
Group V Phospholipase A2 Mediates Endothelial Dysfunction and Acute Lung Injury Caused by Methicillin-Resistant Staphylococcus Aureus

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
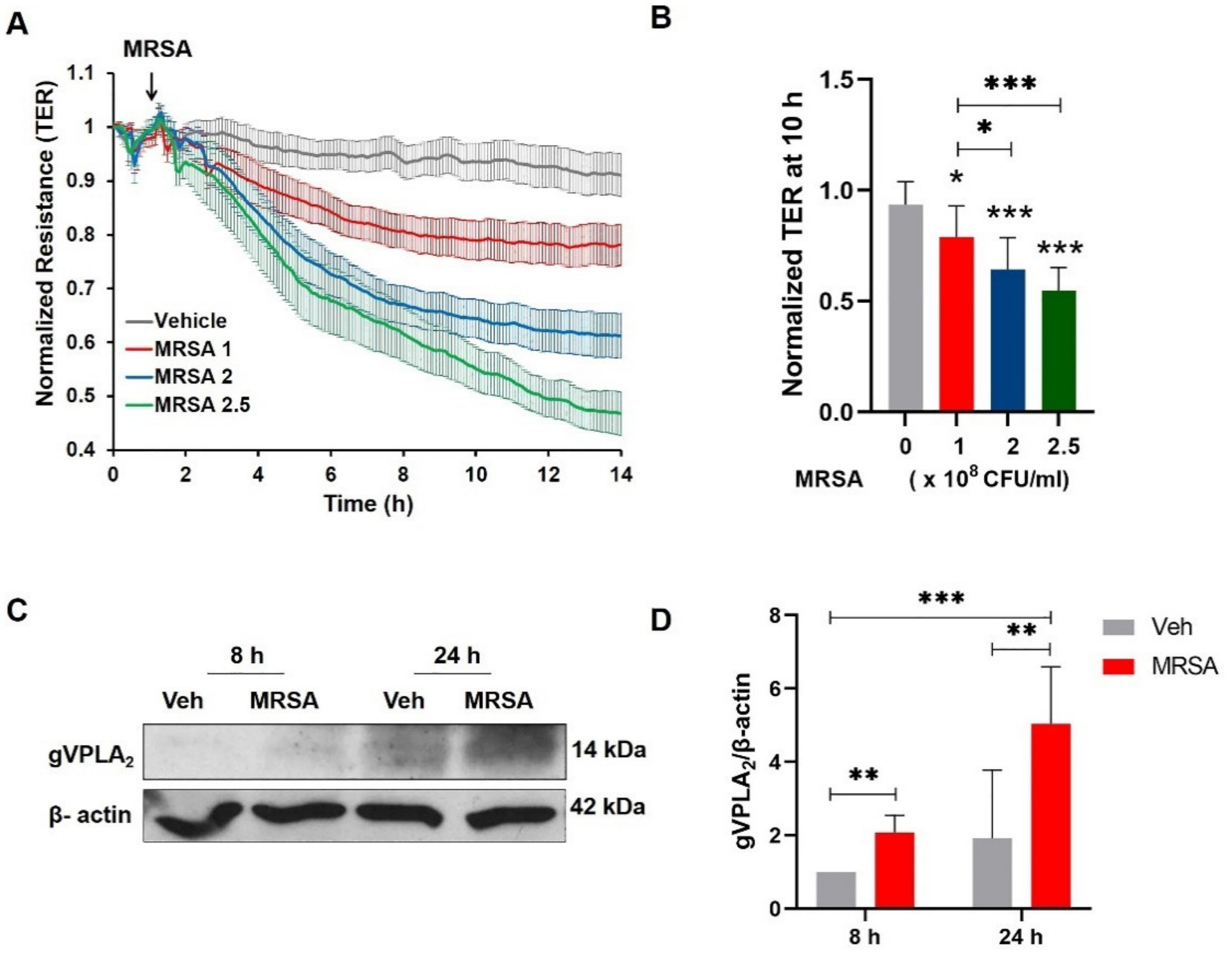
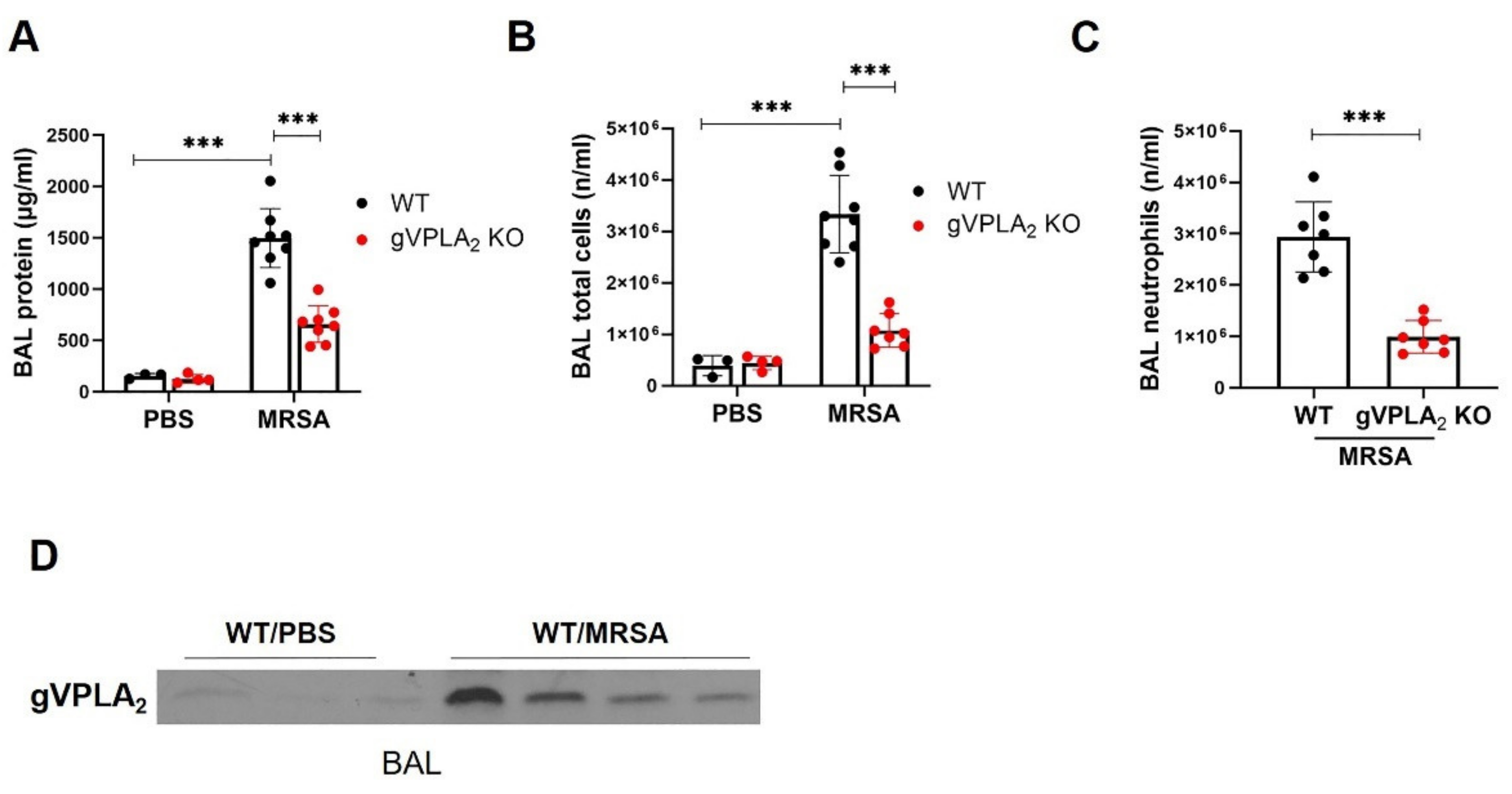
3.1. MRSA Induces EC Barrier Dysfunction and Increases the Expression of gVPLA2
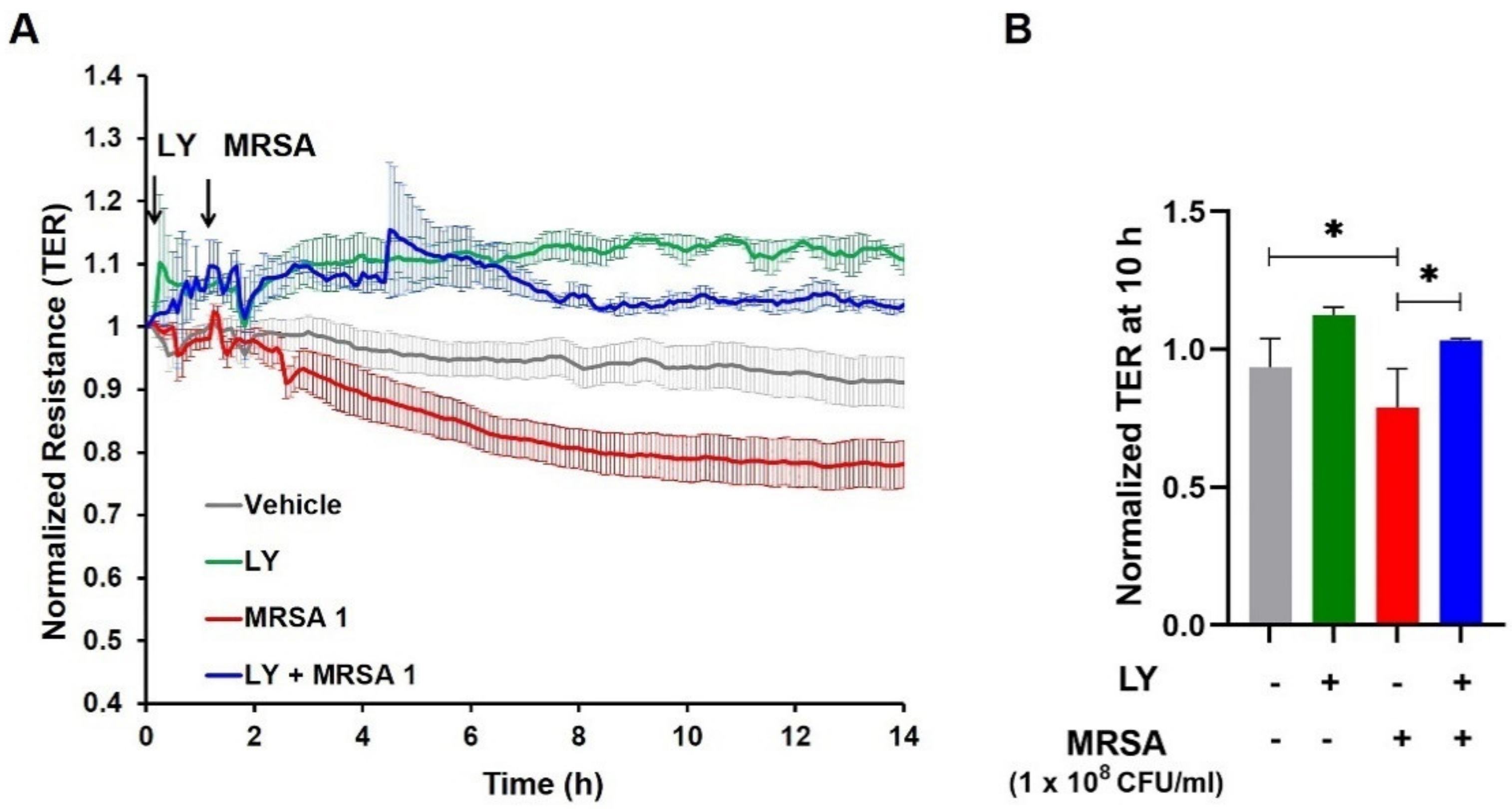
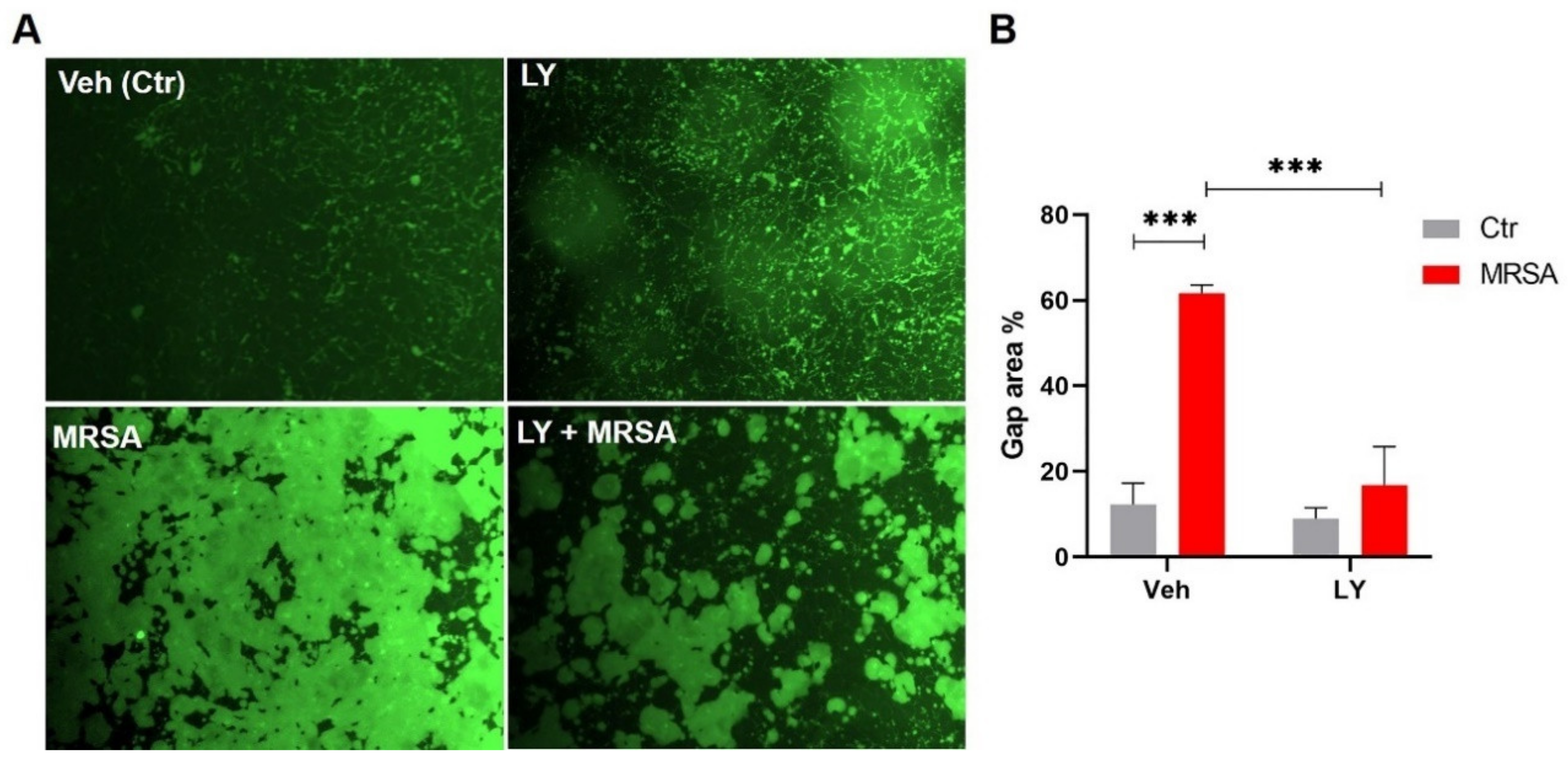
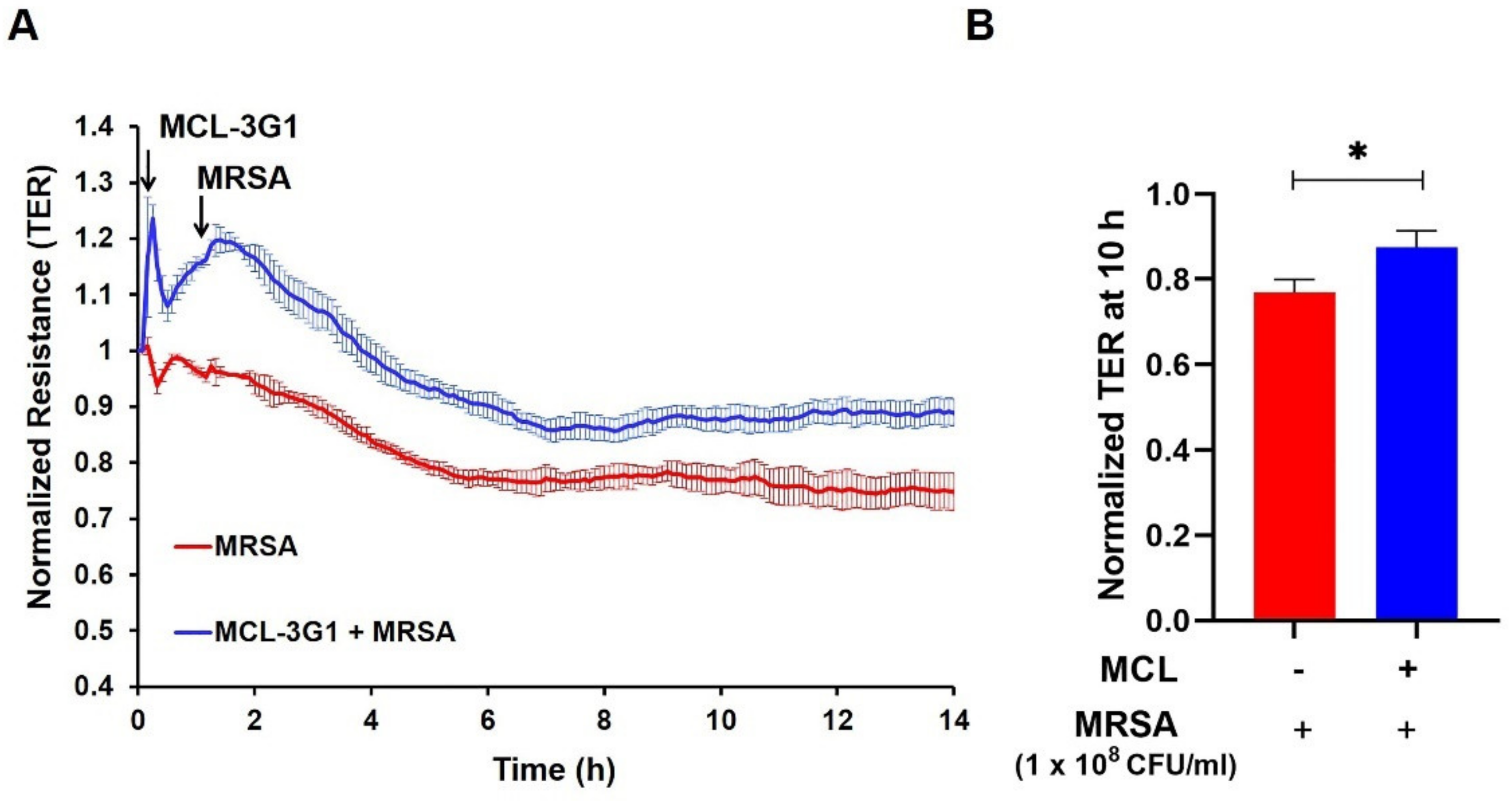
3.2. Inhibition of gVPLA2 Attenuates MRSA-Induced Lung EC Permeability
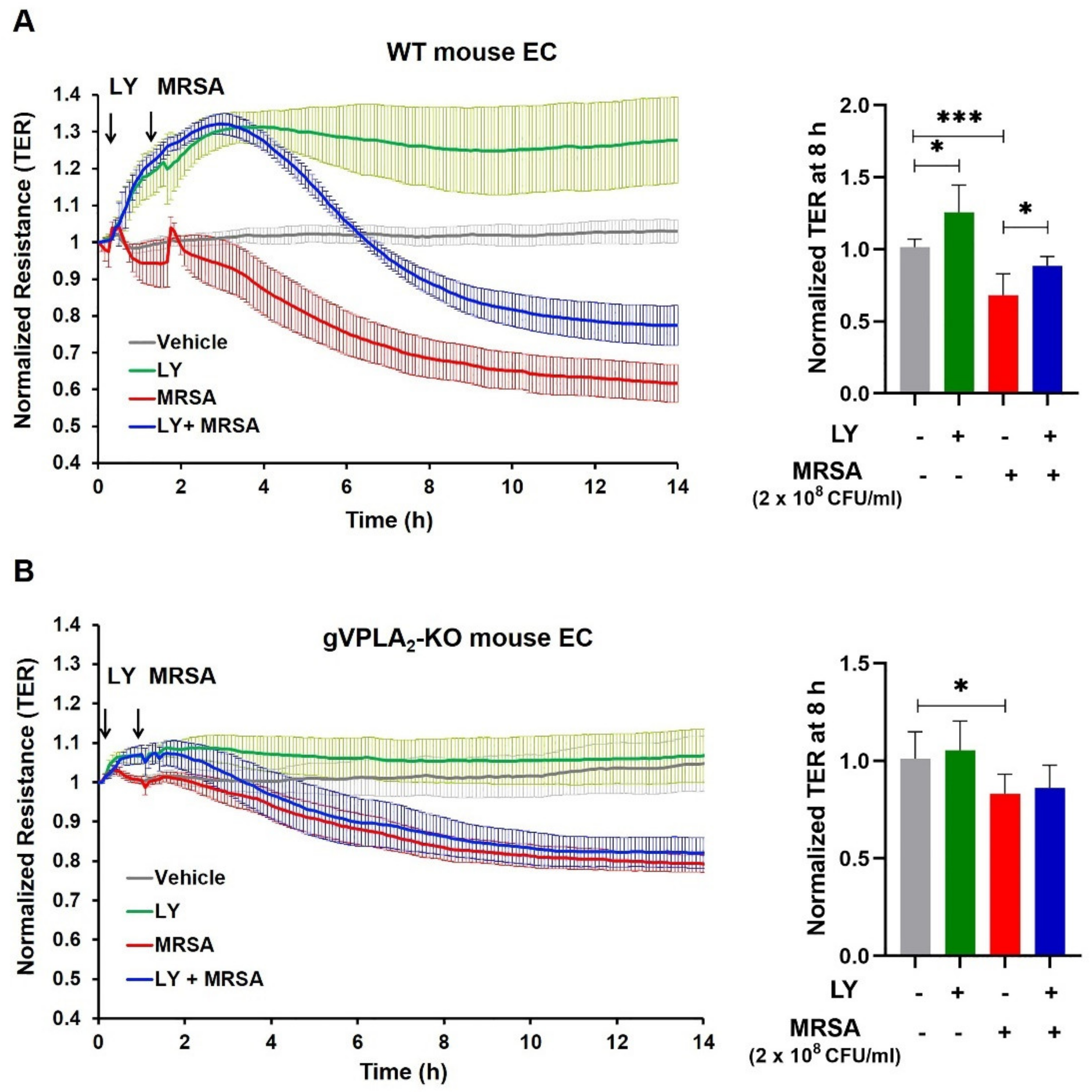
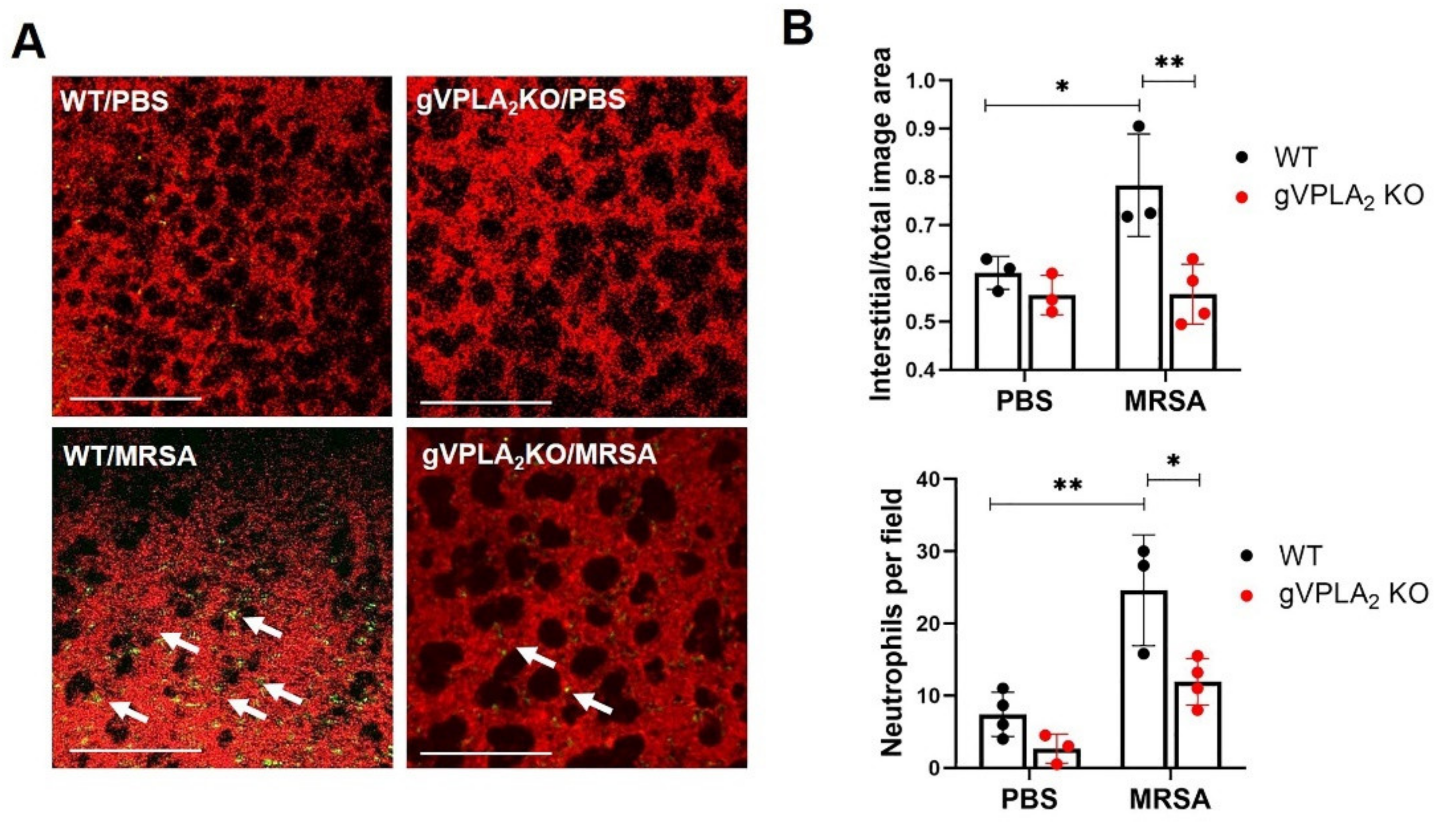
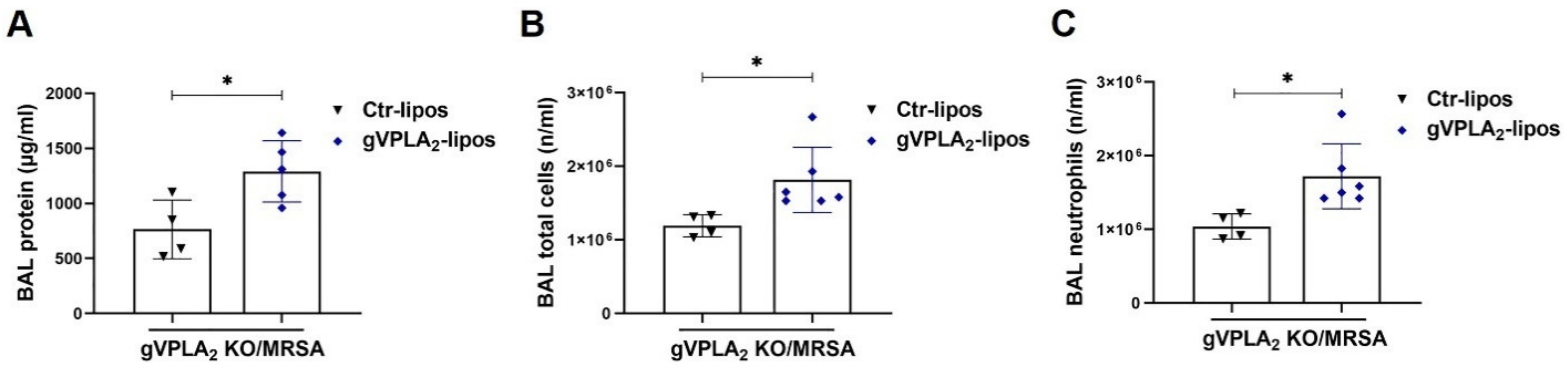
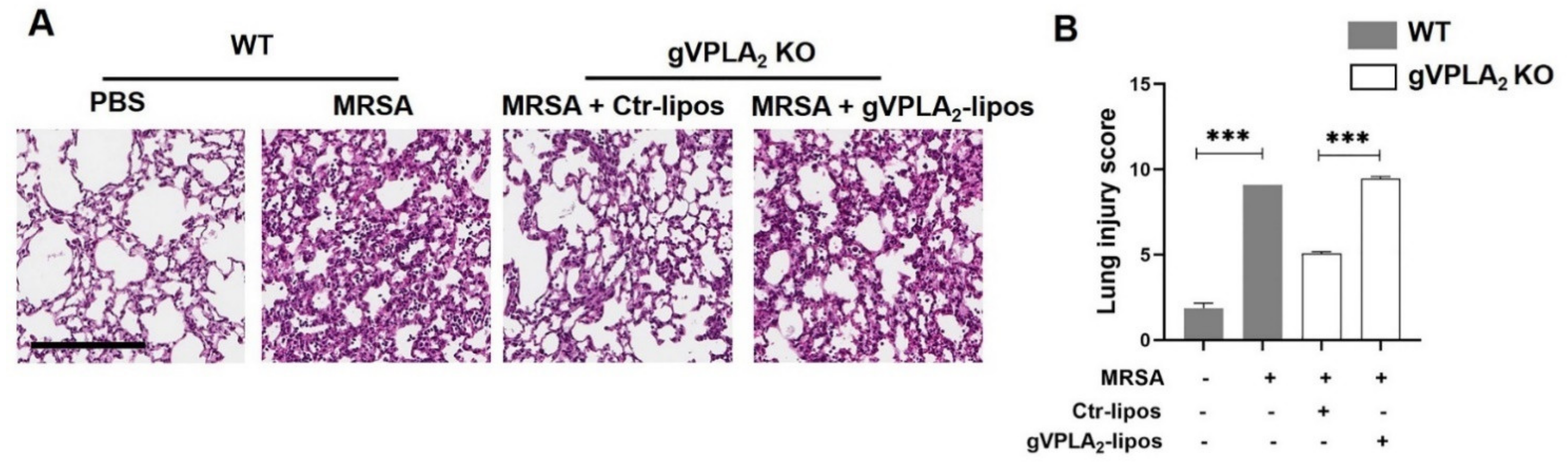
3.3. gVPLA2 Deficiency Protects against ALI In Vivo
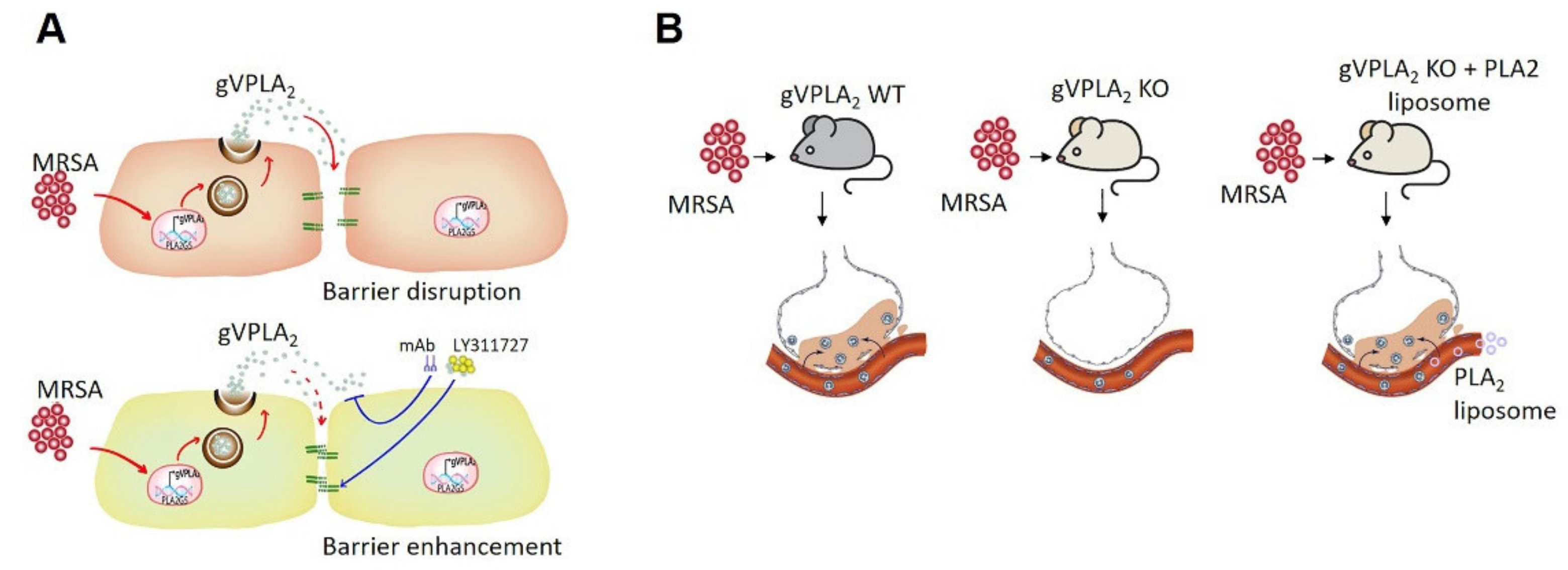
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2017, 377, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthay, M.A.; McAuley, D.F.; Ware, L.B. Clinical trials in acute respiratory distress syndrome: Challenges and opportunities. Lancet Respir. Med. 2017, 5, 524–534. [Google Scholar] [CrossRef]
- Ekren, P.K.; Ranzani, O.T.; Ceccato, A.; Li Bassi, G.; Munoz Conejero, E.; Ferrer, M.; Niederman, M.S.; Torres, A. Evaluation of the 2016 Infectious Diseases Society of America/American Thoracic Society Guideline Criteria for Risk of Multi-drug Resistant Pathogens in Hospital-acquired and Ventilator-associated Pneumonia Patients in the Intensive Care Unit. Am. J. Respir. Crit. Care Med. 2018, 197, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karchmer, A.W.; Bayer, A.S. Methicillin-resistant Staphylococcus aureus: An evolving clinical challenge. Clin. Infect. Dis. 2008, 46, S342–S343. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.C. Risk of death from methicillin-resistant Staphylococcus aureus bacteraemia: A meta-analysis. Med. J. Aust. 2002, 176, 188–189. [Google Scholar] [CrossRef]
- Antonanzas, F.; Lozano, C.; Torres, C. Economic Features of Antibiotic Resistance: The Case of Methicillin-Resistant Staphylococcus aureus. PharmacoEconomics 2015, 33, 285–325. [Google Scholar] [CrossRef]
