi, M.; Yasuhara, T.; Limsirichaikul, S.; Oike, T.; Sato, H.; Held, K.D.; Nakano, T.; et al. 3D-structured illumination microscopy reveals clustered DNA double-strand break formation in widespread γH2AX foci after high LET heavy-ion particle radiation. Oncotarget 2017, 8, 109370–109381. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Oike, T.; Niimi, A.; Yamauchi, M.; Sato, H.; Limsirichaikul, S.; Held, K.D.; Nakano, T.; Shibata, A. Clustered DNA double-strand break formation and the repair pathway following heavy-ion irradiation. J. Radiat. Res. 2019, 60, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef] [Green Version]
- Shikazono, N.; Noguchi, M.; Fujii, K.; Urushibara, A.; Yokoya, A. The yield, processing, and biological consequences of clustered DNA damage induced by ionizing radiation. J. Radiat. Res. 2009, 50, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed repair of radiation induced clustered DNA damage: Friend or foe? Mutat. Res. 2011, 711, 134–141. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Urushibara, A.; Yokoya, A.; O’Neill, P.; Shikazono, N. The mutagenic potential of 8-oxoG/single strand break-containing clusters depends on their relative positions. Mutat. Res. 2012, 732, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Eccles, L.J.; Lomax, M.E.; O’Neill, P. Hierarchy of lesion processing governs the repair, double-strand break formation and mutability of three-lesion clustered DNA damage. Nucleic Acids Res. 2010, 38, 1123–1134. [Google Scholar] [CrossRef] [Green Version]
- Cunniffe, S.; Walker, A.; Stabler, R.; O’Neill, P.; Lomax, M.E. Increased mutability and decreased repairability of a three-lesion clustered DNA-damaged site comprised of an AP site and bi-stranded 8-oxoG lesions. Int. J. Radiat. Biol. 2014, 90, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shikazono, N.; Akamatsu, K. Mutagenic potential of 8-oxo-7,8-dihydroguanine (8-oxoG) is influenced by nearby clustered lesions. Mutat. Res. Fund. Mol. Mech. Mutagen. 2018, 810, 6–12. [Google Scholar] [CrossRef]
- Shikazono, N.; Akamatsu, K. Strand with mutagenic lesion is preferentially used as a template in the region of a bi-stranded clustered DNA damage site in Escherichia coli. Sci. Rep. 2020, 10, 9737. [Google Scholar] [CrossRef]
- Dianov, G.L.; Timchenko, T.V.; Sinitsina, O.I.; Kuzminov, A.V.; Medvedev, O.A.; Salganik, R.I. Repair of uracil residues closely spaced on the opposite strands of plasmid DNA results in double-strand break and deletion formation. Mol. Gen. Genet. 1991, 225, 448–452. [Google Scholar] [CrossRef]
- D’Souza, D.I.; Harrison, L. Repair of clustered uracil DNA damages in Escherichia coli. Nucleic Acids Res. 2003, 31, 4573–4581. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.; Brame, K.L.; Geltz, L.E.; Landry, A.M. Closely opposed apurinic/apyrimidinic sites are converted to double strand breaks in Escherichia coli even in the absence of exonuclease III, endonuclease IV, nucleotide excision repair and AP lyase cleavage. DNA Repair 2006, 5, 324–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedletska, Y.; Radicella, J.P.; Sage, E. Replication fork collapse is a major cause of the high mutation frequency at three-base lesion clusters. Nucleic Acids Res. 2013, 41, 9339–9348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozmin, S.; Sedletska, Y.; Reynaud-Angelin, A.; Gasparutto, D.; Sage, E. The formation of double strand breaks at multiply damaged sites is driven by the kinetics of excision/incision at base damage in eukaryotic cells. Nucleic Acids Res. 2009, 37, 1767–1777. [Google Scholar] [CrossRef] [Green Version]
