
Haloperidol Attenuates Lung Endothelial Cell Permeability In Vitro and In Vivo

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Endothelial Cell Culture
2.3. Immunoblotting
2.4. Transendothelial Electrical Resistance (TER) Measurement
2.5. Transwell Permeability Assay
2.6. Murine ALI Model
2.7. Statistics
3. Results
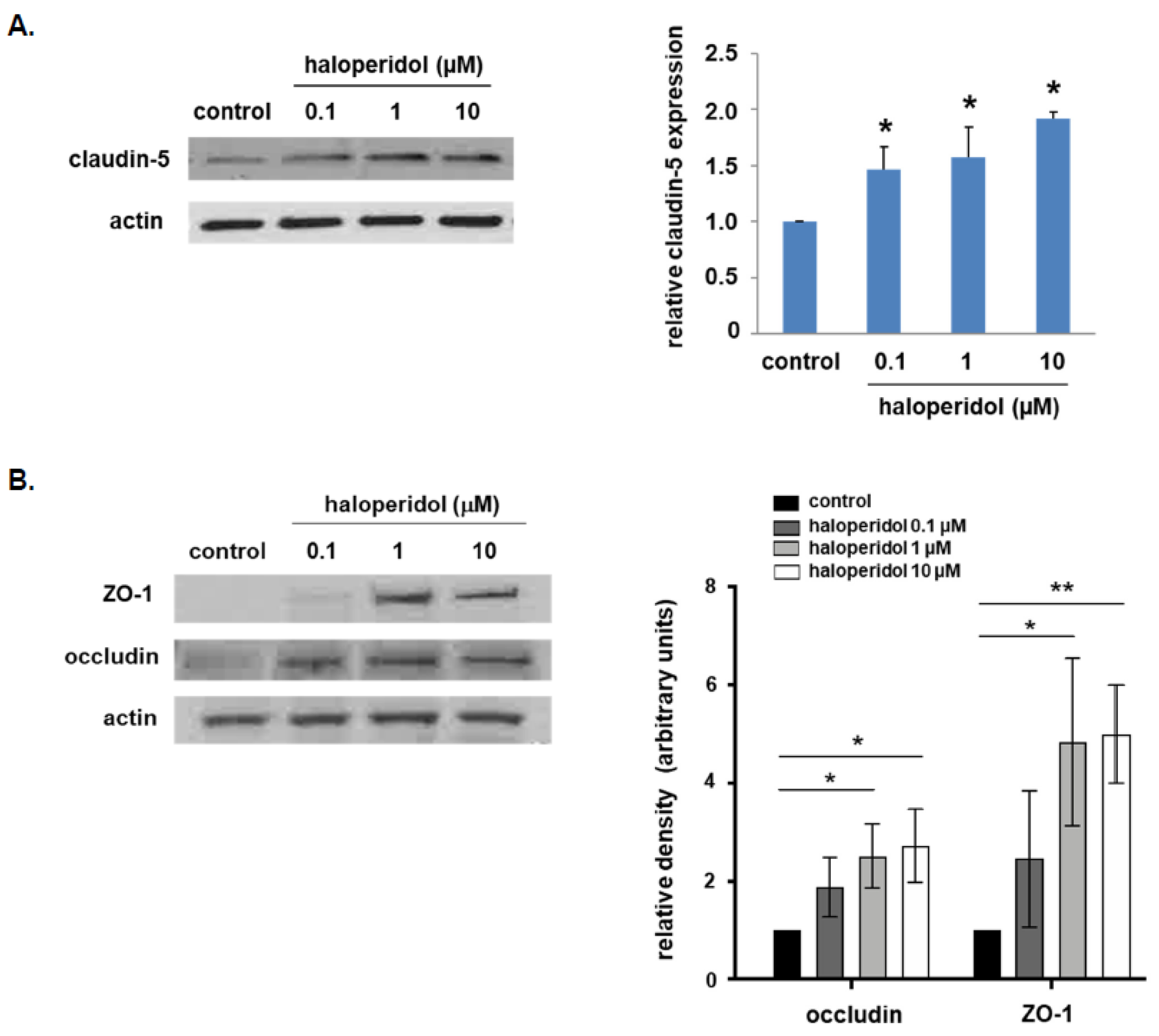
3.1. Effect of Haloperidol on Lung EC Expression of Tight Junctional Proteins
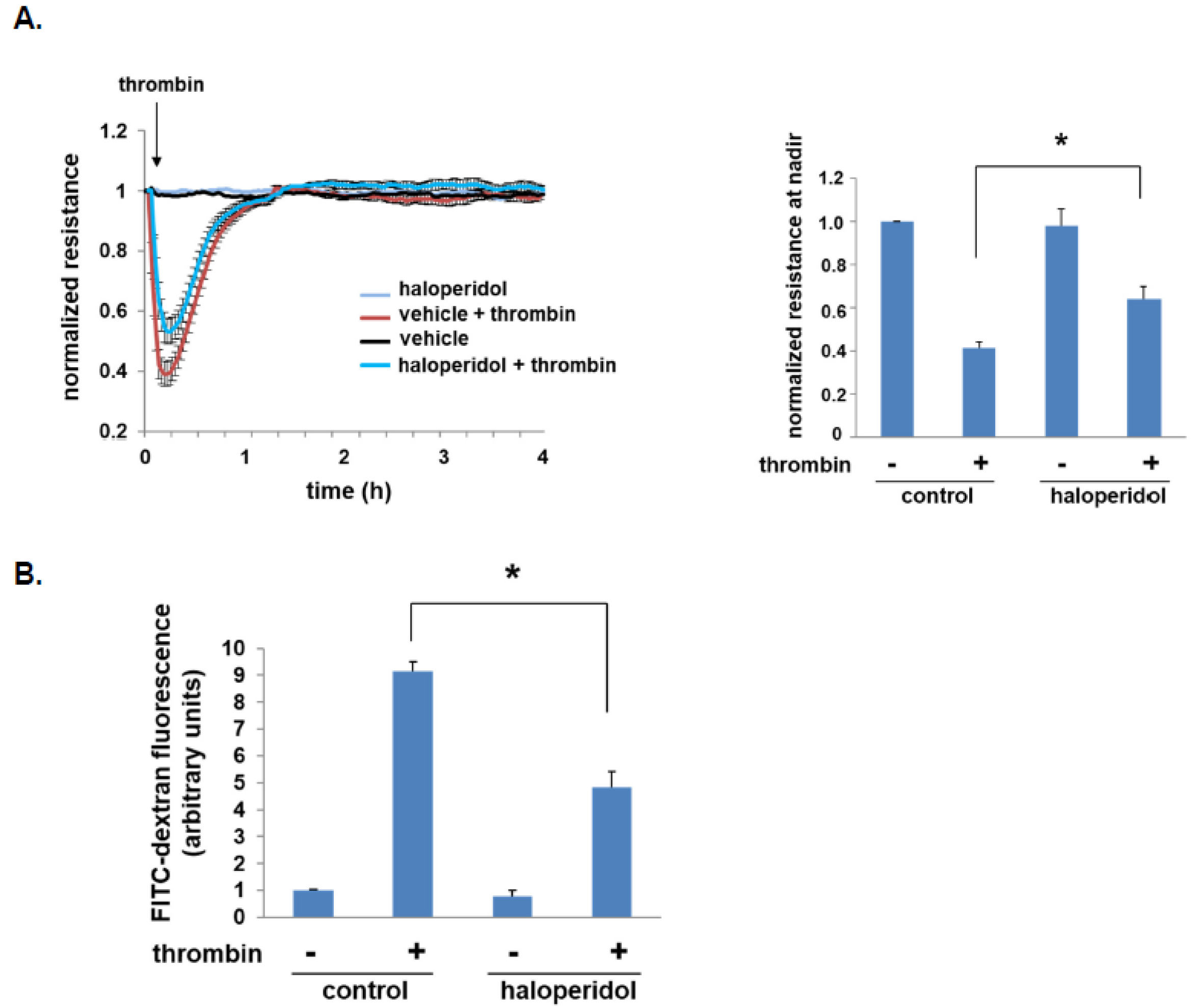
3.2. Effect of Haloperidol on Lung EC Barrier Function
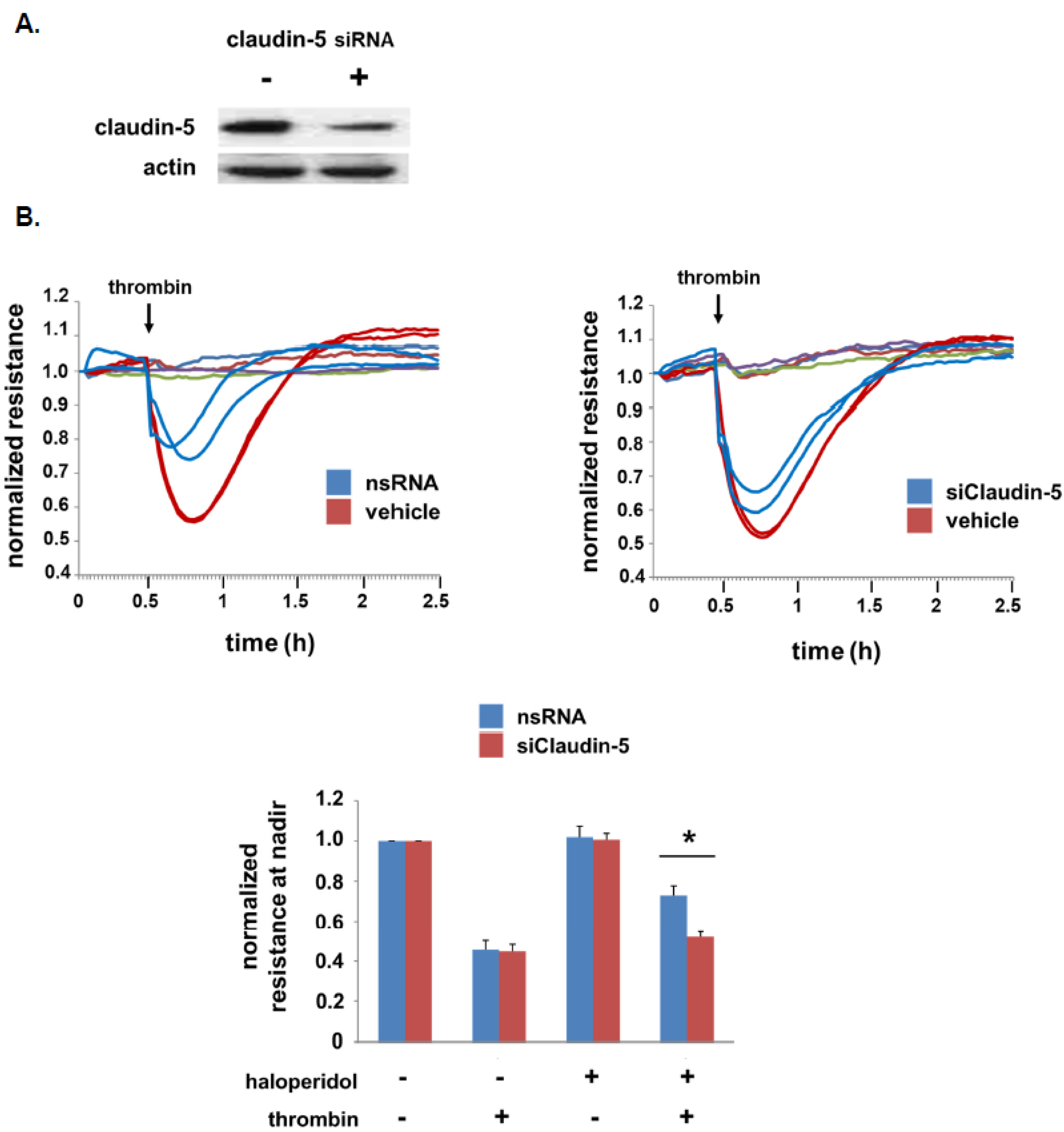
3.3. Role of Claudin-5 in Lung EC Barrier Protection by Haloperidol
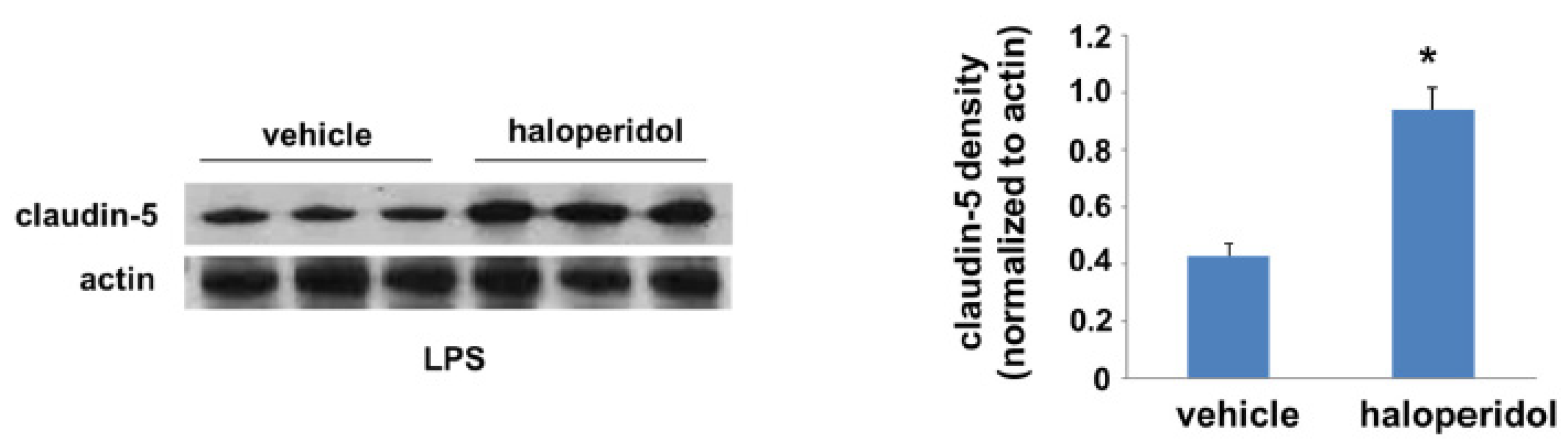
3.4. Effect of Haloperidol on Lung Claudin-5 Expression In Vivo
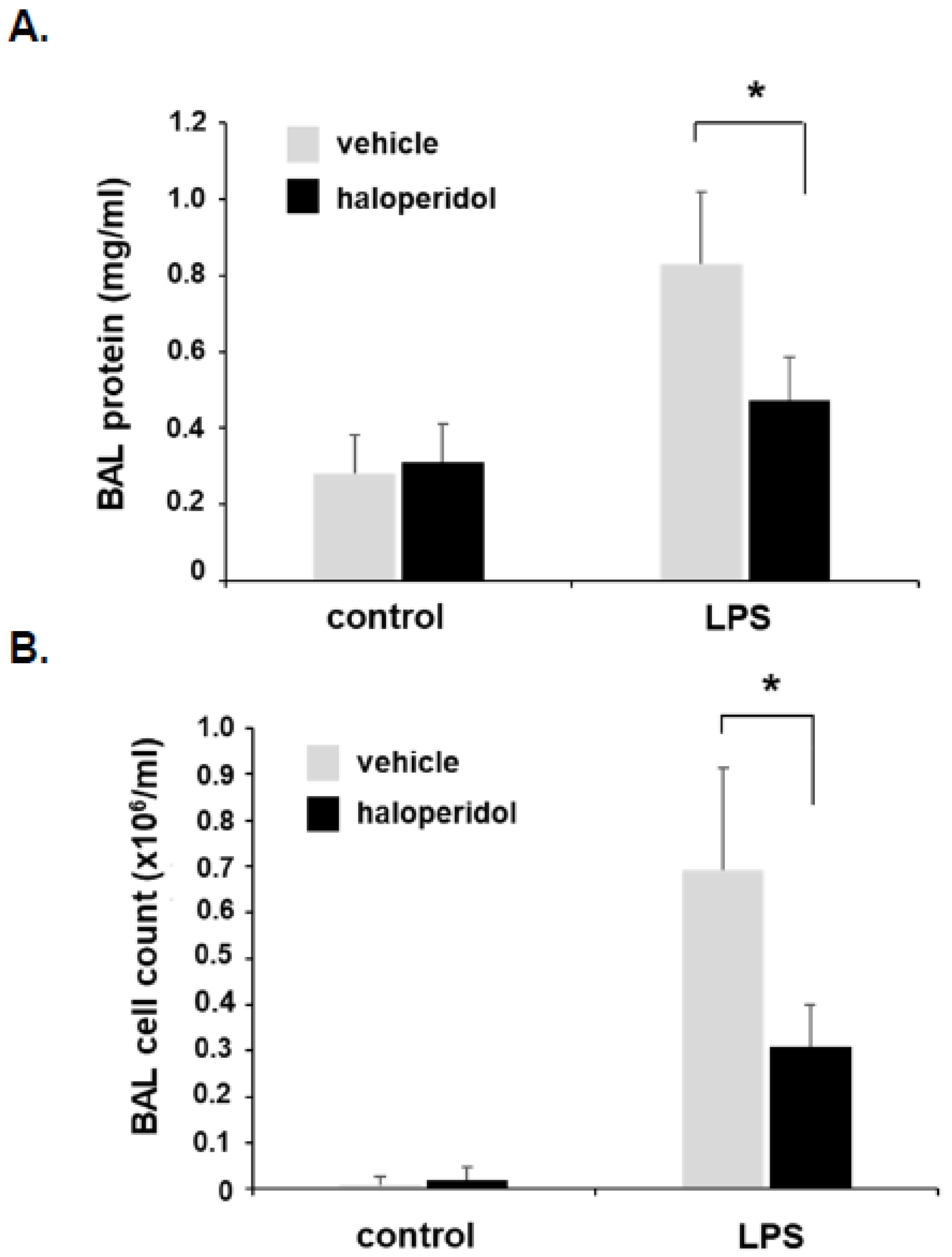
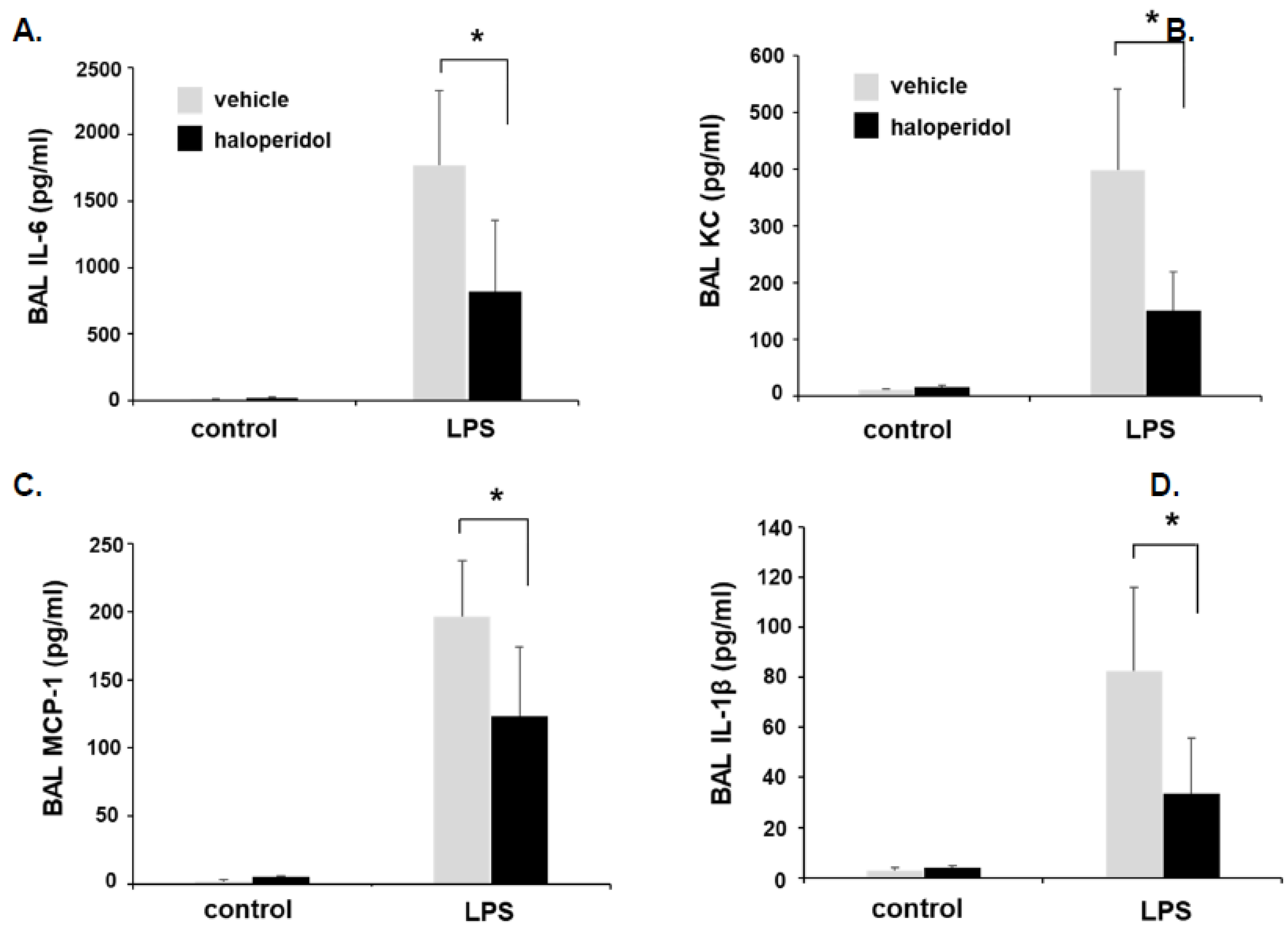
3.5. Effect of Haloperidol on Murine ALI: BAL Fluid Analysis
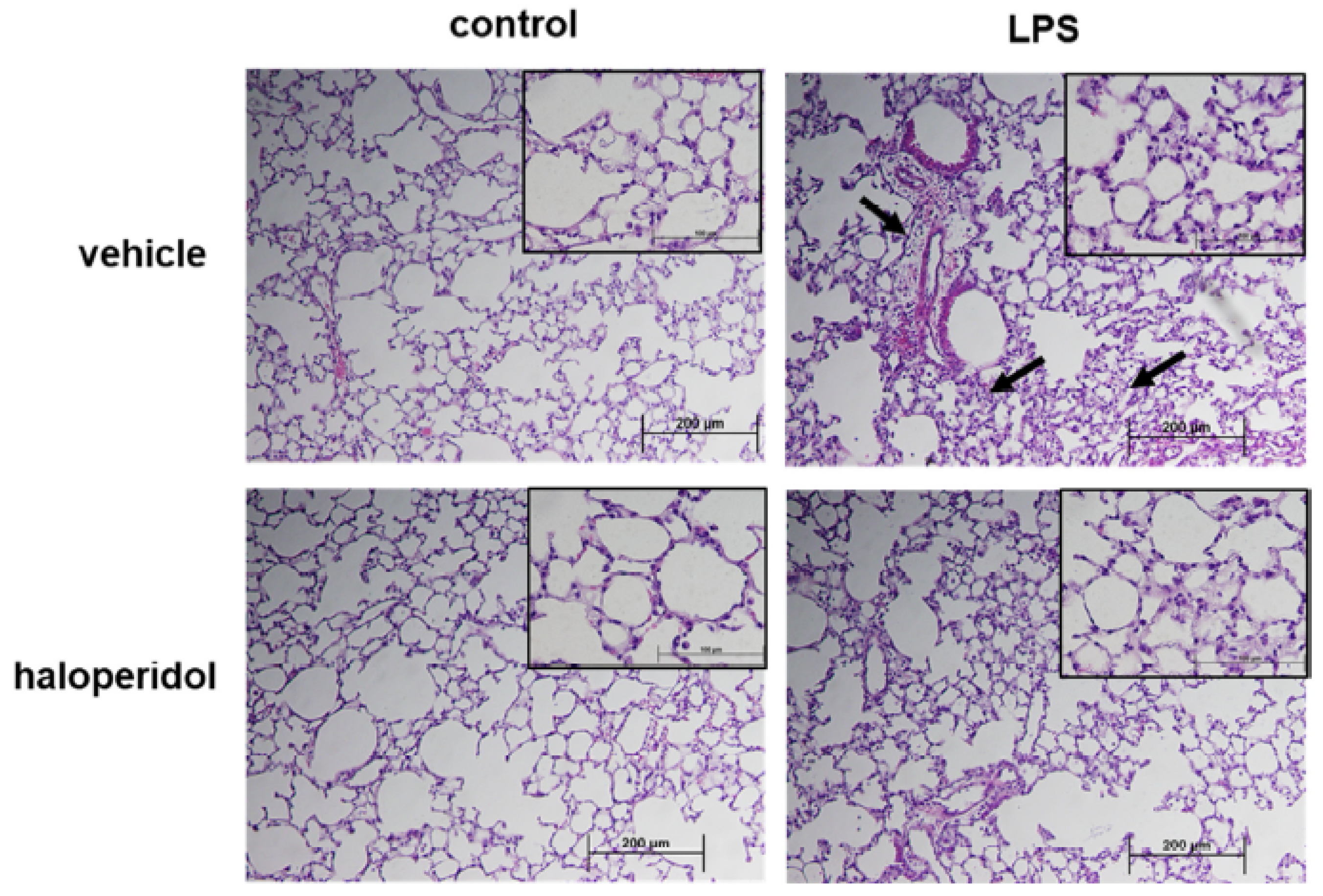
3.6. Effect of Haloperidol on Murine ALI: Lung Histology
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALI | acute lung injury |
