
Processing and Ex Vivo Expansion of Adipose Tissue-Derived Mesenchymal Stem/Stromal Cells for the Development of an Advanced Therapy Medicinal Product for use in Humans

 , and
, and
Abstract
: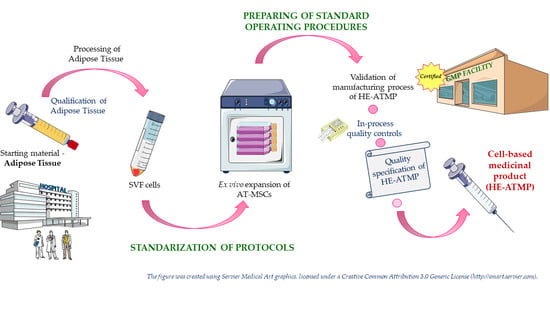
1. Introduction
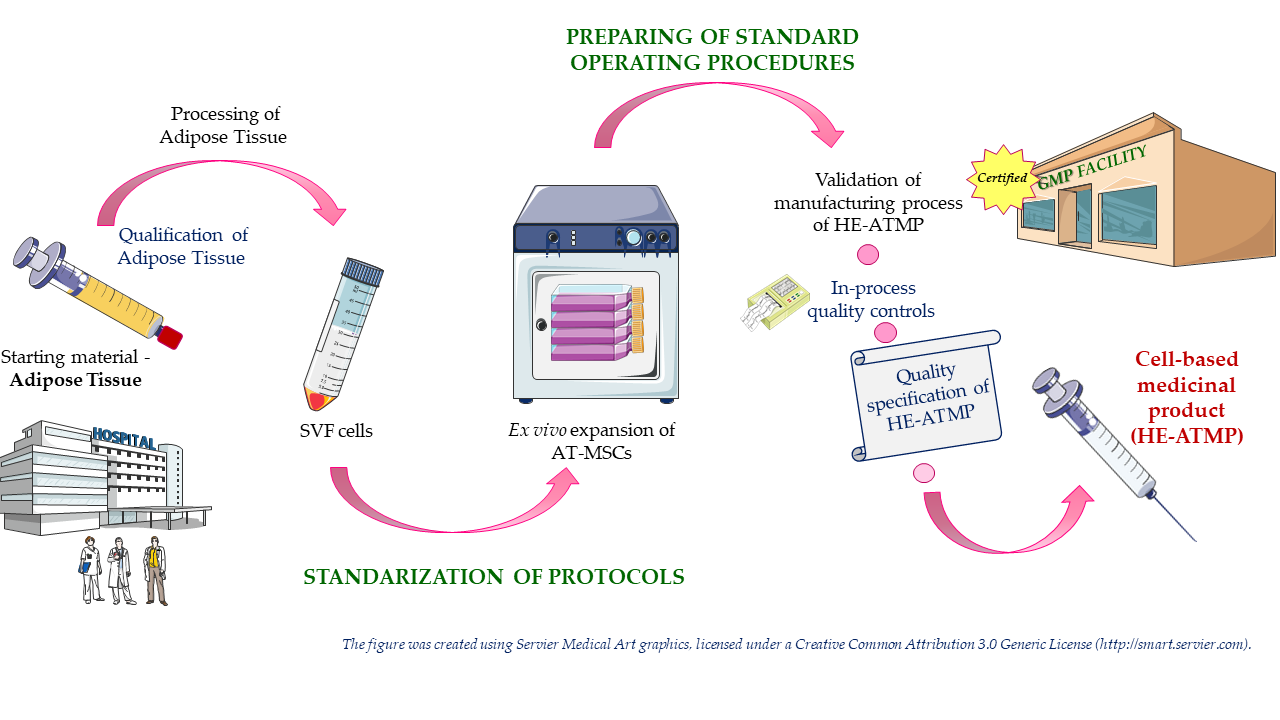
2. Materials and Methods
2.1. Lipoaspirate Collection
2.2. Isolation of SVF Cells
2.3. Modification of SVF Isolation Procedure by Adding the Step of Red Blood Cell Lysis
2.4. Cell Counting and Viability Assessment
2.4.1. RD Phase
2.4.2. Implementation Phase
2.5. Culture of AT-MSCs
2.6. Kinetics of AT-MSCs Growth
2.7. Antigenic Phenotyping by Flow Cytometry
2.8. Trilineage Differentiation of AT-MSCs
2.9. Chondrogenic Differentiation of AT-MSCs in Microenvironment Resembling Conditions in Human Joints
2.10. Histochemical Staining
2.11. Validation of Manufacturing Process of HE-ATMP
2.12. Stability Study of HE-ATMP
2.13. Sterility Testing
2.14. Endotoxins’ Testing
2.15. Statistical Analysis
3. Results
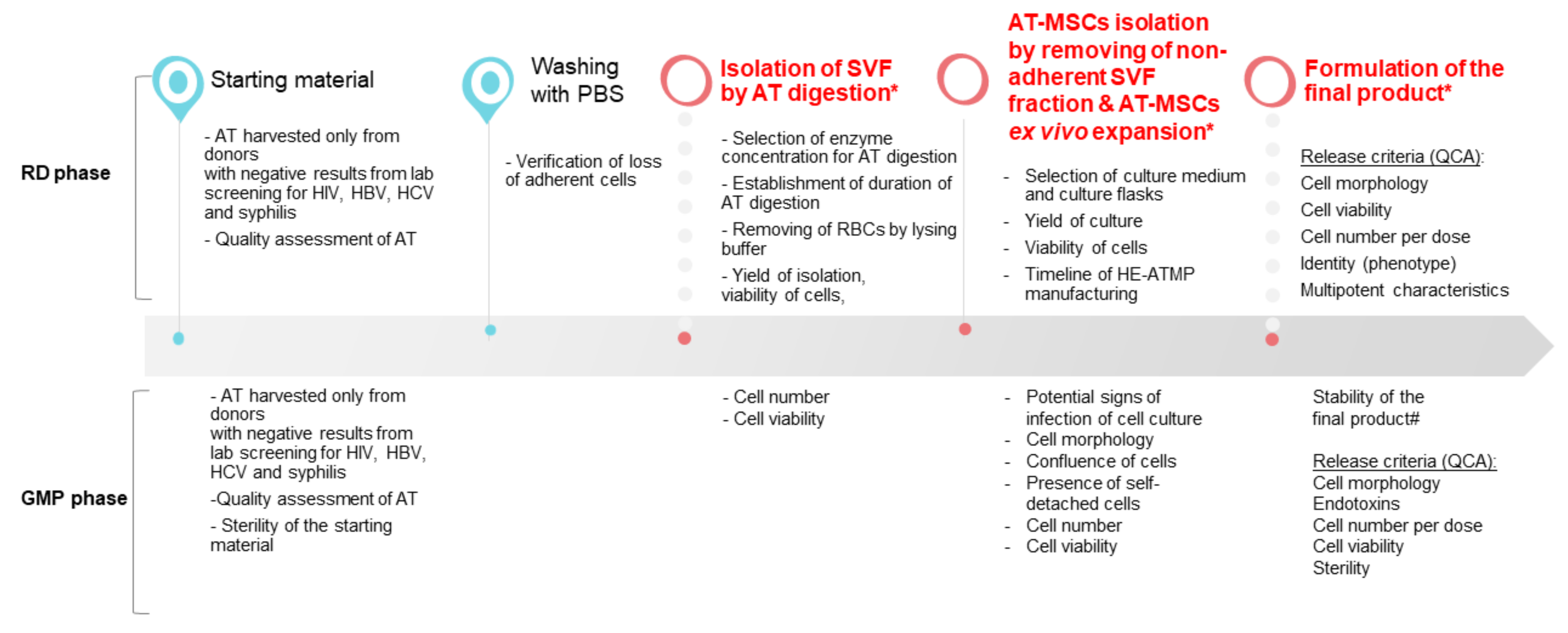
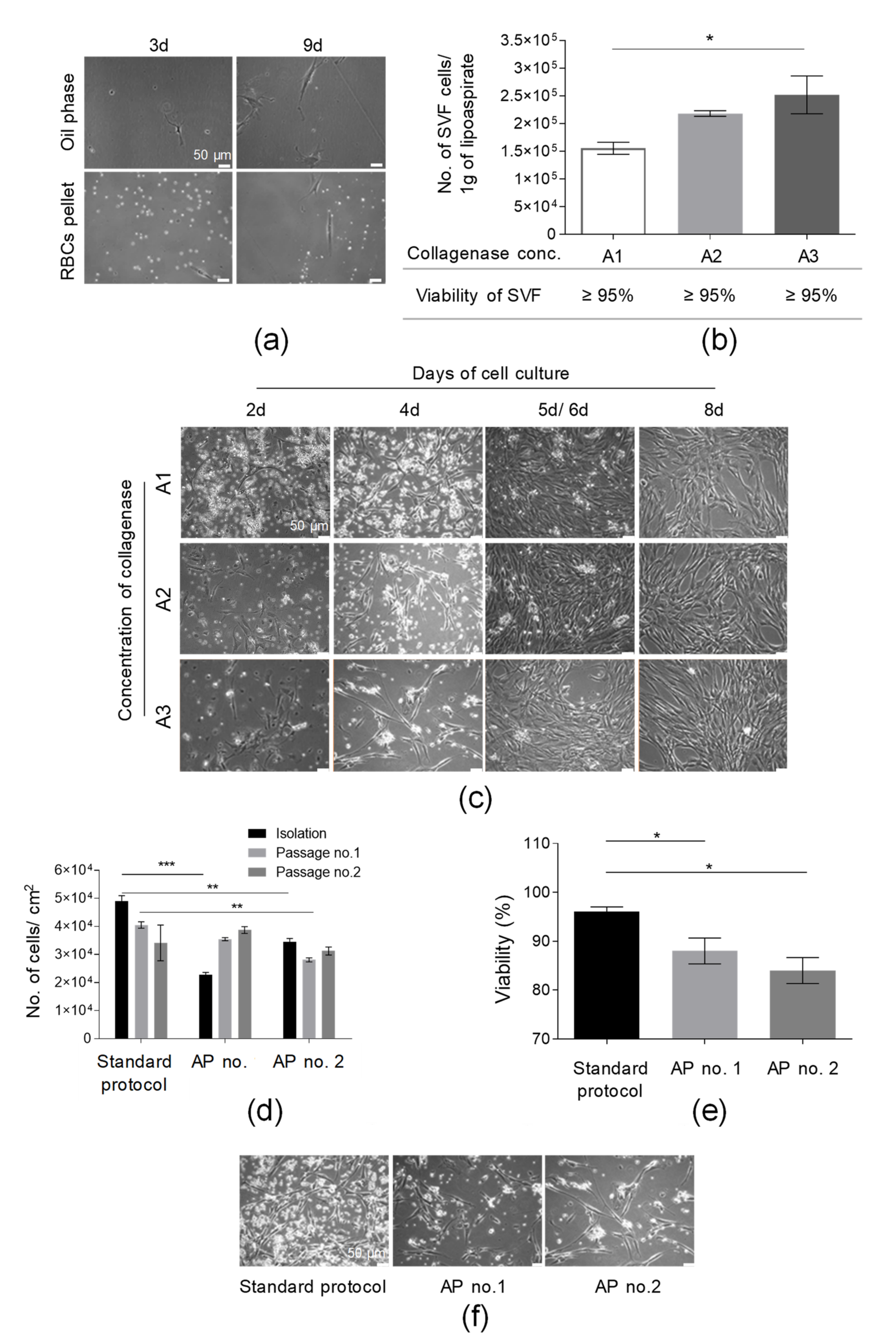
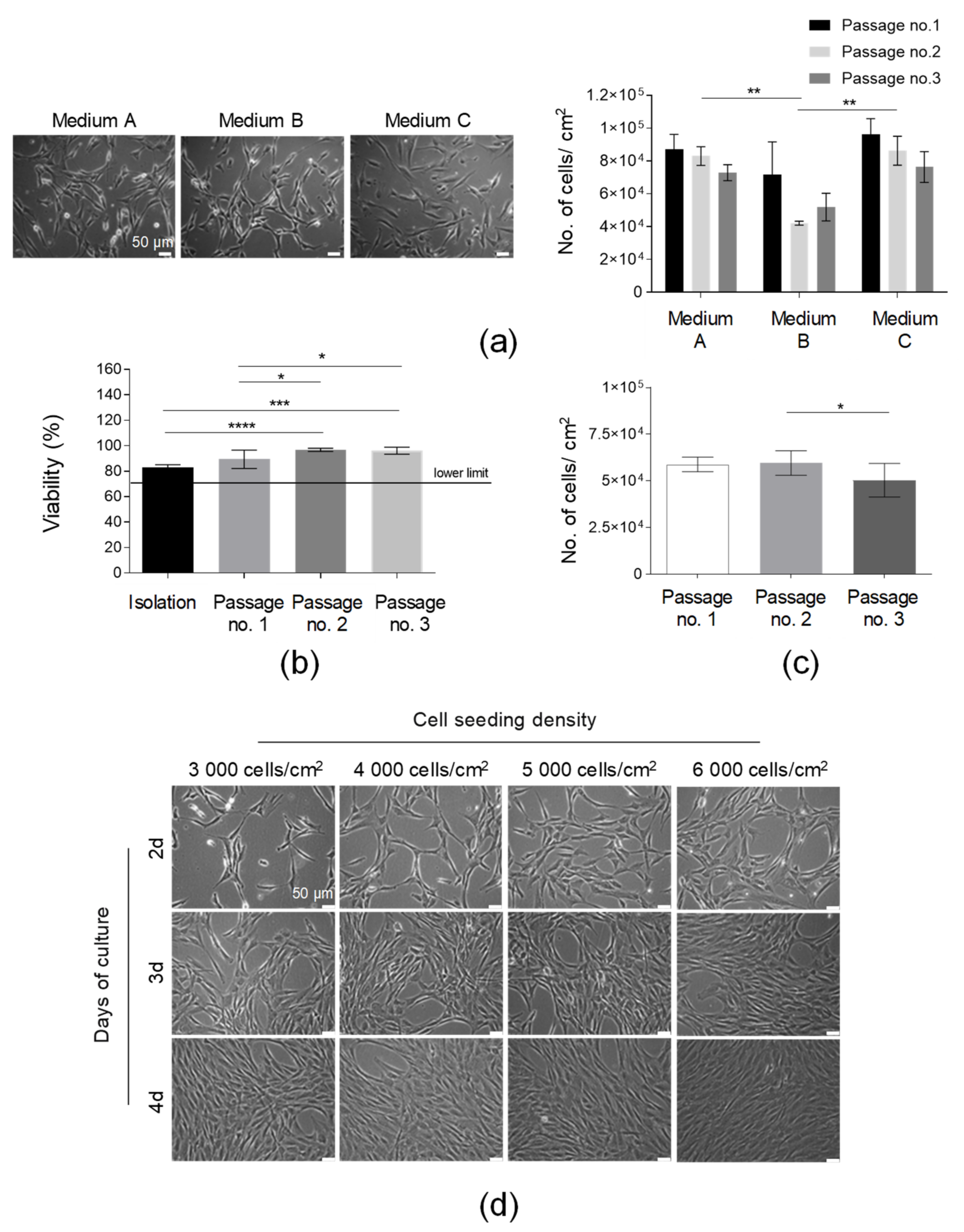
3.1. SVF Cells Were Effectively Isolated from Adipose Tissue
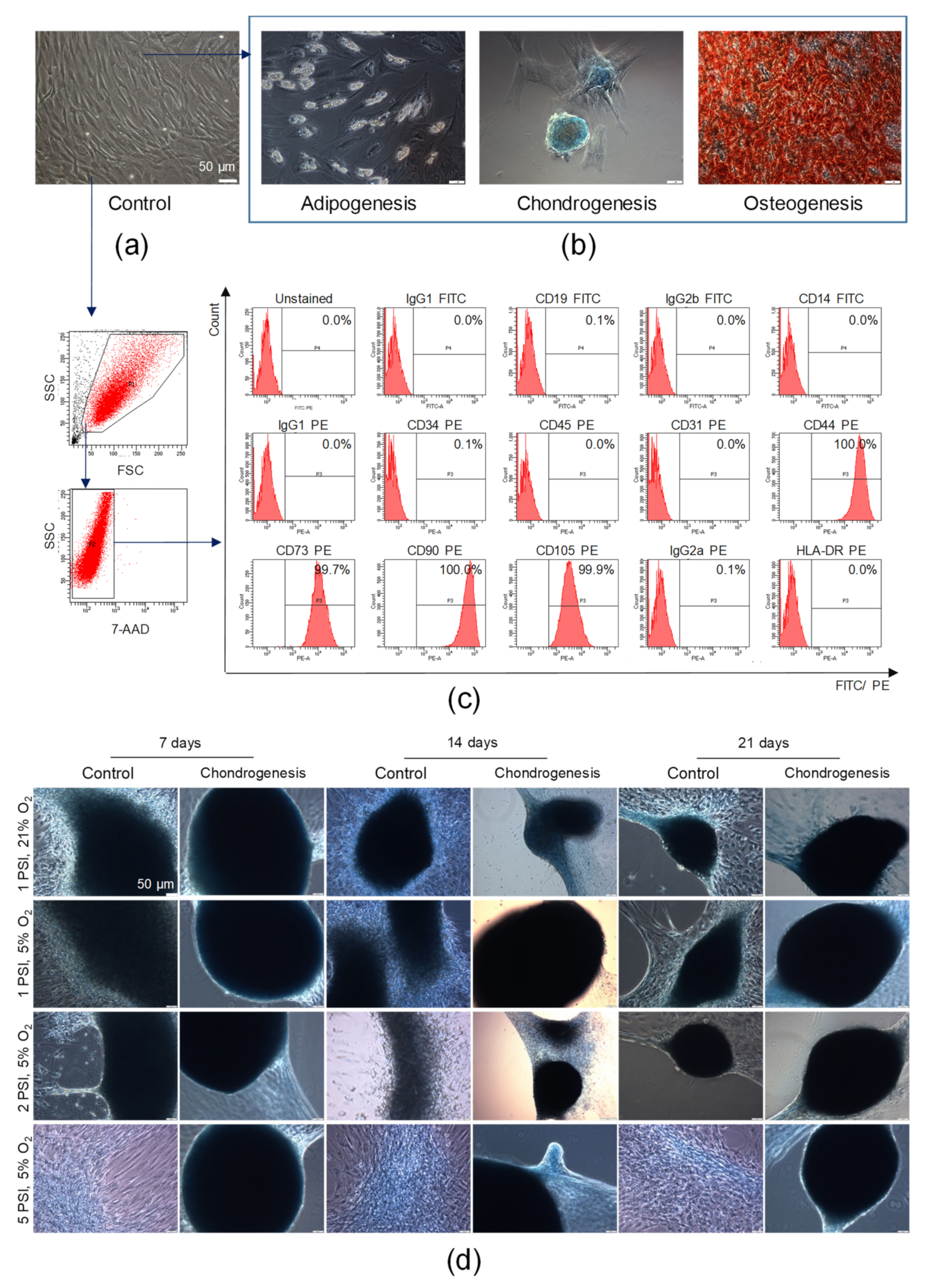
3.2. AT-MSCs Were Efficiently Cultured in In Vitro Conditions in Accordance with GMP Requirements
3.3. The Identity of Isolated AT-MSCs Was Confirmed According to ISCT Recommendations
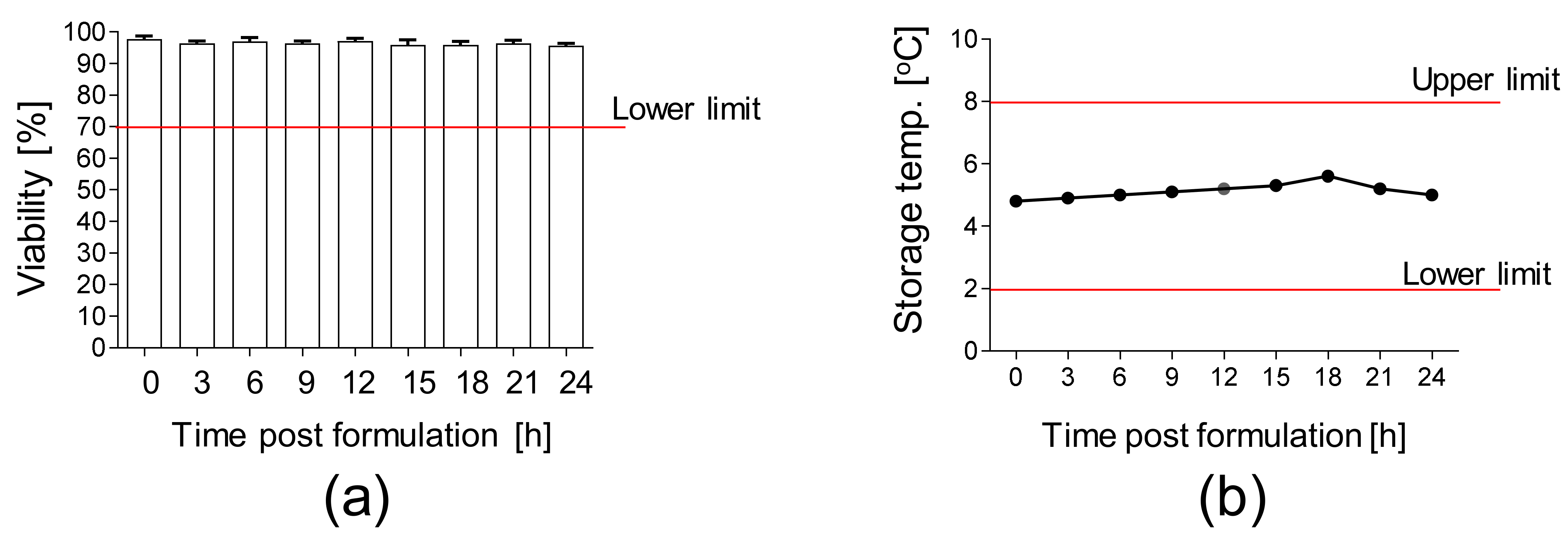
3.4. Validation of Manufacturing Process of HE-ATMP
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berebichez-Fridman, R.; Montero-Olvera, P.R. Sources and clinical applications of mesenchymal stem cells state-of-the-art review. Sultan Qaboos Univ. Med. J. 2018, 18, e264–e277. [Google Scholar] [CrossRef] [Green Version]
- Labedz-Maslowska, A.; Bryniarska, N.; Kubiak, A.; Kaczmarzyk, T.; Sekula-Stryjewska, M.; Noga, S.; Boruczkowski, D.; Madeja, Z.; Zuba-Surma, E. Multilineage differentiation potential of human dental pulp stem cells—Impact of 3d and hypoxic environment on osteogenesis in vitro. Int. J. Mol. Sci. 2020, 21, 6172. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Jimenez-Puerta, G.J.; Marchal, J.A.; López-Ruiz, E.; Gálvez-Martín, P. Role of Mesenchymal Stromal Cells as Therapeutic Agents: Potential Mechanisms of Action and Implications in Their Clinical Use. J. Clin. Med. 2020, 9, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.-J.; Chiu, F.-Y.; Chiu, H.-Y.; Chang, M.-C.; Hung, S.-C. Chondrogenic Differentiation of Mesenchymal Stem Cells in Three-Dimensional Chitosan Film Culture. Cell Transplant. 2016, 26, 417–427. [Google Scholar] [CrossRef]