- Malyarchuk, S.; Castore, R.; Harrison, L. DNA repair of clustered lesions in mammalian cells: Involvement of non-homologous end-joining. Nucleic Acids Res. 2008, 36, 4872–4882. [Google Scholar] [CrossRef] [Green Version]
- Malyarchuk, S.; Castore, R.; Harrison, L. Apex1 can cleave complex clustered DNA lesions in cells. DNA Repair 2009, 8, 1343–1354. [Google Scholar] [CrossRef] [Green Version]
- Malyarchuk, S.; Castore, R.; Shi, R.; Harrison, L. Artemis is required to improve the accuracy of repair of double-strand breaks with 5’-blocked termini generated from non-DSB-clustered lesions. Mutagenesis 2013, 28, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Malyarchuk, S.; Brame, K.L.; Youngblood, R.; Shi, R.; Harrison, L. Two clustered 8-oxo-7,8-dihydroguanine (8-oxodG) lesions increase the point mutation frequency of 8-oxodG, but do not result in double strand breaks or deletions in Escherichia coli. Nucleic Acids Res. 2004, 32, 5721–5731. [Google Scholar] [CrossRef] [Green Version]
- Eot-Houllier, G.; Eon-Marchais, S.; Gasparutto, D.; Sage, E. Processing of a complex multiply damaged DNA site by human cell extracts and purified repair proteins. Nucleic Acids Res. 2005, 33, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Eot-Houllier, G.; Gonera, M.; Gasparutto, D.; Giustranti, C.; Sage, E. Interplay between DNA N-glycosylases/APlyases at multiply damaged sites and biological consequences. Nucleic Acids Res. 2007, 35, 3355–3366. [Google Scholar] [CrossRef] [Green Version]
- Gellon, L.; Barbey, R.; Auffret van der Kemp, P.; Thomas, D.; Boiteux, S. Synergism between base excision repair, mediated by the DNA glycosylases Ntg1 and Ntg2, and nucleotide excision repair in the removal of oxidatively damaged DNA bases in Saccharomyces cerevisiae. Mol. Genet. Genom. 2001, 265, 1087–1096. [Google Scholar] [CrossRef]
- Guillet, M.; Boiteux, S. Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces cerevisiae. EMBO J. 2002, 21, 2833–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, M.D.; Winston, F.; Hieter, P. Methods in Yeast Genetics: A Laboratory Course Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1990. [Google Scholar]
- Miller, J.H. Experiments in Molecular Genetics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1972. [Google Scholar]
- Kamiya, H. Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: Approaches using synthetic oligonucleotides and nucleotides: Survey and summary. Nucleic Acids Res. 2003, 31, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Kreutzer, D.A.; Essigmann, J.M. Oxidized, deaminated cytosines are a source of C to T transitions in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 3578–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyabe, I.; Zhang, Q.M.; Sugiyama, H.; Kino, K.; Yonei, S. Mutagenic effects of 5-formyluracil on a plasmid vector during replication in Escherichia coli. Int. J. Radiat. Biol. 2001, 77, 53–58. [Google Scholar] [CrossRef]
- Boiteux, S.; Gellon, L.; Guibourt, N. Repair of 8-oxoguanine in Saccharomyces cerevisiae: Interplay of DNA repair and replication mechanisms. Free Radic. Biol. Med. 2002, 32, 1244–1253. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Ni, T.T.; Marsischky, G.T.; Kolodner, R.D. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol. Cell 1999, 4, 439–444. [Google Scholar] [CrossRef]
- Kamiya, H.; Murata-Kamiya, N.; Karino, N.; Ueno, Y.; Matsuda, A.; Kasai, H. Induction of T > G and T > A transversions by 5-formyluracil in mammalian cells. Mutat. Res. 2002, 513, 213–222. [Google Scholar] [CrossRef]