- Murakami, M.; Sato, H.; Miki, Y.; Yamamoto, K.; Taketomi, Y. A new era of secreted phospholipase A(2). J. Lipid Res. 2015, 56, 1248–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating Phospholipase A(2) Biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Girard, C.; Yamamoto, K.; Lambeau, G. Emerging roles of secreted phospholipase A2 enzymes: Lessons from transgenic and knockout mice. Biochimie 2010, 92, 561–582. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Yamamoto, K.; Miki, Y.; Murase, R.; Sato, H.; Taketomi, Y. The Roles of the Secreted Phospholipase A2 Gene Family in Immunology. Adv. Immunol. 2016, 132, 91–134. [Google Scholar] [CrossRef]
- Edelson, J.D.; Vadas, P.; Villar, J.; Mullen, J.B.M.; Pruzanski, W. Acute Lung Injury Induced by Phospholipase A2: Structural and Functional Changes. Am. Rev. Respir. Dis. 1991, 143, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Fukuda, T.; Thompson, B.T.; Cockrill, B.; Hales, C.; Bonventre, J.V. Bronchoalveolar lavage fluid phospholipase A2 activities are increased in human adult respiratory distress syndrome. Am. J. Physiol. Content 1995, 269, 109–118. [Google Scholar] [CrossRef]
- Kitsiouli, E.; Nakos, G.; Lekka, M.E. Phospholipase A2 subclasses in acute respiratory distress syndrome. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakos, G.; Kitsiouli, E.; Hatzidaki, E.; Koulouras, V.; Touqui, L.; Lekka, M.E. Phospholipases A2 and platelet-activating-factor acetylhydrolase in patients with acute respiratory distress syndrome*. Crit. Care Med. 2005, 33, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Letsiou, E.; Htwe, Y.M.; Dudek, S.M. Secretory Phospholipase A(2) Enzymes in Acute Lung Injury. Cell Biochem. Biophys. 2021, 1–9. [Google Scholar] [CrossRef]
- Fujioka, D.; Watanabe, Y.; Nakamura, T.; Yokoyama, T.; Miyazawa, K.; Murakami, M.; Kugiyama, K. Group V Secretory Phospholipase A2 Regulates Endocytosis of Acetylated LDL by Transcriptional Activation of PGK1 in RAW264.7 Macrophage Cell Line. J. Atheroscler. Thromb. 2021, 62216. [Google Scholar] [CrossRef]
- Samuchiwal, S.K.; Balestrieri, B. Harmful and protective roles of group V phospholipase A2: Current perspectives and future directions. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2019, 1864, 819–826. [Google Scholar] [CrossRef]