- Barry, F.; Boynton, R.E.; Liu, B.; Murphy, J.M. Chondrogenic differentiation of mesenchymal stem cells from bone marrow: Differentiation-dependent gene expression of matrix components. Exp. Cell Res. 2001, 268, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Fink, D.J.; Hunziker, E.B.; Barry, F.P. Stem Cell Therapy in a Caprine Model of Osteoarthritis. Arthritis Rheum. 2003, 48, 3464–3474. [Google Scholar] [CrossRef]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusuma, G.; Li, A.; Zhu, D.; McDonald, H.; Chambers, D.; Frith, J.; Lim, R. Engineering mesenchymal stem cell paracrine activity with 3D culture. Cytotherapy 2020, 22, S51. [Google Scholar] [CrossRef]
- Zhou, Y.; Yamamoto, Y.; Xiao, Z.; Ochiya, T. The Immunomodulatory Functions of Mesenchymal Stromal/Stem Cells Mediated via Paracrine Activity. J. Clin. Med. 2019, 8, 1025. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Castro, E.; Cunningham, C.; Miller, J.; Martuscelli, L.; Aoulad-Ali, S.; Rothwell, N.J.; Kielty, C.M.; Allan, S.M.; Pinteaux, E. Interleukin-1 primes human mesenchymal stem cells towards an anti-inflammatory and pro-trophic phenotype in vitro. Stem Cell Res. Ther. 2017, 8. [Google Scholar] [CrossRef]
- Han, X.; Yang, Q.; Lin, L.; Xu, C.; Zheng, C.; Chen, X.; Han, Y.; Li, M.; Cao, W.; Cao, K.; et al. Interleukin-17 enhances immunosuppression by mesenchymal stem cells. Cell Death Differ. 2014, 21, 1758–1768. [Google Scholar] [CrossRef]
- Kim, D.S.; Jang, I.K.; Lee, M.W.; Ko, Y.J.; Lee, D.H.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Enhanced Immunosuppressive Properties of Human Mesenchymal Stem Cells Primed by Interferon-γ. EBioMedicine 2018, 28, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Van Megen, K.M.; Van ’t Wout, E.J.T.; Motta, J.L.; Dekker, B.; Nikolic, T.; Roep, B.O. Activated mesenchymal stromal cells process and present antigens regulating adaptive immunity. Front. Immunol. 2019, 10, 694. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.; Gong, L.; Yu, D.; An, C.; Bunpetch, V.; Dai, J.; Huang, H.; Zou, X.; Ouyang, H.; et al. The Plasticity of Mesenchymal Stem Cells in Regulating Surface HLA-I. iScience 2019, 15, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukomska, B.; Stanaszek, L.; Zuba-Surma, E.; Legosz, P.; Sarzynska, S.; Drela, K. Challenges and Controversies in Human Mesenchymal Stem Cell Therapy. Stem Cells Int. 2019, 2019, 9628536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyurkchiev, D. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J. Stem Cells 2014, 6, 552. [Google Scholar] [CrossRef] [PubMed]