- Wood, M.L.; Esteve, A.; Morningstar, M.L.; Kuziemko, G.M.; Essigmann, J.M. Genetic effects of oxidative DNA damage: Comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine in Escherichia coli. Nucleic Acids Res. 1992, 20, 6023–6032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, H.; Miura, H.; Murata-Kamiya, N.; Ishikawa, H.; Sakaguchi, T.; Inoue, H.; Sasaki, T.; Masutani, C.; Hanaoka, F.; Nishimura, S.; et al. 8-Hydroxyadenine (7,8-dihydro-8-oxoadenine) induces misincorporation in in vitro DNA synthesis and mutations in NIH 3T3 cells. Nucleic Acids Res. 1995, 23, 2893–2899. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Grollman, A.P.; Shibutani, S. Comparison of the mutagenic properties of 8-oxo-7,8-dihydro-2’-deoxyadenosine and 8-oxo-7,8-dihydro-2’-deoxyguanosine DNA lesions in mammalian cells. Carcinogenesis 1999, 20, 2287–2292. [Google Scholar] [CrossRef] [PubMed]
- Guschlbauer, W.; Duplaa, A.M.; Guy, A.; Teoule, R.; Fazakerley, G.V. Structure and in vitro replication of DNA templates containing 7,8-dihydro-8-oxoadenine. Nucleic Acids Res. 1991, 19, 1753–1758. [Google Scholar] [CrossRef] [Green Version]
- Koag, M.C.; Jung, H.; Lee, S. Mutagenic Replication of the Major Oxidative Adenine Lesion 7,8-Dihydro-8-oxoadenine by Human DNA Polymerases. J. Am. Chem. Soc. 2019, 141, 4584–4596. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.; Calvayrac, G.; Karahalil, B.; Bohr, V.A.; Stevnsner, T. Mammalian 8-oxoguanine DNA glycosylase 1 incises 8-oxoadenine opposite cytosine in nuclei and mitochondria, while a different glycosylase incises 8-oxoadenine opposite guanine in nuclei. J. Biol. Chem. 2003, 278, 19541–19548. [Google Scholar] [CrossRef] [Green Version]
- Talhaoui, I.; Couvé, S.; Ishchenko, A.A.; Kunz, C.; Schär, P.; Saparbaev, M. 7,8-dihydro-8-oxoadenine, a highly mutagenic adduct, is repaired by Escherichia coli and human mismatch-specific uracil/thymine-DNA glycosylases. Nucleic Acids Res. 2013, 41, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Girard, P.M.; D’Ham, C.; Cadet, J.; Boiteux, S. Opposite base-dependent excision of 7,8-dihydro-8-oxoadenine by the Ogg1 protein of Saccharomyces cerevisiae. Carcinogenesis 1998, 19, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Sentuürker, S.; Auffret van der Kemp, P.; You, H.J.; Doetsch, P.W.; Dizdaroglu, M.; Boiteux, S. Substrate specificities of the Ntg1 and Ntg2 proteins of Saccharomyces cerevisiae for oxidized DNA bases are not identical. Nucleic Acids Res. 1998, 26, 5270–5276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zastawny, T.H.; Doetsch, P.W.; Dizdaroglu, M. A novel activity of E. coli uracil DNA N-glycosylase excision of isodialuric acid (5,6-dihydroxyuracil), a major product of oxidative DNA damage, from DNA. FEBS Lett. 1995, 364, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.L.; Morey, N.J.; Doetsch, P.W.; Jinks-Robertson, S. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 2929–2935. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.E.; Hickson, I.D.; Boiteux, S.; Radicella, J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res. 2001, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Esadze, A.; Rodriguez, G.; Cravens, S.L.; Stivers, J.T. AP-Endonuclease 1 Accelerates Turnover of Human 8-Oxoguanine DNA Glycosylase by Preventing Retrograde Binding to the Abasic- Site Product. Biochemistry 2017, 56, 1974–1986. [Google Scholar] [CrossRef] [PubMed]
- Marenstein, D.R.; Chan, M.K.; Altamirano, A.; Basu, A.K.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Substrate specificity of human endonuclease III (hNTH1). Effect of human APE1 on hNTH1 activity. J. Biol. Chem. 2001, 278, 9005–9012. [Google Scholar] [CrossRef] [Green Version]
