
Genome-Wide Identification and Expression Analysis of SOS Response Genes in Salmonella enterica Serovar Typhimurium

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Culture Media and Growth Conditions
2.2. RNA Isolation, cDNA Library Preparation and Illumina Sequencing
2.3. Read Processing and Alignment
2.4. Quantification of Absolute Gene Expression Levels
2.5. Differential Gene Expression Analysis with Three Biological Replicates
2.6. Identification of LexA Boxes
2.7. Construction of Transcriptional GFP Fusions
2.8. Flow Cytometry
2.9. Statistical Analysis
3. Results
3.1. Identification of LexA Boxes in the Chromosome of S. enterica Serovar Typhimurium
3.2. Identification of SOS Response Loci
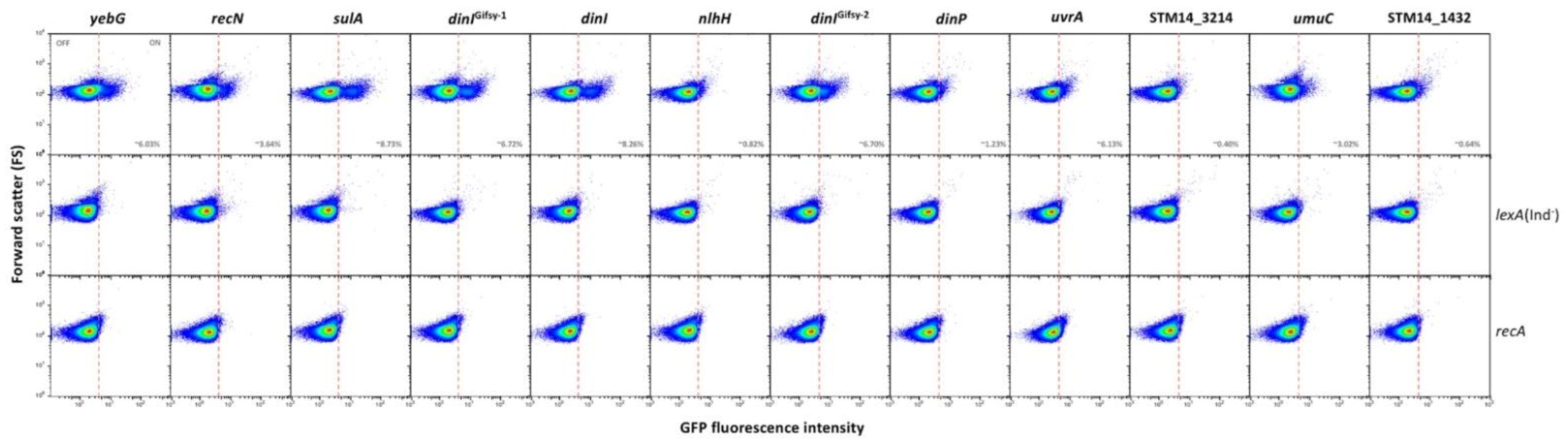
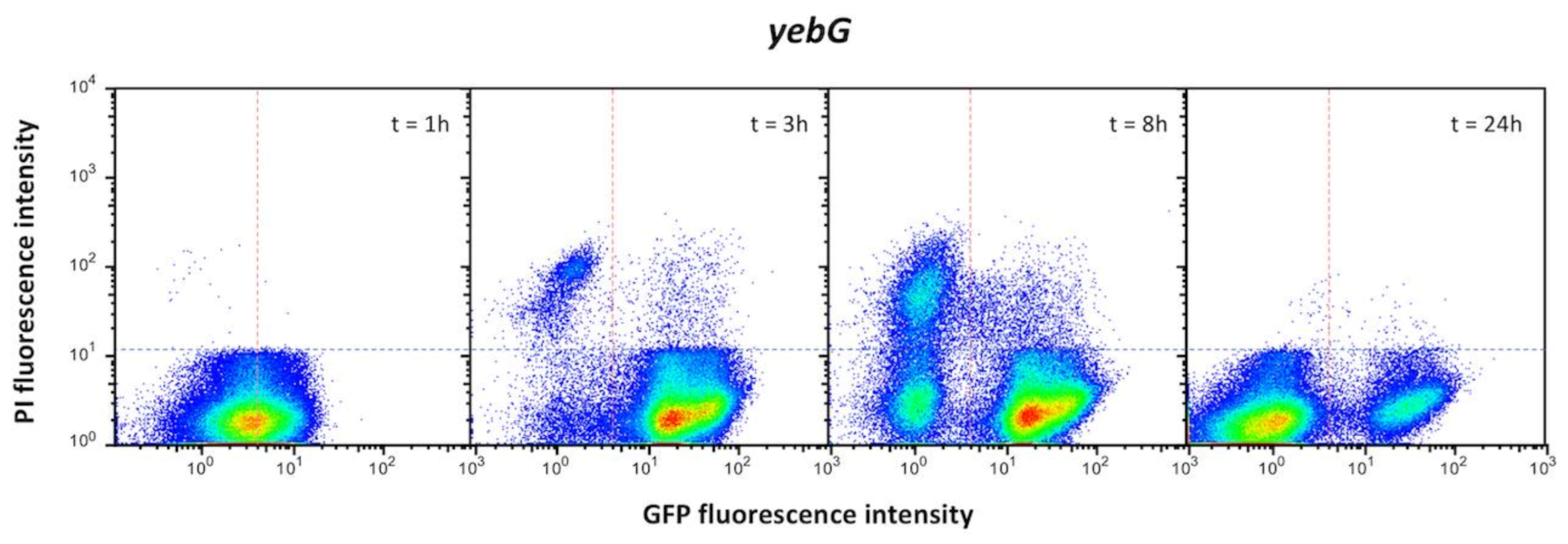
3.3. Single Cell Analysis of SOS Gene Expression in the Absence of SOS Induction
- (i).
- Group I, which is the most numerous (12 loci) presents bistable expression: the bacterial population contains a major subpopulation in which the gene is completely OFF and a smaller subpopulation of cells in which the gene is expressed (ON). The size of the ON subpopulation varies from one gene to another, ranging from ~8.7% in sulA to 1.3% in dinP and 0.8% and nlhH. In all loci, the ON subpopulation is absent in