- Infante, A.; Gener, B.; Vázquez, M.; Olivares, N.; Arrieta, A.; Grau, G.; Llano, I.; Madero, L.; Bueno, A.M.; Sagastizabal, B.; et al. Reiterative infusions of MSCs improve pediatric osteogenesis imperfecta eliciting a pro-osteogenic paracrine response: TERCELOI clinical trial. Clin. Transl. Med. 2021, 11, e265. [Google Scholar] [CrossRef] [PubMed]
- Schu, S.; Nosov, M.; O’Flynn, L.; Shaw, G.; Treacy, O.; Barry, F.; Murphy, M.; O’Brien, T.; Ritter, T. Immunogenicity of allogeneic mesenchymal stem cells. J. Cell. Mol. Med. 2012, 16, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Mirotsou, M.; Jayawardena, T.M.; Schmeckpeper, J.; Gnecchi, M.; Dzau, V.J. Paracrine mechanisms of stem cell reparative and regenerative actions in the heart. J. Mol. Cell. Cardiol. 2011, 50, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—Current trends and future prospective. Biosci. Rep. 2015, 35, 191. [Google Scholar] [CrossRef]
- Kangari, P.; Talaei-Khozani, T.; Razeghian-Jahromi, I.; Razmkhah, M. Mesenchymal stem cells: Amazing remedies for bone and cartilage defects. Stem Cell Res. Ther. 2020, 11, 492. [Google Scholar] [CrossRef]
- Ranjbaran, H.; Mohammadi Jobani, B.; Amirfakhrian, E.; Alizadeh-Navaei, R. Efficacy of mesenchymal stem cell therapy on glucose levels in type 2 diabetes mellitus: A systematic review and meta-analysis. J. Diabetes Investig. 2020, 1–8. [Google Scholar] [CrossRef]
- Li, H.; Deng, Y.; Liang, J.; Huang, F.; Qiu, W.; Zhang, M.; Long, Y.; Hu, X.; Lu, Z.; Liu, W.; et al. Mesenchymal stromal cells attenuate multiple sclerosis via IDO-dependent increasing the suppressive proportion of CD5+ IL-10+ B cells. Am. J. Transl. Res. 2019, 11, 5673–5688. [Google Scholar] [PubMed]
- Richardson, J.D.; Nelson, A.J.; Zannettino, A.C.W.; Gronthos, S.; Worthley, S.G.; Psaltis, P.J. Optimization of the Cardiovascular Therapeutic Properties of Mesenchymal Stromal/Stem Cells-Taking the Next Step. Stem Cell Rev. Rep. 2013, 9, 281–302. [Google Scholar] [CrossRef] [PubMed]