- Tamayo, I.; Velasco, S.E.; Puy, C.; Esmon, C.T.; DiChiara, M.G.; Montes, R.; Hermida, J. Group V secretory phospholipase A2 impairs endothelial protein C receptor-dependent protein C activation and accelerates thrombosis in vivo. J. Thromb. Haemost. 2014, 12, 1921–1927. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.M.; Meliton, A.Y.; Arm, J.P.; Bonventre, J.V.; Cho, W.; Leff, A.R. Deletion of Secretory Group V Phospholipase A2 Attenuates Cell Migration and Airway Hyperresponsiveness in Immunosensitized Mice. J. Immunol. 2007, 179, 4800–4807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabot, S.; Koumanov, K.; Lambeau, G.; Gelb, M.H.; Balloy, V.; Chignard, M.; Whitsett, J.A.; Touqui, L. Inhibitory Effects of Surfactant Protein A on Surfactant Phospholipid Hydrolysis by Secreted Phospholipases A2. J. Immunol. 2003, 171, 995–1000. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuki, M.; Taketomi, Y.; Arata, S.; Masuda, S.; Ishikawa, Y.; Ishii, T.; Takanezawa, Y.; Aoki, J.; Arai, H.; Yamamoto, K.; et al. Transgenic Expression of Group V, but Not Group X, Secreted Phospholipase A2 in Mice Leads to Neonatal Lethality because of Lung Dysfunction. J. Biol. Chem. 2006, 281, 36420–36433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef]
- Dudek, S.M.; Munoz, N.M.; Desai, A.; Osan, C.M.; Meliton, A.Y.; Leff, A.R. Group V phospholipase A2 mediates barrier disruption of human pulmonary endothelial cells caused by LPS in vitro. Am. J. Respir. Cell Mol. Biol. 2011, 44, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.M.; Desai, A.; Meliton, L.N.; Meliton, A.Y.; Zhou, T.; Leff, A.R.; Dudek, S.M. Group V Phospholipase A2 Increases Pulmonary Endothelial Permeability Through Direct Hydrolysis of the Cell Membrane. Pulm. Circ. 2012, 2, 182–192. [Google Scholar] [CrossRef] [Green Version]
- Meliton, A.Y.; Munoz, N.M.; Meliton, L.N.; Birukova, A.A.; Leff, A.R.; Birukov, K.G. Mechanical induction of group V phospholipase A(2) causes lung inflammation and acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L689–L700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, N.M.; Meliton, A.Y.; Meliton, L.N.; Dudek, S.M.; Leff, A.R. Secretory group V phospholipase A2 regulates acute lung injury and neutrophilic inflammation caused by LPS in mice. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L879–L887. [Google Scholar] [CrossRef]