| ARDS | acute respiratory distress syndrome |
| BAL | bronchoalveolar lavage |
| COVID-19 | coronavirus infectious disease 2019 |
| EC | endothelial cell |
| GSK3β | glycogen synthase kinase-3 beta |
| LPS | lipopolysaccharide |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| TER | transendothelial resistance |
| ZO-1 | zona occludens-1 |
References
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Tzotzos, S.J.; Fischer, B.; Fischer, H.; Zeitlinger, M. Incidence of ARDS and outcomes in hospitalized patients with COVID-19: A global literature survey. Crit. Care 2020, 24, 516. [Google Scholar] [CrossRef] [PubMed]
- Acute Respiratory Distress Syndrome, N.; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar]
- Guerin, C.; Reignier, J.; Richard, J.C. Prone positioning in the acute respiratory distress syndrome. N. Engl. J. Med. 2013, 369, 980–981. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 2011, 6, 147–163. [Google Scholar] [CrossRef] [Green Version]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sharma, R.; Rizzo, A.N.; Siegler, J.H.; Garcia, J.G.; Jacobson, J.R. Role of claudin-5 in the attenuation of murine acute lung injury by simvastatin. Am. J. Respir. Cell Mol. Biol. 2014, 50, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Kakogiannos, N.; Ferrari, L.; Giampietro, C.; Scalise, A.A.; Maderna, C.; Rava, M.; Taddei, A.; Lampugnani, M.G.; Pisati, F.; Malinverno, M.; et al. JAM-A Acts via C/EBP-alpha to Promote Claudin-5 Expression and Enhance Endothelial Barrier Function. Circ. Res. 2020, 127, 1056–1073. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Kealy, J.; Humphries, M.M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.M.; Martiniano, R.; Shashi, V.; Hooper, S.R.; et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef]
- Chen, W.; Sammani, S.; Mitra, S.; Ma, S.F.; Garcia, J.G.; Jacobson, J.R. Critical role for integrin-beta4 in the attenuation of murine acute lung injury by simvastatin. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L279–L285. [Google Scholar] [CrossRef] [Green Version]