- Oikonomopoulos, A.; Van Deen, W.K.; Manansala, A.R.; Lacey, P.N.; Tomakili, T.A.; Ziman, A.; Hommes, D.W. Optimization of human mesenchymal stem cell manufacturing: The effects of animal/xeno-free media. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Pacelli, L.; Bassi, G.; Dulong, J.; Bifari, F.; Bezier, I.; Zanoncello, J.; Ricciardi, M.; Latour, M.; Bourin, P.; et al. Clinical-grade mesenchymal stromal cells produced under various good manufacturing practice processes differ in their immunomodulatory properties: Standardization of immune quality controls. Stem Cells Dev. 2013, 22, 1789–1801. [Google Scholar] [CrossRef]
- Haque, N.; Abu Kasim, N.H.; Rahman, M.T. Optimization of pre-transplantation conditions to enhance the efficacy of mesenchymal stem cells. Int. J. Biol. Sci. 2015, 11, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Bourin, P.; Bunnell, B.A.; Casteilla, L.; Dominici, M.; Katz, A.J.; March, K.L.; Redl, H.; Rubin, J.P.; Yoshimura, K.; Gimble, J.M. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: A joint statement of the International Federation for Adipose Therapeutics (IFATS) and Science and the International S. Cytotherapy 2013, 15, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barekzai, J.; Petry, F.; Zitzmann, J.; Czermak, P.; Salzig, D. Bioprocess Development for Human Mesenchymal Stem Cell Therapy Products. In New Advances on Fermentation Processes; IntechOpen: London, UK, 2020; pp. 1–25. [Google Scholar]
- Thitilertdecha, P.; Lohsiriwat, V.; Poungpairoj, P.; Tantithavorn, V.; Onlamoon, N. Extensive Characterization of Mesenchymal Stem Cell Marker Expression on Freshly Isolated and In Vitro Expanded Human Adipose-Derived Stem Cells from Breast Cancer Patients. Stem Cells Int. 2020, 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; El-Jawhari, J.J.; Burska, A.N.; Ponchel, F.; Giannoudis, P.V.; Jones, E.A. The Analysis of in Vivo Aging in Human Bone Marrow Mesenchymal Stromal Cells Using Colony-Forming Unit-Fibroblast Assay and the CD45lowCD271+ Phenotype. Stem Cells Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Herberts, C.A.; Kwa, M.S.G.; Hermsen, H.P.H. Risk factors in the development of stem cell therapy. J. Transl. Med. 2011, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Svalgaard, J.D.; Juul, S.; Vester-Glovinski, P.V.; Haastrup, E.K.; Ballesteros, O.R.; Lynggaard, C.D.; Jensen, A.K.; Fischer-Nielsen, A.; Herly, M.; Munthe-Fog, L. Lipoaspirate Storage Time and Temperature: Effects on Stromal Vascular Fraction Quality and Cell Composition. Cells Tissues Organs 2020, 209, 54–63. [Google Scholar] [CrossRef]
