
Learning Yeast Genetics from Miro Radman


Department of Biology, Rosenstiel Basic Medical Sciences Research Center, Brandeis University, Waltham, MA 02454-9110, USA
Cells 2021, 10(4), 945; https://doi.org/10.3390/cells10040945
Submission received: 29 March 2021
/
Revised: 13 April 2021
/
Accepted: 13 April 2021
/
Published: 20 April 2021
(This article belongs to the Special Issue DNA Repair, Genome Stability/Diversity, and Oxidative Stress and Aging, from Bacteria to Human Cells: A Themed Issue in Honor of Prof. Miroslav Radman)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Miroslav Radman’s far-sighted ideas have penetrated many aspects of our study of the repair of broken eukaryotic chromosomes. For over 35 years my lab has studied different aspects of the repair of chromosomal breaks in the budding yeast, Saccharomyces cerevisiae. From the start, we have made what we thought were novel observations that turned out to have been predicted by Miro’s extraordinary work in the bacterium Escherichia coli and then later in the radiation-resistant Dienococcus radiodurans. In some cases, we have been able to extend some of his ideas a bit further.
1. When Mismatch Repair Makes the Situation Worse
One of Miro’s stunning discoveries was that dam recA double mutants were extremely sensitive to base-pair substitution mutagens such as 2-amino purine (2-AP) [1]. These strains lack the deoxyadenine methyltransferase, which modifies DNA at GATC sites and directs mismatch repair to correct the newly copied DNA strand, and also lack homologous recombination mediated by the RecA protein. In fact, when treated with high doses of 2-AP, these double mutants could produce cells lacking any visible DNA at all [2,3]. However, this mismatch-stimulated killing was blocked by mutations in MutS, MutL, and MutH. The explanation was that—without the hemimethylation of GATC to direct the mismatch repair machinery to the newly replicated strand—MutS/L/H could initiate nicks on either strand and then two 5′ to 3′ exonucleases could converge, producing a double-strand break that required RecA for its repair.
The pioneering work of Bob Mortimer and Sy Fogel [4] had defined the key elements of gene conversion and mismatch repair in budding yeast meiosis. In the early-1980s there were no mismatch repair mutations yet identified in yeast, although alleles of some biosynthetic genes showed post-meiotic segregation (PMS), suggesting that they had avoided mismatch repair of heteroduplex DNA during meiotic recombination. At this time, my lab had begun to study meiotic recombination and figured out how to monitor meiotic recombination in real-time using Southern blots [5]. We realized we could also determine the length of gene conversion tracts by inserting a small number of sequence alterations into the homologous DNA segment that we were monitoring [6,7]. The region was created by integrating a pBR322 plasmid containing both a URA3 gene and a MAT sequence into the MAT locus, thus creating MATa------URA3---MATa and MATα-----URA3----MATα regions, such that a crossover would produce MATa-----URA3---MATα and MATα-----URA3---MATa reciprocal recombinants that were easily identified because they were non-mating (Figure 1). We introduced about 1 mismatched site each 1 kb along the 9-kb region so that we could estimate gene conversion tract lengths associated with crossing-over. Crossovers occurred in approximately 24% of tetrads, with an average gene conversion tract length of 1.5 kb. What we did not expect were tetrads in which one segregant was Ura3− MATa or MATα and the other 3 segregants were Ura3+, one of which had apparently undergone a crossover and was thus non-mating. Further analysis revealed that the Ura3− segregants had lost URA3 and the pBR322 sequences and had only a single copy of MATa or MATα (Figure 1). These exceptional segregants were generally not the result of an unequal crossover that would have created a segregant with three MAT loci and a duplication of pBR322 segments. We suggested that these exceptions could arise if crossing-over had occurred in the interval between the flanking MAT loci, creating heteroduplex that was subject to mismatch repair [6]. However, if mismatch repair initiated at two separate sites and then two exonucleases, on opposite strands, resected toward each other, they could produce a secondary DSB that could be most easily repaired by single-strand annealing between the flanking MAT loci. In our initial paper [5] we made this proposal in complete ignorance of Glickman and Radman’s [1] paper (and also similar results from McGraw and Marinus [8]). By 1990, the first yeast mismatch repair genes, PMS1, had been gene cloned and studied [9]; in collaboration with Seymour Fogel’s lab, we showed that knocking out PMS1 resulted in a suppression of the mismatch-dependent deletions arising in meiosis [10]. By then we had come to appreciate and properly cite Miro’s important insight. In meiosis, recombination between two homologous chromosomes occurs long after replication has been completed, so there are no strand-specific signals that would confine mismatch repair to one or the other chromatid.
All of these results led us to what we called the “Borts uncertainty principle”: To study the details of crossing-over in meiosis we had to introduce heterologies; but those heterologies altered the outcomes.