- Kim, K.P.; Rafter, J.D.; Bittova, L.; Han, S.K.; Snitko, Y.; Munoz, N.M.; Leff, A.R.; Cho, W. Mechanism of Human Group V Phospholipase A2(PLA2)-induced Leukotriene Biosynthesis in Human Neutrophils. J. Biol. Chem. 2001, 276, 11126–11134. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kim, K.P.; Han, S.K.; Munoz, N.M.; Zhu, X.; Sano, H.; Leff, A.R.; Cho, W. Group V Phospholipase A2 Induces Leukotriene Biosynthesis in Human Neutrophils through the Activation of Group IVA Phospholipase A2. J. Biol. Chem. 2002, 277, 36479–36488. [Google Scholar] [CrossRef] [Green Version]
- Sato, R.; Yamaga, S.; Watanabe, K.; Hishiyama, S.; Kawabata, K.; Kobayashi, T.; Fujioka, D.; Saito, Y.; Yano, T.; Watanabe, K.; et al. Inhibition of secretory phospholipase A2 activity attenuates lipopolysaccharide-induced acute lung injury in a mouse model. Exp. Lung Res. 2010, 36, 191–200. [Google Scholar] [CrossRef]
- Kawasaki, T.; Chen, W.; Htwe, Y.M.; Tatsumi, K.; Dudek, S.M. DPP4 inhibition by sitagliptin attenuates LPS-induced lung injury in mice. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L834–L845. [Google Scholar] [CrossRef]
- Meliton, L.N.; Zhu, X.; Brown, M.; Epshtein, Y.; Kawasaki, T.; Letsiou, E.; Dudek, S.M. Degradation of group V secretory phospholipase A2 in lung endothelium is mediated by autophagy. Microvasc. Res. 2020, 129, 103954. [Google Scholar] [CrossRef]
- Chen, J.; Feng, G.; Guo, Q.; Wardenburg, J.B.; Lin, S.; Inoshima, I.; Deaton, R.; Yuan, J.X.J.; Garcia, J.G.N.; Machado, R.F.; et al. Transcriptional Events during the Recovery from MRSA Lung Infection: A Mouse Pneumonia Model. PLoS ONE 2013, 8, e70176. [Google Scholar] [CrossRef]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Dubrovskyi, O.; Birukova, A.A.; Birukov, K.G. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab. Investig. 2012, 93, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Belvitch, P.; Brown, M.E.; Brinley, B.N.; Letsiou, E.; Rizzo, A.N.; Garcia, J.G.N.; Dudek, S.M. The ARP 2/3 complex mediates endothelial barrier function and recovery. Pulm. Circ. 2017, 7, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.N.; Belvitch, P.; Demeritte, R.; Garcia, J.G.; Letsiou, E.; Dudek, S.M. Arg mediates LPS-induced disruption of the pulmonary endothelial barrier. Vasc. Pharmacol. 