- Li, S.H.; Liao, X.; Zhou, T.E.; Xiao, L.L.; Chen, Y.W.; Wu, F.; Wang, J.R.; Cheng, B.; Song, J.X.; Liu, H.W. Evaluation of 2 Purification Methods for Isolation of Human Adipose-Derived Stem Cells Based on Red Blood Cell Lysis with Ammonium Chloride and Hypotonic Sodium Chloride Solution. Ann. Plast. Surg. 2017, 78, 83–90. [Google Scholar] [CrossRef]
- Markarian, C.F.; Frey, G.Z.; Silveira, M.D.; Chem, E.M.; Milani, A.R.; Ely, P.B.; Horn, A.P.; Nardi, N.B.; Camassola, M. Isolation of adipose-derived stem cells: A comparison among different methods. Biotechnol. Lett. 2014, 36, 693–702. [Google Scholar] [CrossRef]
- Zimmerlin, L.; Donnenberg, V.S.; Pfeifer, M.E.; Meyer, E.M.; Péault, B.; Rubin, J.P.; Donnenberg, A.D. Stromal vascular progenitors in adult human adipose tissue. Cytom. Part A 2010, 77, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.S.; Wu, X.; Dietrich, M.; Rood, J.; Gimble, J.M. A non-enzymatic method for isolating human adipose tissue-derived stromal stem cells. Cytotherapy 2013, 15, 979–985. [Google Scholar] [CrossRef]
- Becherucci, V.; Piccini, L.; Casamassima, S.; Bisin, S.; Gori, V.; Gentile, F.; Ceccantini, R.; De Rienzo, E.; Bindi, B.; Pavan, P.; et al. Human platelet lysate in mesenchymal stromal cell expansion according to a GMP grade protocol: A cell factory experience. Stem Cell Res. Ther. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Kandoi, S.; Praveen kumar, L.; Patra, B.; Vidyasekar, P.; Sivanesan, D.; Vijayalakshmi, S.; Rajagopal, K.; Verma, R.S. Evaluation of platelet lysate as a substitute for FBS in explant and enzymatic isolation methods of human umbilical cord MSCs. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeller, M.; Laner-plamberger, S.; Krisch, L.; Rohde, E.; Strunk, D.; Schallmoser, K. Human platelet lysate for good manufacturing practice-compliant cell production. Int. J. Mol. Sci. 2021, 22, 5178. [Google Scholar] [CrossRef] [PubMed]
- Nikolits, I.; Nebel, S.; Egger, D.; Kreß, S.; Kasper, C. Towards Physiologic Culture Approaches to Improve Standard Cultivation of Mesenchymal Stem Cells. Cells 2021, 10, 886. [Google Scholar] [CrossRef]
- Almeida-Porada, G.; Atala, A.J.; Porada, C.D. Therapeutic Mesenchymal Stromal Cells for Immunotherapy and for Gene and Drug Delivery. Mol. Ther. Methods Clin. Dev. 2020, 16, 204–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A new mesenchymal stem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or an immunosuppressive MSC2 phenotype. PLoS One 2010, 5, e10088. [Google Scholar] [CrossRef]