2. A Tale of Rejection
Recombination between mismatched DNA is suppressed, as Miro’s lab spectacularly demonstrated for the case of exchanges between Escherichia coli and Salmonella thyphimurium, whose genomes are 20% divergent [11,12,13]. This barrier can be overcome by removing the MutS/L/H mismatch repair proteins [11,14]. Later work suggested there might be two pathways to defeat homeologous recombination. One was independent of the MutH endonuclease but required the helicase UvrD, suggesting that heteroduplexes were recognized by MutS and MutL and then unwound [15].
In budding yeast, heterologous substrates are also less efficient in recombination than fully homologous sequences, both in meiotic and mitotic cells [16]. We studied heteroduplex rejection in a Rad51-independent assay, involving single-strand annealing (SSA) [17] (Figure 2). With 3% divergence, SSA was less than 50% efficient; but this barrier could be overcome by deleting the MutS homolog, Msh6. Similarly, repair was restored by a separation-of-function mutation in Msh2 that did not recognize single base pair mismatches but still allowed Msh2 to participate in the removal of nonhomologous single-strand DNA tails that are created during SSA [18]. Tail removal requires not only the Rad1-Rad10 endonuclease that is also needed in nucleotide excision repair, but also Msh2 and Msh3, which apparently recognize and stabilize 3′ flap structures attacked by Rad1-Rad10; moreover, clipping needs the scaffold protein, Slx4 and Saw1 [18,19,20]. So, in this process, Msh2 must associate both with Msh6 and with Msh3 and indeed the competition between rejection and tail-clipping can be tilted by overexpressing Msh6 [17,21].
Unlike the heteroduplex rejection process in E. coli, this discouragement of strand annealing is independent of the MutL homologs, Mlh1 and Pms1 [17]. However, the subsequent correction of mismatches in the hetDNA still needs the MutL homologs; in their absence, a colony contains both parental alleles.
In addition, heteroduplex rejection requires the BLM helicase homolog, Sgs1, and its associated proteins Rmi1 and Top3 [17,21]. Sgs1-Rmi1-Top3 were discovered to be a double-Holliday junction “dissolvase”, in which Top3 was needed to resolve the linked strands [22,23]. However, in SSA, Rmi1 and Top3 are still required, perhaps because unwinding starts at both ends and their resolution still needs TopIIIα topoisomerase activity or perhaps simply because Top3 is an essential structural element. Sgs1’s role in heteroduplex rejection has also been well-documented for spontaneous recombination between homeologous sequences [16].
But the 3′-ended nonhomologous tail at the end of heteroduplex DNA proves to be more than a nuisance; it is an essential part of heteroduplex rejection. We constructed a series of strains that can repair an HO-induced DSB by break-induced replication (BIR). We designed these strains so that all the homology shared between the DSB end and the repair template occurred within a 108-bp synthetic intron and in which there was a perfect match between the DSB end and the donor [24]. As a control, we used Cas9 to generate ends with as much as a 34-bp nonhomologous end. With the tailed strain, even a single mismatch in the 108-bp region was sufficient to reduce BIR to about 50% of the level seen in a msh2∆ derivative; but in the tailless strain, there was no reduction compared to msh2∆ even when every 9th base was mismatched. We have confirmed these results by creating strains in which SSA occurs between 3% mismatched flanking sequences either with long honhomologous tails or such that cleavage occurred between two directly repeated sequences; again, the tailless strain was insensitive to the presence of mismatches (E. Sapède and J.E.H., MS in preparation). How the tail plays such a critical role in recruiting the STR complex and links this to Msh2-Msh6 that detects the mismatches will be important to figure out.
We should not lose sight of the key insight by Radman’s lab that sequence divergence plays a central role in speciation, by preventing recombination between “cousins” as they evolve away from each other [11,12,13]. This is also the case in eukaryotes, as crosses between highly divergent strains—though still both Saccharomyces cerevisiae—display marked inviability in meiosis; but this barrier can be greatly reduced by disabling mismatch repair [25].
3. BIR and E-SDSA
One of the most stunning instances of DSB repair is the ability of D. radiodurans to reassemble its genome after astonishingly high levels of ionizing radiation [26,27]. Radman and his colleagues showed that the restitution depended on homologous recombination and on the fact that D. radiodurans carries multiple copies of its genome (it would be impossible to stitch together a shattered genome from a single copy). They considered a number of possible mechanisms that would allow the restitution of an intact genome and concluded that repair would demand a combination of strand invasion between a resected end and a homologous segment, followed by new DNA synthesis, then dissociation of the newly copied DNA and then either another round of invasion/synthesis with another fragmented template or the annealing of two such extended, newly copied DNAs. From the perspective of 2021, the distinction between an incomplete synthesis-dependent strand annealing (SDSA) event and a BIR event has become more difficult to articulate, especially because we have shown that SDSA and BIR are alternative outcomes of a common initiation event in budding yeast [28]. How often second-end capture occurs (for the completion of SDSA) or fails (resulting in BIR) depends on a number of factors. For example, two 3′ to 5′ helicases, Sgs1 (BLM) and Mph1 (FANCM) play opposing roles in assuring second-end capture: while mph1∆ drives nearly all the events into BIR, sgs1∆ results in almost all outcomes as gene conversions through SDSA. Likely, Sgs1 promotes heteroduplex rejection when second-end capture is attempted, even if there are no mismatches.