- Van den Boogaard, M.; Slooter, A.J.C.; Bruggemann, R.J.M.; Schoonhoven, L.; Beishuizen, A.; Vermeijden, J.W.; Pretorius, D.; de Koning, J.; Simons, K.S.; Dennesen, P.J.W.; et al. Effect of Haloperidol on Survival Among Critically Ill Adults With a High Risk of Delirium: The REDUCE Randomized Clinical Trial. JAMA 2018, 319, 680–690. [Google Scholar] [CrossRef]
- Riker, R.R.; Fraser, G.L.; Cox, P.M. Continuous infusion of haloperidol controls agitation in critically ill patients patients. Crit. Care Med. 1994, 22, 433–440. [Google Scholar] [CrossRef]
- Page, V.J.; Ely, E.W.; Gates, S.; Zhao, X.B.; Alce, T.; Shintani, A.; Jackson, J.; Perkins, G.D.; McAuley, D.F. Effect of intravenous haloperidol on the duration of delirium and coma in critically ill patients (Hope-ICU): A randomised, double-blind, placebo-controlled trial. Lancet. Respir. Med. 2013, 1, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Girard, T.D.; Exline, M.C.; Carson, S.S.; Hough, C.L.; Rock, P.; Gong, M.N.; Douglas, I.S.; Malhotra, A.; Owens, R.L.; Feinstein, D.J.; et al. Haloperidol and Ziprasidone for Treatment of Delirium in Critical Illness. N. Engl. J. Med. 2018, 379, 2506–2516. [Google Scholar] [CrossRef]