- Lombardo, E.; Delarosa, O.; Mancheño-Corvo, P.; Menta, R.; Ramírez, C.; Büscher, D. Toll-like receptor-mediated signaling in human adipose-derived stem cells: Implications for immunogenicity and immunosuppressive potential. Tissue Eng. Part A 2009, 15, 1579–1589. [Google Scholar] [CrossRef]
- Lechanteur, C.; Briquet, A.; Bettonville, V.; Baudoux, E. MSC Manufacturing for Academic Clinical Trials: From a Clinical-Grade to a Full GMP-Compliant Process. Cells 2021, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Ścieżyńska, A.; Soszyńska, M.; Szpak, P.; Krześniak, N.; Malejczyk, J.; Kalaszczyńska, I. Influence of Hypothermic Storage Fluids on Mesenchymal Stem Cell Stability: A Comprehensive Review and Personal Experience. Cells 2021, 10, 1043. [Google Scholar] [CrossRef]
- Mazini, L.; Ezzoubi, M.; Malka, G. Overview of current adipose-derived stem cell (ADSCs) processing involved in therapeutic advancements: Flow chart and regulation updates before and after COVID-19. Stem Cell Res. Ther. 2021, 12, 113. [Google Scholar] [CrossRef]
- Aghayan, H.R.; Goodarzi, P.; Arjmand, B. GMP-compliant human adipose tissue-derived mesenchymal stem cells for cellular therapy. Methods Mol. Biol. 2015, 1283, 93–107. [Google Scholar] [PubMed]
- Godthardt, K.; Heifer, C.; Jüngerkes, F.; Bosio, A.; Knöbel, S. Efficient GMP compliant expansion of mesenchymal stromal cells (MSCs) from umbilical cord, bone marrow and adipose tissue using a closed cultivation system. Cytotherapy 2019, 21, S90–S91. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage I: Isolation of SVF Cells | |||
| Parameter | Correct Result | Incorrect Result | Method |
| (1) Cell number | Presence of SVF cells (successful isolation) | Lack of SVF cells (unsuccessful isolation) | Propidium iodide staining method combined with advanced image analysis (ADAM-MCTM automated cell counter) |
| (2) Cell viability | ≥60% → seeding SVF cells at a density 5000–11,000 cells/cm2 | <60% → decrease in seeding growth area | Propidium iodide staining method combined with advanced image analysis (ADAM-MCTM automated cell counter) |
| (3) Sterility of the starting material | Sterile → culture of cells | Non-sterile → utilization of cell culture | Direct inoculation (BD Bactec FX400 system) |
| Stage II: Culture of AT-MSCs | |||
| Parameter | Correct Result | Incorrect Result | Method |