lexA (Ind−) and recA backgrounds. Expression of lexA, recA, umuDC and colicin genes in non-inducing conditions has been described previously in E. coli [52,53,54], and the formation of ON subpopulations under such conditions has been proposed to be triggered by activation of the SOS response upon spontaneous DNA strand breakage [53]. In agreement with this view, our analysis of gfp fusions did not detect ON cells in a recA background (Figure 1), which seems to rule out the possibility that the subpopulation might be produced by spontaneous alleviation of LexA repression (if that were the case, an ON subpopulation should be still detected in a recA mutant). Furthermore, the size of the ON subpopulation increased in a recD background and to a lesser extent in a recF background (Supplementary Figure S3), in agreement with the major role played by the RecBCD recombination pathway in double-strand break repair [55,56].
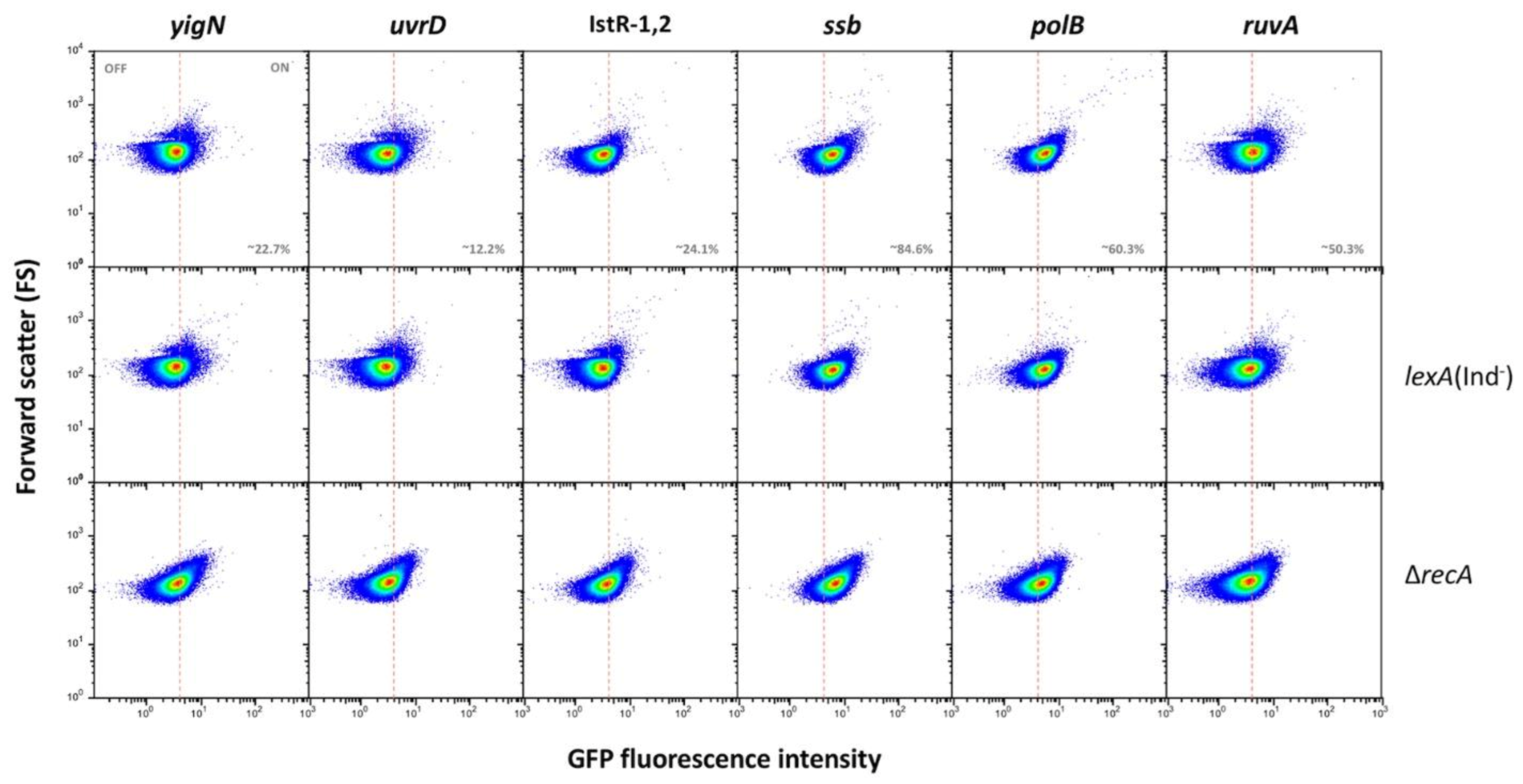
- (ii).
- Group II genes show “noisy”, heterogeneous expression that splits the population into OFF and ON subpopulations. The percentages of ON cells are larger than in genes of group I (from ~13% in uvrD up to ~85% in ssb). However, the main difference with group I is that formation of ON cells remains unaltered in lexA (Ind−) and