The efficiency of second-end capture is also dependent on the strand-annealing functions of Rad52 and its cousin, Rad59 [29]. If the region within the donor sequence that is required for second-end annealing indeed harbors mismatches, the frequency of BIR also goes up significantly at the cost of gene conversion (M.F. Afreen, R. Anand and J.E.H., unpublished).
At least in ectopic recombination, second-end capture may also involve a nonhomologous extension if the region copied from the donor goes past the limits of shared homology; such extensions would need to be clipped off.
A model for the events of E-SDSA (named BIR-SSA in yeast [30,31]) involves the repair of inverted repeats which there is a DSB in one of two repeats sitting in inverted orientation [32] or on a chromosome (Figure 3). When the extent of shared homology is only 33 bp on either side of an HO-induced DSB, Rad51 actually impairs repair; apparently, the amount of homology within the filament is too short to effectuate strand invasion [32]. In the absence of Rad51, repair by BIR-SSA is surprisingly robust, but now dependent on several recombination factors not required for Rad51-dependent gene conversion, including Rad59, Rdh54/Tid1, and Pol32, a non-essential subunit of DNA polymerase δ. In Dienococcus, there is also a RecA-independent aspect to restoring genome integrity [33].
Though hardly in the same league as D. radiodurans, budding yeast is actually quite adept in repairing a heavily irradiated diploid. Ref. [34] introduced about 250 DSBs into a diploid cell arrested in G2. Thus among 32 chromosomes, with 4 chromatids, there were about 2 breaks per average-sized chromosome. Survival was measured to be 7% and 28% in two separate experiments. About 2% of the repair events resulted in chromosome aberrations. It would be interesting to determine what proportion of these events required BIR and SSA.
4. SOS
And then of course there’s the SOS response to DNA damage. Miro’s ideas shaped everyone’s thinking about the inducibility of DNA repair functions and the cellular responses to damage [35,36]. Yeast is exquisitely sensitive to DNA damage since a single DSB will trigger prolonged checkpoint-mediated arrest. Miro’s ideas as well as those of Evelyn Witkin [37,38] prompted Weinert and Hartwell [39,40,41] and Elledge [42,43] to identify the key elements of the DNA damage checkpoint in budding yeast. Budding yeast will arrest prior to mitosis in response to a single HO-induced DSB but then will adapt after 12–15 h; but two DSBs were sufficient to prevent cell cycle progression entirely [44,45]. The DNA damage checkpoint, governed by the ATR and ATM kinases, results in the induction of many genes and the post-translational modification of many proteins involved in DSB repair [46].
And as with E. coli, damage-induced DNA repair in budding yeast is surprisingly mutagenic; recombination-associated DNA repair exhibits a 1000-fold increase in mutations in the newly-copied sequences compared to normal replication [47,48]. However, different from E. coli, where much of the mutagenic repair can be attributed to error-prone alternative DNA polymerases, in yeast, the same polymerases, but acting outside the reassuring confines of the intact replication fork—most notably DNA polymerase δ—are responsible for the increased error rate. In contrast, translesion DNA polymerases appear to play only a minor role during DSB repair. Many of the errors suggest that the DNA polymerase frequently dissociates from its template and results in -1 frameshifts in homonucleotide runs, quasipalindrome mutations, and template jumps to homeologous sequences even on different chromosomes [47,49]. These events are also largely independent of the mismatch repair machinery. We speculate that Msh/Mlh proteins normally track behind the replication fork and correct newly generated errors [50], but in repair DNA synthesis this intimate association appears to be lost.
5. Summing up
Miro Radman’s contributions to our understanding of DNA repair and mutagenesis have spanned 4 decades. As experimental techniques became more and more precise, Miro’s insights have gone deeper and deeper. We honor his longevity and his passion.
Funding
Research in the Haber lab was funded by NIH grant R35GM127029 and previous grants.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data mentioned in this article are published or available from the author.
Acknowledgments
Work in my lab has been supported for 49 years by the NIH, most recently by NIH grant R35GM127029 and has been a labor of love by dozens of dedicated postdocs, grad students, undergraduates and technicians. Interactions with Miro Radman have been fueled by good food, good wine and, on several occasions, good gypsy music.
Conflicts of Interest
The author declares no conflict of interest.