- Gros, L.; Ishchenko, A.A.; Ide, H.; Elder, R.H.; Saparbaev, M.K. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res. 2004, 32, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.M.; Hashiguchi, K.; Kino, K.; Sugiyama, H.; Yonei, S. Ntg1 and Ntg2 proteins as 5-formyluracil-DNA glycosylases/AP lyases in Saccharomyces cerevisiae. Int. J. Radiat. Biol. 2003, 79, 341–349. [Google Scholar] [CrossRef]
- Kelley, M.R.; Kow, Y.W.; Wilson, D.M., 3rd. Disparity between DNA base excision repair in yeast and mammals: Translational implications. Cancer Res. 2003, 63, 549–554. [Google Scholar]
- Caldecott, K.W. Mammalian DNA single-strand break repair: An X-ra(y)ted affair. Bioessays 2001, 23, 447–455. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S.D.; Kokoska, R.J.; Garg, P.; Burgers, P.M.; Kunkel, T.A. The efficiency and fidelity of 8-oxo-guanine bypass by DNA polymerases δ and ε. Nucleic Acids Res. 2009, 37, 2830–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budworth, H.; Dianova, I.I.; Podust, V.N.; Dianov, G.L. Repair of clustered DNA lesions Sequence-specific inhibition of long-patch base excision repair be 8-oxoguanine. J. Biol. Chem. 2002, 277, 21300–21305. [Google Scholar] [CrossRef] [Green Version]
- Moscariello, M.; Sutherland, B. Saccharomyces cerevisiae-based system for studying clustered DNA damages. Radiat. Environ. Biophys. 2010, 49, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Cam, E.; Fack, F.; Ménissier-de Murcia, J.; Cognet, J.; Barbin, A.; Sarantoglou, V.; Revet, B.; Delain, E.; de Murcia, G. Conformational analysis of a 139 base-pair DNA fragment containing a single-stranded break and its interaction with human Poly(ADP-ribose) polymerase. J. Mol. Biol. 1994, 235, 1062–1071. [Google Scholar] [CrossRef]
- Imoto, S.; Bransfield, L.A.; Croteau, D.L.; Van Houten, B.; Greenberg, M.M. DNA tandem lesion repair by strand displacement synthesis and nucleotide excision repair. Biochemistry 2008, 47, 4306–4316. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ding, S.; Kropachev, K.; Jia, L.; Amin, S.; Broyde, S.; Geacintov, N.E. Resistance to Nucleotide Excision Repair of Bulky Guanine Adducts Opposite Abasic Sites in DNA Duplexes and Relationships between Structure and Function. PLoS ONE 2015, 10, e0137124. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Cunniffe, S.; O’Neill, P. 8-OxoG retards the activity of the ligase III/XRCC1 complex during the repair of a single-strand break, when present within a clustered DNA damage site. DNA Repair 2004, 3, 289–299. [Google Scholar] [CrossRef]
- Wardle, J.; Burgers, P.M.; Cann, I.K.; Darley, K.; Heslop, P.; Johansson, E.; Lin, L.J.; McGlynn, P.; Sanvoisin, J.; Stith, C.M.; et al. Uracil recognition by replicative DNA polymerases is limited to the archaea, not occurring with bacteria and eukarya. Nucleic Acids Res. 2008, 36, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, S.M.; Lomax, M.E.; O’Neill, P. An AP site can protect against the mutagenic potential of 8-oxoG when present within a tandem clustered site in E. coli. DNA Repair 2007, 6, 1839–1849. [Google Scholar] [CrossRef]
- Cappelli, E.; Hazra, T.; Hill, J.W.; Slupphaug, G.; Bogliolo, M.; Frosina, G. Rates of base excision repair are not solely dependent on levels of initiating enzymes. Carcinogenesis 2001, 22, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Shikazono, N.; Pearson, C.; O’Neill, P.; Thacker, J. The roles of specific glycosylases in determining the mutagenic consequences of clustered DNA base damage. Nucleic Acids Res. 2006, 34, 3722–3730. [Google Scholar] [CrossRef]