2020, 128–129, 106677. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, B.; Hsu, V.W.; Gilbert, H.; Leslie, C.C.; Han, W.K.; Bonventre, J.V.; Arm, J.P. Group V Secretory Phospholipase A2 Translocates to the Phagosome after Zymosan Stimulation of Mouse Peritoneal Macrophages and Regulates Phagocytosis. J. Biol. Chem. 2006, 281, 6691–6698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letsiou, E.; Rizzo, A.N.; Sammani, S.; Naureckas, P.; Jacobson, J.R.; Garcia, J.G.N.; Dudek, S.M. Differential and opposing effects of imatinib on LPS- and ventilator-induced lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L259–L269. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.L.; Sammani, S.; Kempf, C.; Saadat, L.; Letsiou, E.; Wang, T.; Moreno-Vinasco, L.; Rizzo, A.N.; Fortman, J.D.; Garcia, J.G. Pleiotropic effects of interleukin-6 in a “two-hit” murine model of acute respiratory distress syndrome. Pulm. Circ. 2014, 4, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzapoiazova, T.; Moitra, J.; Moreno-Vinasco, L.; Sammani, S.; Turner, J.R.; Chiang, E.T.; Evenoski, C.; Wang, T.; Singleton, P.A.; Huang, Y.; et al. Non-muscle myosin light chain kinase isoform is a viable molecular target in acute inflammatory lung injury. Am. J. Respir. Cell Mol. Biol. 2011, 44, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Looney, M.R.; Bhattacharya, J. Live imaging of the lung. Annu. Rev. Physiol. 2014, 76, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Maniatis, N.A.; Orfanos, S.E. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 2008, 14, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Fleisch, J.H.; Armstrong, C.T.; Roman, C.R.; Mihelich, E.D.; Spaethe, S.M.; Jackson, W.T.; Bobbitt, J.L.; Draheim, S.; Bach, N.J.; Dillard, R.D.; et al. Recombinant human secretory phospholipase A2 released thromboxane from guinea pig bronchoalveolar lavage cells: In vitro and ex vivo evaluation of a novel secretory phospholipase A2 inhibitor. J. Pharmacol. Exp. Ther. 1996, 278, 252–257. [Google Scholar] [PubMed]