References
- Glickman, B.W.; Radman, M. Escherichia coli mutator mutants deficient in methylation-instructed DNA mismatch correction. Proc. Natl. Acad. Sci. USA 1980, 77, 1063–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doutriaux, M.P.; Wagner, R.; Radman, M. Mismatch-stimulated killing. Proc. Natl. Acad. Sci. USA 1986, 83, 2576–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matic, I.; Ekiert, D.; Radman, M.; Kohiyama, M. Generation of DNA-free Escherichia coli cells by 2-aminopurine requires mismatch repair and nonmethylated DNA. J. Bacteriol. 2006, 188, 339–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogel, S.; Mortimer, R.; Lusnak, K.; Tavares, F. Meiotic gene conversion: A signal of the basic recombination event in yeast. Cold Spring Harb. Symp. Quant. Biol. 1979, 43 Pt 2, 1325–1341. [Google Scholar] [CrossRef]
- Borts, R.H.; Lichten, M.; Haber, J.E. Analysis of meiosis-defective mutations in yeast by physical monitoring of recombination. Genetics 1986, 113, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Borts, R.H.; Haber, J.E. Meiotic recombination in yeast: Alteration by multiple heterozygosities. Science 1987, 237, 1459–1465. [Google Scholar] [CrossRef]
- Borts, R.H.; Haber, J.E. Length and distribution of meiotic gene conversion tracts and crossovers in Saccharomyces cerevisiae. Genetics 1989, 123, 69–80. [Google Scholar] [CrossRef]
- McGraw, B.R.; Marinus, M.G. Isolation and characterization of Dam+ revertants and suppressor mutations that modify secondary phenotypes of dam-3 strains of Escherichia coli K-12. Mol. Genet. Genom. 1980, 178, 309–315. [Google Scholar] [CrossRef]
- Kramer, W.; Kramer, B.; Williamson, M.S.; Fogel, S. Cloning and nucleotide sequence of DNA mismatch repair gene PMS1 from Saccharomyces cerevisiae: Homology of PMS1 to procaryotic MutL and HexB. J. Bacteriol. 1989, 171, 5339–5346. [Google Scholar] [CrossRef] [Green Version]
- Borts, R.H.; Leung, W.Y.; Kramer, W.; Kramer, B.; Williamson, M.; Fogel, S.; Haber, J.E. Mismatch repair-induced meiotic recombination requires the pms1 gene product. Genetics 1990, 124, 573–584. [Google Scholar] [CrossRef]
- Rayssiguier, C.; Thaler, D.S.; Radman, M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 1989, 342, 396–401. [Google Scholar] [CrossRef]
- Matic, I.; Radman, M.; Rayssiguier, C. Structure of recombinants from conjugational crosses between Escherichia coli donor and mismatch-repair deficient Salmonella typhimurium recipients. Genetics 1994, 136, 17–26. [Google Scholar] [CrossRef]
- Matic, I.; Rayssiguier, C.; Radman, M. Interspecies gene exchange in bacteria: The role of SOS and mismatch repair systems in evolution of species. Cell 1995, 80, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Huang, H.V. Effect of base pair mismatches on recombination via the RecBCD pathway. Mol. Genet. Genom. 1989, 218, 358–360. [Google Scholar] [CrossRef]
- Tham, K.C.; Hermans, N.; Winterwerp, H.H.; Cox, M.M.; Wyman, C.; Kanaar, R.; Lebbink, J.H. Mismatch repair inhibits homeologous recombination via coordinated directional unwinding of trapped DNA structures. Mol. Cell 2013, 51, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Adjiri, A.; New, L.; Crouse, G.F.; Robertson, S.J. Mitotic crossovers between diverged sequences are regulated by mismatch repair proteins in Saccaromyces cerevisiae. Mol. Cell. Biol. 1996, 16, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, N.; Goldfarb, T.; Studamire, B.; Alani, E.; Haber, J.E. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. USA 2004, 101, 9315–9320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studamire, B.; Price, G.; Sugawara, N.; Haber, J.E.; Alani, E. Separation-of-function mutations in Saccharomyces cerevisiae MSH2 that confer mismatch repair defects but do not affect nonhomologous-tail removal during recombination. Mol. Cell. Biol. 1999, 19, 7558–7567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, G.W.; Sugawara, N.; Dong, J.; Toth, R.; Lee, S.E.; Haber, J.E.; Rouse, J. Mec1/Tel1-dependent phosphorylation of Slx4 stimulates Rad1-Rad10-dependent cleavage of non-homologous DNA tails. DNA Repair 2010, 9, 718–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flott, S.; Alabert, C.; Toh, G.W.; Toth, R.; Sugawara, N.; Campbell, D.G.; Haber, J.E.; Pasero, P.; Rouse, J. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol. Cell. Biol. 2007, 27, 6433–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, U.; George, C.M.; Lyndaker, A.M.; Alani, E. A Delicate Balance between Repair and Replication Factors Regulates Recombination between Divergent DNA Sequences in Saccharomyces cerevisiae. Genetics 2016, 202, 525–540. [Google Scholar] [CrossRef] [Green Version]
- Ira, G.; Malkova, A.; Liberi, G.; Foiani, M.; Haber, J.E. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 2003, 115, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Beach, A.; Li, K.; Haber, J. Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 2017, 544, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Hunter, N.; Chambers, S.R.; Louis, E.J.; Borts, R.H. The mismatch repair system contributes to meiotic sterility in an interspecific yeast hybrid. EMBO J. 1996, 15, 1726–1733. [Google Scholar] [CrossRef]
- Repar, J.; Cvjetan, S.; Slade, D.; Radman, M.; Zahradka, D.; Zahradka, K. RecA protein assures fidelity of DNA repair and genome stability in Deinococcus radiodurans. DNA Repair 2010, 9, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Zahradka, K.; Slade, D.; Bailone, A.; Sommer, S.; Averbeck, D.; Petranovic, M.; Lindner, A.B.; Radman, M. Reassembly of shattered chromosomes in Deinococcus radiodurans. Nature 2006, 443, 569–573. [Google Scholar] [CrossRef]
- Mehta, A.; Beach, A.; Haber, J.E. Homology Requirements and Competition between Gene Conversion and Break-Induced Replication during Double-Strand Break Repair. Mol. Cell 2017, 65, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, D.N.; Pham, N.; Tsai, A.M.; Janto, A.N.; Choi, J.; Ira, G.; Haber, J.E. A Rad51-independent pathway promotes single-strand template repair in gene editing. PLoS Genet. 2020, 16, e1008689. [Google Scholar] [CrossRef]
- Kang, L.E.; Symington, L.S. Aberrant double-strand break repair in rad51 mutants of Saccharomyces cerevisiae. Mol. Cell. Biol. 2000, 20, 9162–9172. [Google Scholar] [CrossRef] [Green Version]
- Spell, R.M.; Jinks-Robertson, S. Role of mismatch repair in the fidelity of RAD51- and RAD59-dependent recombination in Saccharomyces cerevisiae. Genetics 2003, 165, 1733–1744. [Google Scholar] [CrossRef] [Green Version]
- Ira, G.; Haber, J.E. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol. Cell. Biol. 2002, 22, 6384–6392. [Google Scholar] [CrossRef] [Green Version]
- Slade, D.; Lindner, A.B.; Paul, G.; Radman, M. Recombination and replication in DNA repair of heavily irradiated Deinococcus radiodurans. Cell 2009, 136, 1044–1055. [Google Scholar] [CrossRef] [Green Version]
- Argueso, J.L.; Westmoreland, J.; Mieczkowski, P.A.; Gawel, M.; Petes, T.D.; Resnick, M.A. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. USA 2008, 105, 11845–11850. [Google Scholar] [CrossRef] [Green Version]