recA backgrounds (Figure 2). We thus conclude that expression of these genes under non-inducing conditions is not under LexA control. Because this group contains housekeeping genes, a tentative interpretation is that the genes may be active in a subpopulation of cells in the absence of DNA damage. It is also possible that cells in the OFF state might have expression levels below the threshold for experimental detection.
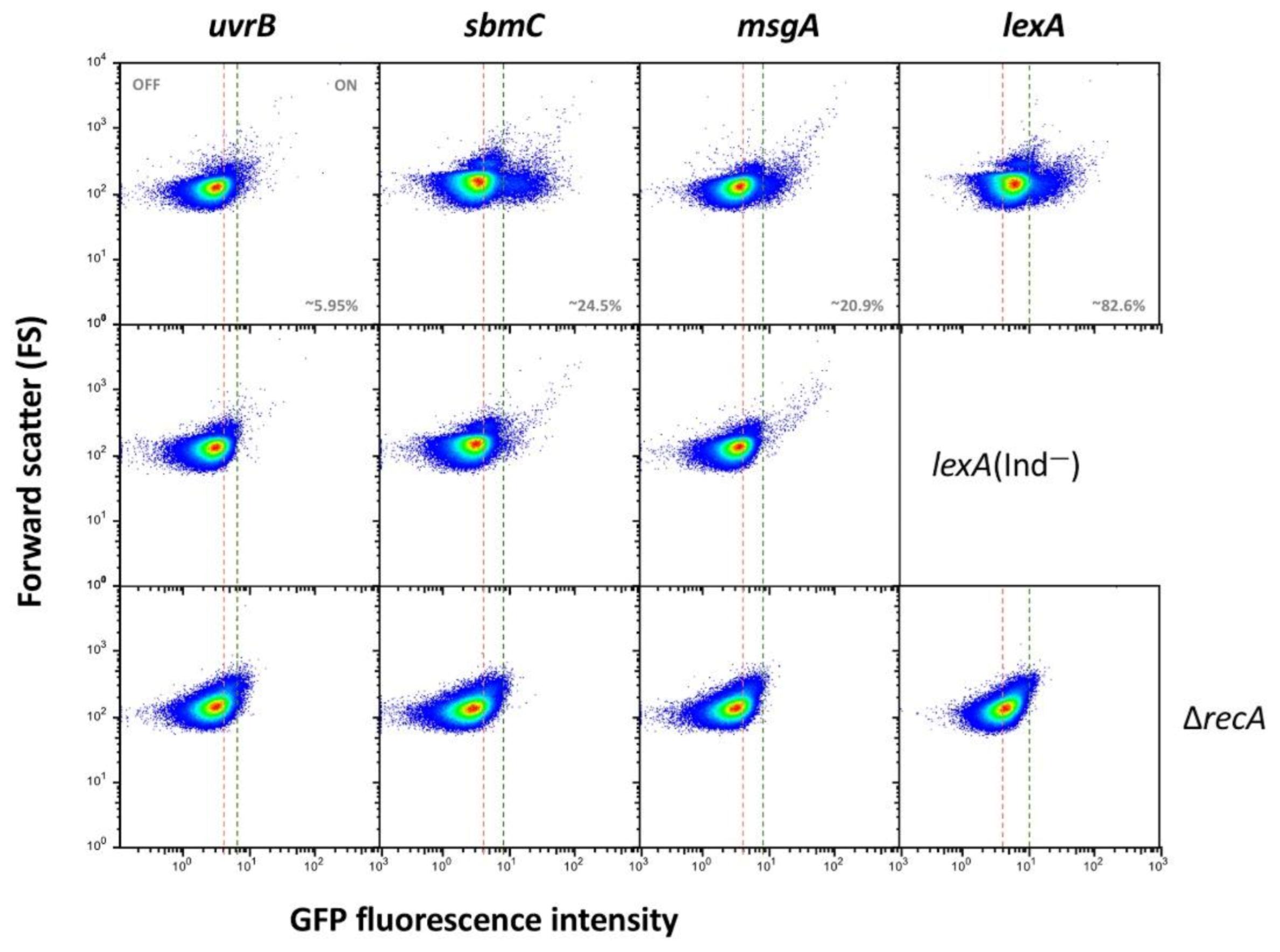
- (iii).
- Genes classified into group III show heterogeneous expression that is not bimodal, and two ON subpopulations are detected. However, these subpopulations differ in their