- Muñoz, N.M.; Kim, K.P.; Han, S.K.; Boetticher, E.; Sperling, A.I.; Sano, H.; Han, K.; Zhu, X.; Leff, A.R.; Cho, W. Characterization of Monoclonal Antibodies Specific for 14-kDa Human Group V Secretory Phospholipase A2 (hVPLA2). Hybridoma 2000, 19, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hippensteel, J.A.; Schmidt, E.P. Intravital Microscopy in the Mouse Lung. Adv. Struct. Saf. Stud. 2018, 1809, 331–339. [Google Scholar] [CrossRef]
- Ashbaugh, D.; Bigelow, D.B.; Petty, T.; Levine, B. Acute respiratory distress in adults. Lancet 1967, 290, 319–323. [Google Scholar] [CrossRef]
- Shaver, C.M.; Bastarache, J.A. Clinical and biological heterogeneity in acute respiratory distress syndrome: Direct versus indirect lung injury. Clin. Chest Med. 2014, 35, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Font, M.D.; Thyagarajan, B.; Khanna, A.K. Sepsis and Septic Shock–Basics of diagnosis, pathophysiology and clinical decision making. Med. Clin. N. Am. 2020, 104, 573–585. [Google Scholar] [CrossRef]
- Defres, S.; Marwick, C.; Nathwani, D. MRSA as a cause of lung infection including airway infection, community-acquired pneumonia and hospital-acquired pneumonia. Eur. Respir. J. 2009, 34, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Lomaestro, B.; et al. Therapeutic monitoring of vancomycin for serious methicillin-resistant Staphylococcus aureus infections: A revised consensus guideline and review by the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists. Am. J. Health Syst. Pharm. 2020, 77, 835–864. [Google Scholar] [PubMed] [Green Version]
- Köck, R.; Becker, K.; Cookson, B.; Van Gemert-Pijnen, J.E.; Harbarth, S.; Kluytmans, J.; Mielke, M.; Peters, G.; Skov, R.L.; Struelens, M.J.; et al. Methicillin-resistant Staphylococcus aureus (MRSA): Burden of disease and control challenges in Europe. Eurosurveillance 2010, 15, 19688. [Google Scholar] [CrossRef]