- Bjedov, I.; Tenaillon, O.; Gerard, B.; Souza, V.; Denamur, E.; Radman, M.; Taddei, F.; Matic, I. Stress-induced mutagenesis in bacteria. Science 2003, 300, 1404–1409. [Google Scholar] [CrossRef]
- Radman, M. SOS repair hypothesis: Phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci. 1975, 5A, 355–367. [Google Scholar] [CrossRef]
- Witkin, E.M. Ultraviolet mutagenesis in bacteria: The inducible nature of error-prone repair. An. Acad. Bras. Ciências 1973, 45, 185–191. [Google Scholar]
- Witkin, E.M. Radiation-induced mutations and their repair. Science 1966, 152, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.A.; Kiser, G.L.; Hartwell, L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994, 8, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, T.A.; Hartwell, L.H. Characterization of RAD9 of Saccharomyces cerevisiae and evidence that its function acts posttranslationally in cell cycle arrest after DNA damage. Mol. Cell. Biol. 1990, 10, 6554–6564. [Google Scholar] [CrossRef] [Green Version]
- Weinert, T.A.; Hartwell, L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 1988, 241, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.B.; Zhou, Z.; Siede, W.; Friedberg, E.C.; Elledge, S.J. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 1994, 8, 2401–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, Y.; Desany, B.A.; Jones, W.J.; Liu, Q.; Wang, B.; Elledge, S.J. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 1996, 271, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Sandell, L.L.; Zakian, V.A. Loss of a yeast telomere: Arrest, recovery, and chromosome loss. Cell 1993, 75, 729–739. [Google Scholar] [CrossRef]
- Lee, S.E.; Moore, J.K.; Holmes, A.; Umezu, K.; Kolodner, R.D.; Haber, J.E. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 1998, 94, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Waterman, D.P.; Haber, J.E.; Smolka, M.B. Checkpoint Responses to DNA Double-Strand Breaks. Annu. Rev. Biochem. 2020, 89, 103–133. [Google Scholar] [CrossRef] [Green Version]
- Hicks, W.M.; Kim, M.; Haber, J.E. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 2010, 329, 82–85. [Google Scholar] [CrossRef] [Green Version]
- Deem, A.; Keszthelyi, A.; Blackgrove, T.; Vayl, A.; Coffey, B.; Mathur, R.; Chabes, A.; Malkova, A. Break-induced replication is highly inaccurate. PLoS Biol. 2011, 9, e1000594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsaponina, O.; Haber, J.E. Frequent Interchromosomal Template Switches during Gene Conversion in S. cerevisiae. Mol. Cell 2014, 55, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombauer, H.; Srivatsan, A.; Putnam, C.D.; Kolodner, R.D. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 2011, 334, 1713–1716. [Google Scholar] [CrossRef] [Green Version]
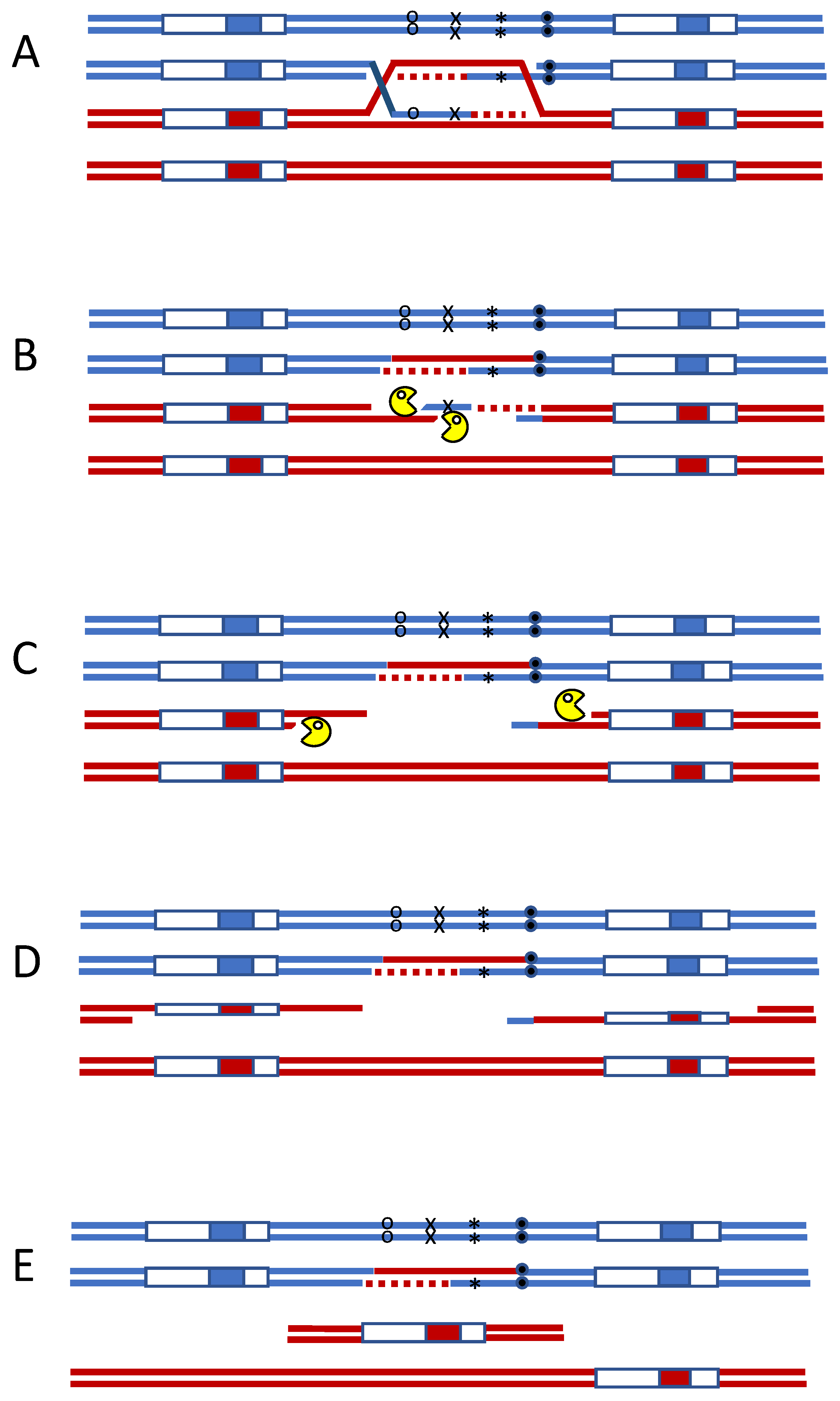
Figure 1.
Mismatch repair initiates secondary DSBs during meiotic recombination. Heteroduplex DNA formed during meiotic recombination. (A) can be attacked by two independent mismatch repair complexes, oriented on opposite strands (B). Exonuclease-dependent removal of adjacent DNA can lead to secondary DSBs (C). When the region is flanked by repeated sequences, single-strand annealing (SSA) will result in the deletion of the intervening DNA (D,E).
Figure 1.
Mismatch repair initiates secondary DSBs during meiotic recombination. Heteroduplex DNA formed during meiotic recombination. (A) can be attacked by two independent mismatch repair complexes, oriented on opposite strands (B). Exonuclease-dependent removal of adjacent DNA can lead to secondary DSBs (C). When the region is flanked by repeated sequences, single-strand annealing (SSA) will result in the deletion of the intervening DNA (D,E).