- Crowley, J.J.; Adkins, D.E.; Pratt, A.L.; Quackenbush, C.R.; van den Oord, E.J.; Moy, S.S.; Wilhelmsen, K.C.; Cooper, T.B.; Bogue, M.A.; McLeod, H.L.; et al. Antipsychotic-induced vacuous chewing movements and extrapyramidal side effects are highly heritable in mice. Pharm. J. 2012, 12, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Zaworski, J.; Delannoy, P.Y.; Boussekey, N.; Thellier, D.; Georges, H.; Leroy, O. Lithium: One drug, five complications. J. Intensive Care 2017, 5, 70. [Google Scholar] [CrossRef] [Green Version]
- Vodovar, D.; El Balkhi, S.; Curis, E.; Deye, N.; Megarbane, B. Lithium poisoning in the intensive care unit: Predictive factors of severity and indications for extracorporeal toxin removal to improve outcome. Clin. Toxicol. (Phila.) 2016, 54, 615–623. [Google Scholar] [CrossRef]
- Burek, M.; Arias-Loza, P.A.; Roewer, N.; Forster, C.Y. Claudin-5 as a novel estrogen target in vascular endothelium. Arter. Thromb. Vasc. Biol. 2010, 30, 298–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breithaupt-Faloppa, A.C.; Thais Fantozzi, E.; Romero, D.C.; Rodrigues, A.d.S.; de Sousa, P.T.; Lino Dos Santos Franco, A.; Oliveira-Filho, R.M.; Boris Vargaftig, B.; Tavares de Lima, W. Acute effects of estradiol on lung inflammation due to intestinal ischemic insult in male rats. Shock 2014, 41, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Speyer, C.L.; Rancilio, N.J.; McClintock, S.D.; Crawford, J.D.; Gao, H.; Sarma, J.V.; Ward, P.A. Regulatory effects of estrogen on acute lung inflammation in mice. Am. J. Physiol. Cell Physiol. 2005, 288, C881–C890. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; He, J.; Wang, D.; Deng, W.; Zhao, Y.; Ye, Y.; Feng, L. 17beta-estradiol suppresses lipopolysaccharide-induced acute lung injury through PI3K/Akt/SGK1 mediated up-regulation of epithelial sodium channel (ENaC) in vivo and in vitro. Respir Res. 2014, 15, 159. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, S.A.; Dickman, K.G.; Berisha, H.; Said, S.I. 17beta-estradiol protects the lung against acute injury: Possible mediation by vasoactive intestinal polypeptide. Endocrinology 2011, 152, 4729–4737. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Zhao, P.; Li, J.; Xie, X.; Xu, M.; Zhang, Y.; Mu, D.; Li, W.; Sun, R.; Liu, W.; et al. 17beta-Estradiol administration attenuates seawater aspiration-induced acute lung injury in rats. Pulm. Pharmacol. Ther. 2011, 24, 673–681. [Google Scholar] [CrossRef]
- Moss, M.; Mannino, D.M. Race and gender differences in acute respiratory distress syndrome deaths in the United States: An analysis of multiple-cause mortality data (1979–1996). Crit. Care Med. 2002, 30, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Huang, X.F.; Deng, C. Aripiprazole and Haloperidol Activate GSK3beta-Dependent Signalling Pathway Differentially in Various Brain Regions of Rats. Int. J. Mol. Sci. 2016, 17, 459. [Google Scholar] [CrossRef]
- Koppel, J.; Jimenez, H.; Adrien, L.; Greenwald, B.S.; Marambaud, P.; Cinamon, E.; Davies, P. Haloperidol inactivates AMPK and reduces tau phosphorylation in a tau mouse model of Alzheimer’s disease. Alzheimers Dement. (NY) 2016, 2, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.W.; Jiang, S.; Liu, Y.; Siegal, G.P.; Inoki, K.; Abraham, E.; Zmijewski, J.W. GSK3beta-dependent inhibition of AMPK potentiates activation of neutrophils and macrophages and enhances severity of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L735–L745. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colamonici, M.A.; Epshtein, Y.; Chen, W.; Jacobson, J.R. Haloperidol Attenuates Lung Endothelial Cell Permeability In Vitro and In Vivo. Cells 2021, 10, 2186. https://doi.org/10.3390/cells10092186
Colamonici MA, Epshtein Y, Chen W, Jacobson JR. Haloperidol Attenuates Lung Endothelial Cell Permeability In Vitro and In Vivo. Cells. 2021; 10(9):2186. https://doi.org/10.3390/cells10092186
Chicago/Turabian StyleColamonici, Marco A., Yulia Epshtein, Weiguo Chen, and Jeffrey R. Jacobson. 2021. "Haloperidol Attenuates Lung Endothelial Cell Permeability In Vitro and In Vivo" Cells 10, no. 9: 2186. https://doi.org/10.3390/cells10092186