| (1) Potential signs of infection of cell culture | Orange or red-raspberry clear culture medium → culture of cells | Yellow and/or turbid culture medium → evaluation of cell morphology | Macroscopic observation, light microscopy |
| (2) Cell morphology | Characteristic for MSCs (spindle-shaped, elongated cells possessing fibroblast-like morphology) | Morphology different than characteristic for MSCs → utilization of cell culture | Light microscopy |
| (3) Confluence of cells | ≥60% → passage of AT-MSCs <60% → further cell culture | <10% → evaluation of presence of self-detached cells | Light microscopy |
| (4) Presence of self-detached cells (qualitative assessment) | Lack of self-detached cells | Presence of a high amount of self-detached cells → utilization of cell culture | Light microscopy |
| (5) Cell number | At least 2× higher than number of seeding cells → continuation of manufacturing process | Less than 2× higher than number of seeding cells → utilization of cells | Propidium iodide staining method combined with advanced image analysis (ADAM-MCTM automated cell counter) |
| (6) Cell viability | ≥70% → continuation of the manufacturing process | <70% → utilization of cell culture | Propidium iodide staining method combined with advanced image analysis (ADAM-MCTM automated cell counter) |
| Stage III: Termination of AT-MSCs Culture and Formulation of the Final Product | |||
| Parameter | Correct Result | Incorrect Result | Method |
| Parameters (1)–(4) from Stage II: Culture of AT-MSCs | |||
| Cell number | 12 × 106 viable cells | >12 × 106 viable cells → the final product is not released | Propidium iodide staining method combined with advanced image analysis (ADAM-MCTM automated cell counter) |
| Cell viability | ≥70% → preparation of the final product | <70% → the final product is not released | Propidium iodide staining method combined with advanced image analysis (ADAM-MCTM automated cell counter) |
| Specification of the HE-ATMP | |||
|---|---|---|---|
| Parameter | Limit or Range | Method | Additional Information |
| (1) Cell morphology | Characteristic for MSCs (spindle-shaped, elongated cells possessing fibroblast-like morphology) | Light microscopy | Parameter tested before the last passage |
| (2) Endotoxins | <2.5 UI/mL | LAL method (Endosafe®-PTS™ system) | Parameter tested before the last passage; the product is released before completion of the test |