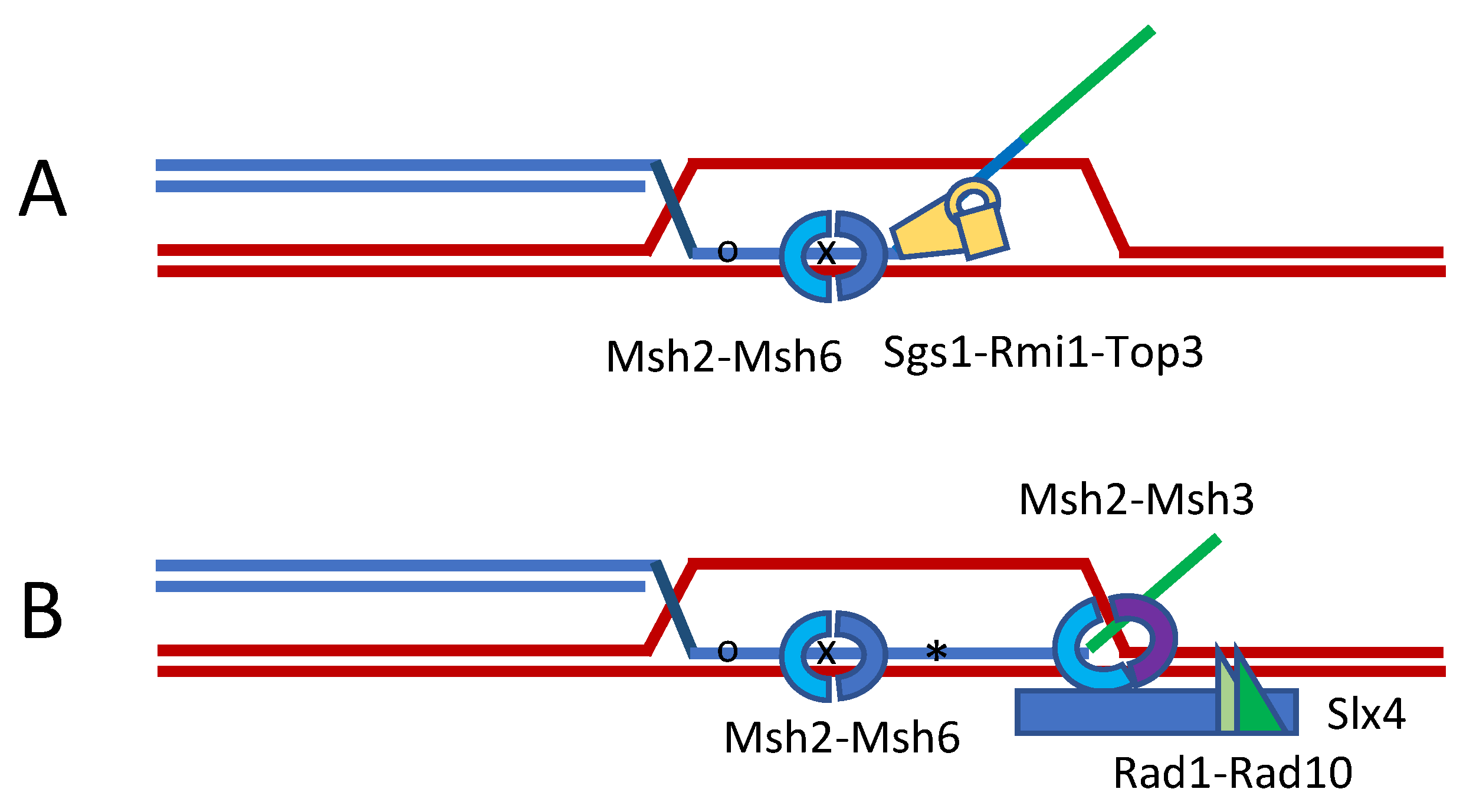
Figure 2.
Heteroduplex rejection during strand invasion. The formation of heteroduplex DNA during strand invasion or in SSA will stimulate its unwinding. (A) This anti-recombination mechanism is greatly stimulated by the presence of a nonhomologous 3′ tail, which attracts Slx4 and Msh2-Msh3 to enhance the tail’s removal by Rad1-Rad10 (B). Recognition of the mismatches requires Msh2-Msh6 and is unwound by the 3′ to 5′ helicase Sgs1 (BLM)-Rmi1-Top3. Heteroduplex rejection does not require MutL homologs, but these are needed to repair the mismatches.
Figure 2.
Heteroduplex rejection during strand invasion. The formation of heteroduplex DNA during strand invasion or in SSA will stimulate its unwinding. (A) This anti-recombination mechanism is greatly stimulated by the presence of a nonhomologous 3′ tail, which attracts Slx4 and Msh2-Msh3 to enhance the tail’s removal by Rad1-Rad10 (B). Recognition of the mismatches requires Msh2-Msh6 and is unwound by the 3′ to 5′ helicase Sgs1 (BLM)-Rmi1-Top3. Heteroduplex rejection does not require MutL homologs, but these are needed to repair the mismatches.