- Bellon, S.; Shikazono, N.; Cunniffe, S.; Lomax, M.; O’Neill, P. Processing of thymine glycol in a clustered DNA damage site: Mutagenic or cytotoxic. Nucleic Acids Res. 2009, 37, 4430–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, S.S. Molecular radiobiology and the origins of the base excision repair pathway: An historical perspective. Int. J. Radiat. Biol. 2021, 72, 1–12. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A). Mutations at oG. | ||||
| Sequence at oG•C Site | SDS-oG | IMDS-oG/hU | MDS1 | MDS2 |
| oG•C -> G•C (“WT”) | 89 | 23 | 63 | 26 |
| oG•C -> T•A | 2 | 3 | 13 | 0 |
| oG•C -> Δ | 2 | 5 | 5 | 0 |
| Total mutations | 4 | 8 | 18 | 0 |
| Total clones | 93 | 31 | 84 | 26 |
| Mutation frequency | 4% | 26% | 21% | <4% |
| (B). Mutations at hU. | ||||
| Sequence at hU•G Site | SDS-hU | IMDS-oG/hU | MDS1 | MDS2 |
| hU•G -> C•G (“WT”) | 83 | 28 | 74 | 8 |
| hU•G -> T•A | 9 | 3 | 9 | 17 |
| hU•G -> Δ | 1 | 0 | 1 | 0 |
| hU•G -> G•C | 0 | 0 | 0 | 1 |
| Total mutations | 10 | 3 | 10 | 18 |
| Total clones | 93 | 31 | 84 | 26 |
| Mutation frequency | 11% | 10% | 12% | 69% |
| (C). Mutations at fU. | ||||
| Sequence at fU•A Site | SDS-fU | MDS1 | MDS2 | |
| fU•A -> T•A (“WT”) | 89 | 76 | 26 | |
| fU•A -> A•T | 1 | 5 | 0 | |
| fU•A -> Δ | 4 | 3 | 0 | |
| Total mutations | 5 | 8 | 0 | |
| Total clones | 94 | 84 | 26 | |
| Mutation frequency | 5% | 10% | <4% | |
| (A). Mutations at oG. | |||||||
| MDS1 | SDS-oG | ||||||
| Sequence at oG•C Site | WT | BER- | ntg1 ntg2 | rad14 | rad14 ntg1 ntg2 | rev3 | WT |
| oG•C -> G•C (“WT”) | 66 | 68 | 70 | 77 | 44 | 58 | 89 |
| oG•C -> T•A | 13 | 4 | 6 | 8 | 4 | 9 | 2 |
| oG•C -> Δ | 5 | 7 | 5 | 7 | 2 | 6 | 2 |
| oG•C -> C•G | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Total mutations | 18 | 12 | 11 | 15 | 6 | 15 | 4 |
| Total clones | 84 | 80 | 81 | 92 | 50 | 73 | 93 |
| Mutation frequency | 21% | 15% | 14% | 16% | 12% | 21% | 4% |
| (B). Mutations at hU. | |||||||
| MDS1 | SDS-hU | ||||||
| Sequence at hU•G Site | WT | BER- | ntg1 ntg2 | rad14 | rad14 ntg1 ntg2 | rev3 | WT |
| hU•G -> C•G (“WT”) | 74 | 60 | 62 | 81 | 41 | 66 | 83 |
| hU•G -> T•A | 9 | 20 | 18 | 7 | 9 | 5 | 9 |
| hU•G -> Δ | 1 | 0 | 0 | 4 | 0 | 2 | 1 |
| hU•G -> G•C | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Total mutations | 10 | 20 | 19 | 11 | 9 | 7 | 10 |
| Total clones | 84 | 80 | 81 | 92 | 50 | 73 | 93 |
| Mutation frequency | 12% | 25% | 24% | 18% | 18% | 10% | 11% |
| (C). Mutations at fU. | |||||||
| MDS1 | SDS-fU | ||||||
| Sequence at fU•A Site | WT | BER- | ntg1 ntg2 | rad14 | rad14 ntg1 ntg2 | rev3 | WT |
| fU•A -> T•A (“WT”) | 76 | 72 | 73 | 88 | 44 | 72 | 89 |
| fU•A -> A•T | 5 | 6 | 6 | 3 | 4 | 0 | 1 |
| fU•A -> Δ | 3 | 2 | 2 | 1 | 1 | 1 | 4 |
| fU•A -> G•C | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Total mutations | 8 | 8 | 8 | 4 | 6 | 1 | 5 |
| Total clones | 84 | 80 | 81 | 92 | 50 | 73 | 94 |
| Mutation frequency | 10% | 10% | 10% | 4% | 12% | 1% | 5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozmin, S.G.; Eot-Houllier, G.; Reynaud-Angelin, A.; Gasparutto, D.; Sage, E. Dissecting Highly Mutagenic Processing of Complex Clustered DNA Damage in Yeast Saccharomyces cerevisiae. Cells 2021, 10, 2309. https://doi.org/10.3390/cells10092309
Kozmin SG, Eot-Houllier G, Reynaud-Angelin A, Gasparutto D, Sage E. Dissecting Highly Mutagenic Processing of Complex Clustered DNA Damage in Yeast Saccharomyces cerevisiae. Cells. 2021; 10(9):2309. https://doi.org/10.3390/cells10092309
Chicago/Turabian StyleKozmin, Stanislav G., Gregory Eot-Houllier, Anne Reynaud-Angelin, Didier Gasparutto, and Evelyne Sage. 2021. "Dissecting Highly Mutagenic Processing of Complex Clustered DNA Damage in Yeast Saccharomyces cerevisiae" Cells 10, no. 9: 2309. https://doi.org/10.3390/cells10092309