- Diekema, D.J.; Richter, S.S.; Heilmann, K.P.; Dohrn, C.L.; Riahi, F.; Tendolkar, S.; McDanel, J.S.; Doern, G.V. Continued emergence of USA300 methicillin-resistant Staphylococcus aureus in the United States: Results from a nationwide surveillance study. Infect. Control. Hosp. Epidemiol. 2014, 35, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.; Eichenberger, E.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Genet. 2019, 17, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, S.; Kazepidou, E.; Antonelou, M.H.; Leondaritis, G.; Tsapinou, A.; Koulouras, V.P.; Avgeropoulos, A.; Nakos, G.; Lekka, M.E. Secretory phospholipase A(2)-IIA protein and mRNA pools in extracellular vesicles of bronchoalveolar lavage fluid from patients with early acute respiratory distress syndrome: A new perception in the dissemination of inflammation? Pharmaceuticals 2020, 13, 415. [Google Scholar] [CrossRef]
- Degousee, N.; Kelvin, D.J.; Geisslinger, G.; Hwang, D.M.; Stefanski, E.; Wang, X.-H.; Danesh, A.; Angioni, C.; Schmidt, H.; Lindsay, T.F.; et al. Group V Phospholipase A2 in Bone Marrow-derived Myeloid Cells and Bronchial Epithelial Cells Promotes Bacterial Clearance after Escherichia coli Pneumonia*. J. Biol. Chem. 2011, 286, 35650–35662. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-C.; Tsai, Y.-F.; Pan, Y.-L.; Hwang, T.-L. Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Murakami, M.; Mitsuishi, M.; Komiyama, K.; Ishikawa, Y.; Ishii, T.; Kudo, I. Expression of secretory phospholipase A2 enzymes in lungs of humans with pneumonia and their potential prostaglandin-synthetic function in human lung-derived cells. Biochem. J. 2005, 387, 27–38. [Google Scholar] [CrossRef] [Green Version]
- De Luca, D.; Lopez-Rodriguez, E.; Minucci, A.; Vendittelli, F.; Gentile, L.; Stival, E.; Conti, G.; Piastra, M.; Antonelli, M.; Echaide, M.; et al. Clinical and biological role of secretory phospholipase A2 in acute respiratory distress syndrome infants. Crit. Care. 2013, 17, R163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Chen, Y.; Zhang, P.; Lin, P.; Xie, N.; Wu, M. Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases. Cells 2019, 8, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hite, R.D.; Seeds, M.C.; Safta, A.M.; Jacinto, R.B.; Gyves, J.I.; Bass, D.A.; Waite, B.M. Lysophospholipid generation and phosphatidylglycerol depletion in phospholipase A2-mediated surfactant dysfunction. Am. J. Physiol. Cell. Mol. Physiol. 2005, 288, L618–L624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.K.; Kim, K.P.; Koduri, R.; Bittova, L.; Munoz, N.M.; Leff, A.R.; Wilton, D.C.; Gelb, M.H.; Cho, W. Roles of Trp31 in High Membrane Binding and Proinflammatory Activity of Human Group V Phospholipase A2. J. Biol. Chem. 1999, 274, 11881–11888. [Google Scholar] [CrossRef] [Green Version]