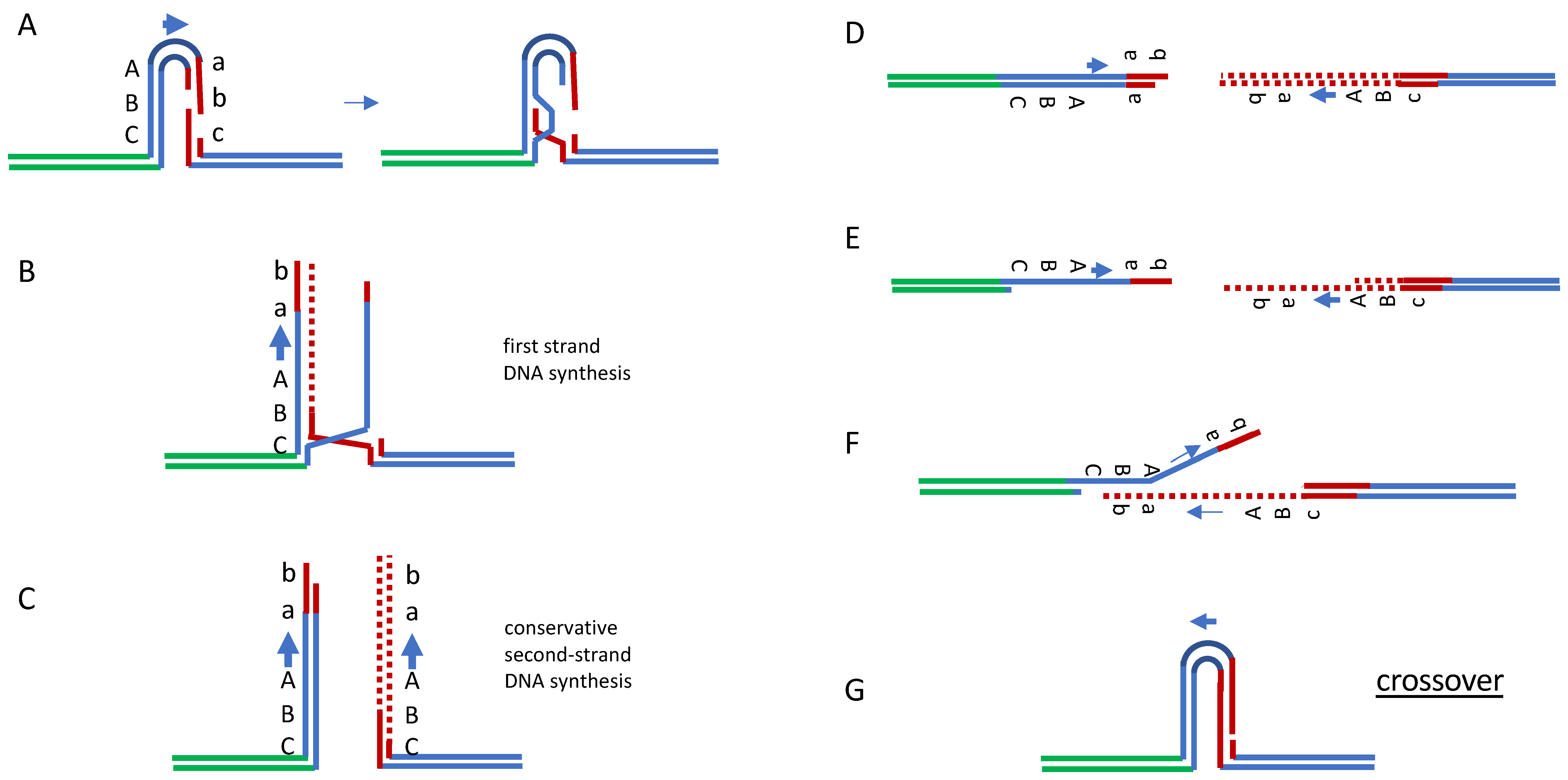
Figure 3.
Rad51-independent repair by break-induced replication and single-strand annealing. Extended SDSA can be modeled in yeast by recombination between inverted repeated sequences in which one repeat suffers a DSB. Break-indued replication (A–C), followed by resection of the ends (D,E) and SSA (F), will complete repair that can result either in a crossover (shown, (G)) or noncrossover with equal probability.
Figure 3.
Rad51-independent repair by break-induced replication and single-strand annealing. Extended SDSA can be modeled in yeast by recombination between inverted repeated sequences in which one repeat suffers a DSB. Break-indued replication (A–C), followed by resection of the ends (D,E) and SSA (F), will complete repair that can result either in a crossover (shown, (G)) or noncrossover with equal probability.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Haber, J.E. Learning Yeast Genetics from Miro Radman. Cells 2021, 10, 945. https://doi.org/10.3390/cells10040945
AMA Style
Haber JE. Learning Yeast Genetics from Miro Radman. Cells. 2021; 10(4):945. https://doi.org/10.3390/cells10040945
Chicago/Turabian StyleHaber, James E. 2021. "Learning Yeast Genetics from Miro Radman" Cells 10, no. 4: 945. https://doi.org/10.3390/cells10040945
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.