| (3) Sterility of the final product | Sterile | Direct inoculation (BD Bactec FX400 system) | Sample is collected at the last passage and after the final product formulation; because of the short life-time of the product, the product is released before completion of the test |
| (4) Cell number per dose | 10 × 106 viable cells | Propidium iodide staining method combined with advanced image analysis (ADAM-MCTM automated cell counter) | Parameter tested after the last passage |
| (5) Cell viability | ≥70% | Propidium iodide staining method combined with advanced image analysis (ADAM-MCTM automated cell counter) | Parameter tested after the last passage |
| Validation of Manufacturing Process of HE-ATMP | ||
|---|---|---|
| Parameter | Limit or Range | Results |
| (1) Cell morphology | Characteristic for MSCs (spindle-shaped, elongated cells possessing fibroblast-like morphology) | Batch no. 1: Correct |
| Batch no. 2: Correct | ||
| Batch no. 3: Correct | ||
| (2) Endotoxins | <2.5 UI/mL | Batch no. 1: Correct |
| Batch no. 2: Correct | ||
| Batch no. 3: Correct | ||
| (3) Sterility of the final product | Sterile | Batch no. 1: Sterile |
| Batch no. 2: Sterile | ||
| Batch no. 3: Sterile | ||
| (4) Cell number per dose | 10 × 106 viable cells | Batch no. 1: 10 × 106 viable cells |
| Batch no. 2: 10 × 106 viable cells | ||
| Batch no. 3: 10 × 106 viable cells | ||
| (5) Cell viability | ≥70% | Batch no. 1: 98% |
| Batch no. 2: 96% | ||
| Batch no. 3: 98% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labedz-Maslowska, A.; Szkaradek, A.; Mierzwinski, T.; Madeja, Z.; Zuba-Surma, E. Processing and Ex Vivo Expansion of Adipose Tissue-Derived Mesenchymal Stem/Stromal Cells for the Development of an Advanced Therapy Medicinal Product for use in Humans. Cells 2021, 10, 1908. https://doi.org/10.3390/cells10081908
Labedz-Maslowska A, Szkaradek A, Mierzwinski T, Madeja Z, Zuba-Surma E. Processing and Ex Vivo Expansion of Adipose Tissue-Derived Mesenchymal Stem/Stromal Cells for the Development of an Advanced Therapy Medicinal Product for use in Humans. Cells. 2021; 10(8):1908. https://doi.org/10.3390/cells10081908
Chicago/Turabian StyleLabedz-Maslowska, Anna, Agnieszka Szkaradek, Tomasz Mierzwinski, Zbigniew Madeja, and Ewa Zuba-Surma. 2021. "Processing and Ex Vivo Expansion of Adipose Tissue-Derived Mesenchymal Stem/Stromal Cells for the Development of an Advanced Therapy Medicinal Product for use in Humans" Cells 10, no. 8: 1908. https://doi.org/10.3390/cells10081908