- Loffredo, S.; Borriello, F.; Iannone, R.; Ferrara, A.L.; Galdiero, M.R.; Gigantino, V.; Esposito, P.; Varricchi, G.; Lambeau, G.; Cassatella, M.A.; et al. Group V Secreted Phospholipase A2 Induces the Release of Proangiogenic and Antiangiogenic Factors by Human Neutrophils. Front. Immunol. 2017, 8, 443. [Google Scholar] [CrossRef] [Green Version]
- Karki, P.; Ke, Y.; Tian, Y.; Ohmura, T.; Sitikov, A.; Sarich, N.; Montgomery, C.P.; Birukova, A.A. Staphylococcus aureus-induced endothelial permeability and inflammation are mediated by microtubule destabilization. J. Biol. Chem. 2019, 294, 3369–3384. [Google Scholar] [CrossRef] [Green Version]
- Meliton, A.Y.; Meng, F.; Tian, Y.; Sarich, N.; Mutlu, G.M.; Birukova, A.A.; Birukov, K.G. Oxidized phospholipids protect against lung injury and endothelial barrier dysfunction caused by heat-inactivated Staphylococcus aureus. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L550–L562. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Htwe, Y.M.; Wang, H.; Belvitch, P.; Meliton, L.; Bandela, M.; Letsiou, E.; Dudek, S.M. Group V Phospholipase A2 Mediates Endothelial Dysfunction and Acute Lung Injury Caused by Methicillin-Resistant Staphylococcus Aureus. Cells 2021, 10, 1731. https://doi.org/10.3390/cells10071731
Htwe YM, Wang H, Belvitch P, Meliton L, Bandela M, Letsiou E, Dudek SM. Group V Phospholipase A2 Mediates Endothelial Dysfunction and Acute Lung Injury Caused by Methicillin-Resistant Staphylococcus Aureus. Cells. 2021; 10(7):1731. https://doi.org/10.3390/cells10071731
Chicago/Turabian StyleHtwe, Yu Maw, Huashan Wang, Patrick Belvitch, Lucille Meliton, Mounica Bandela, Eleftheria Letsiou, and Steven M. Dudek. 2021. "Group V Phospholipase A2 Mediates Endothelial Dysfunction and Acute Lung Injury Caused by Methicillin-Resistant Staphylococcus Aureus" Cells 10, no. 7: 1731. https://doi.org/10.3390/cells10071731