expression level, and only the ON subpopulation with higher expression level disappears in a lexA (Ind−) mutant (Figure 3). The latter observation has a paradoxical side as formation of the “high ON” subpopulation appears to be LexA-dependent. Interestingly, the lexA gene belongs to this group. Expression of lexA in a lexA (Ind−) background was not tested because a lexA::gfp fusion could not be constructed.
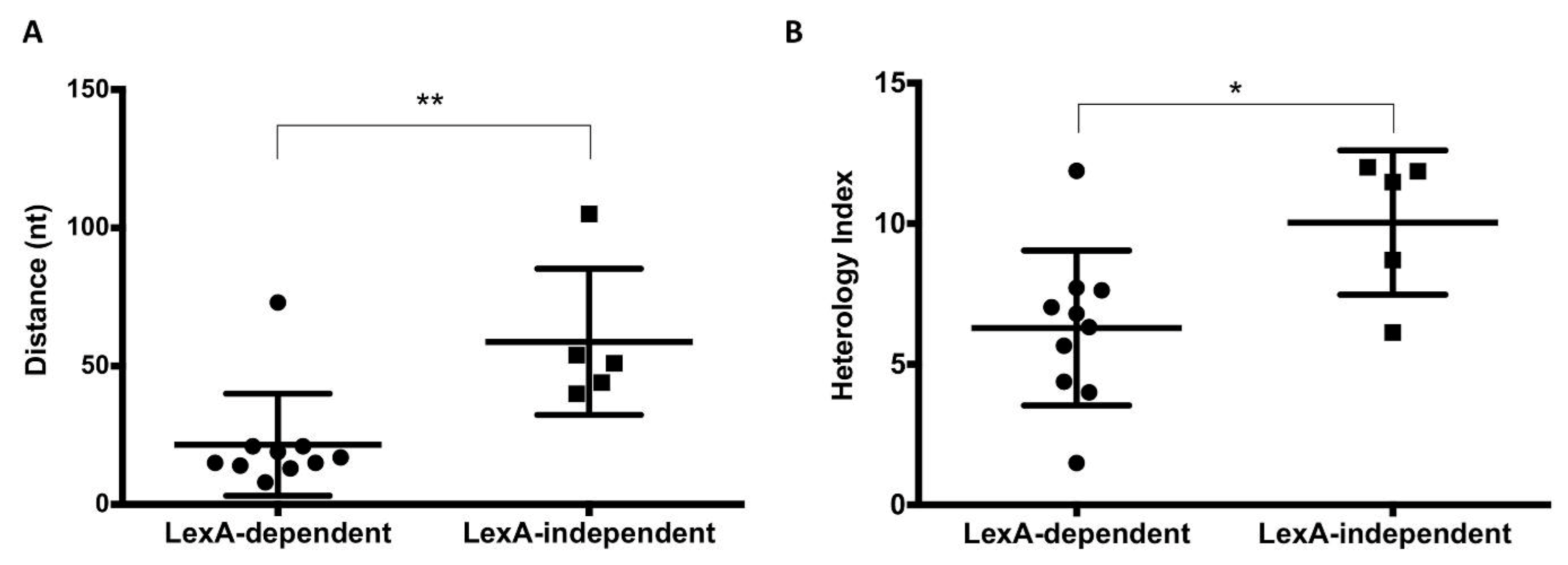
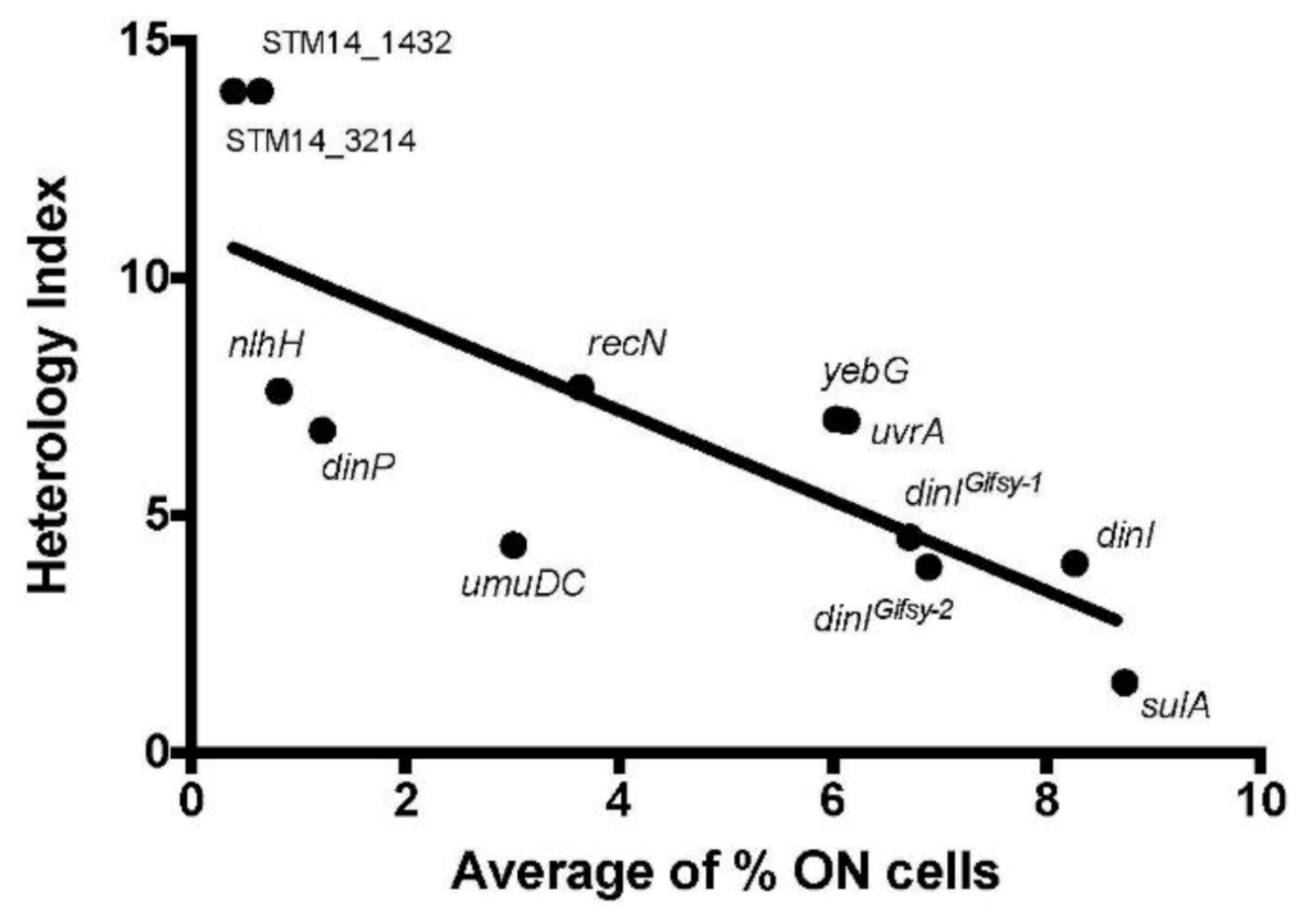
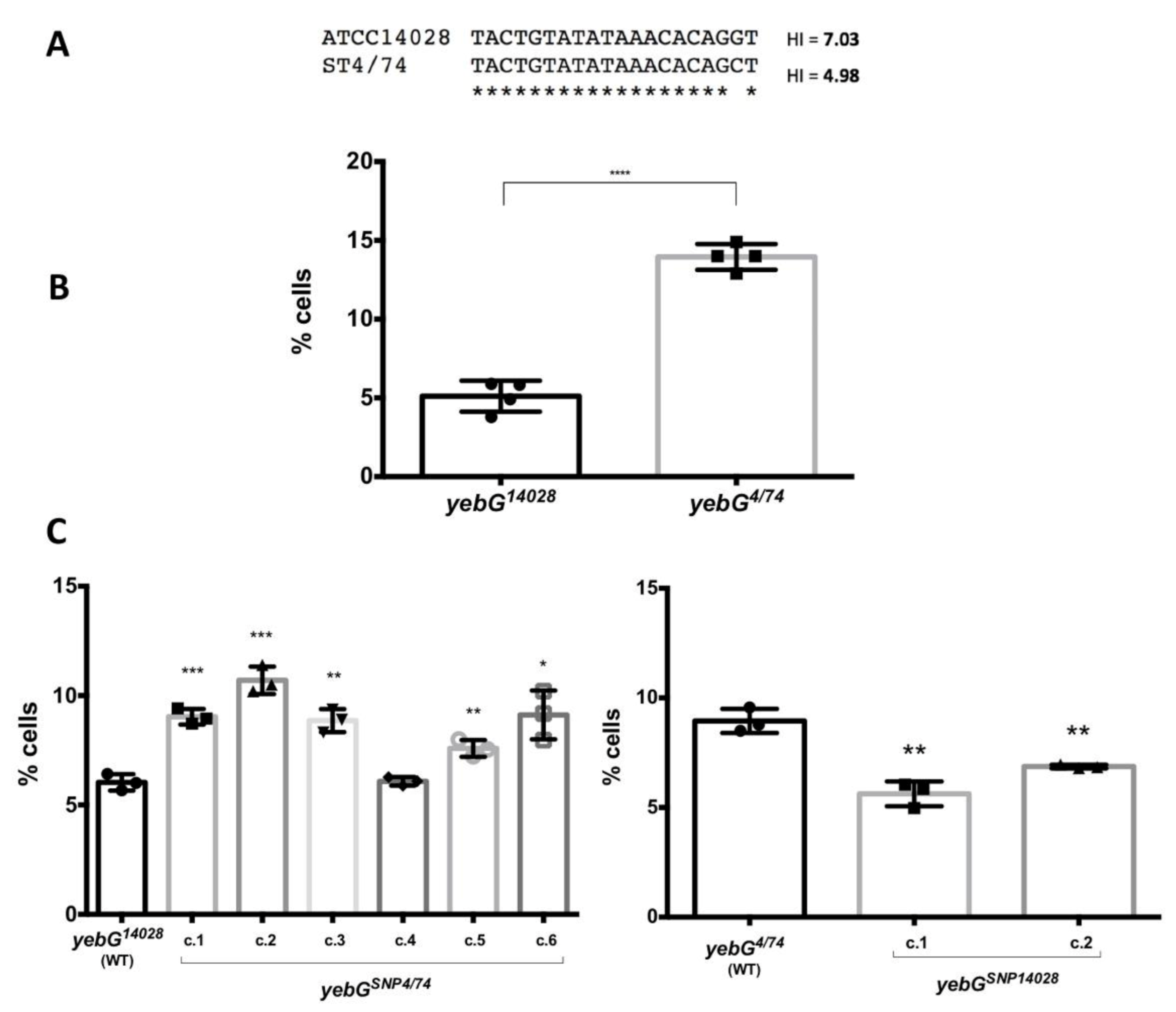
3.4. Influence of the Distance and the Nucleotide Sequence of the LexA Box on the Expression Patterns of SOS Genes
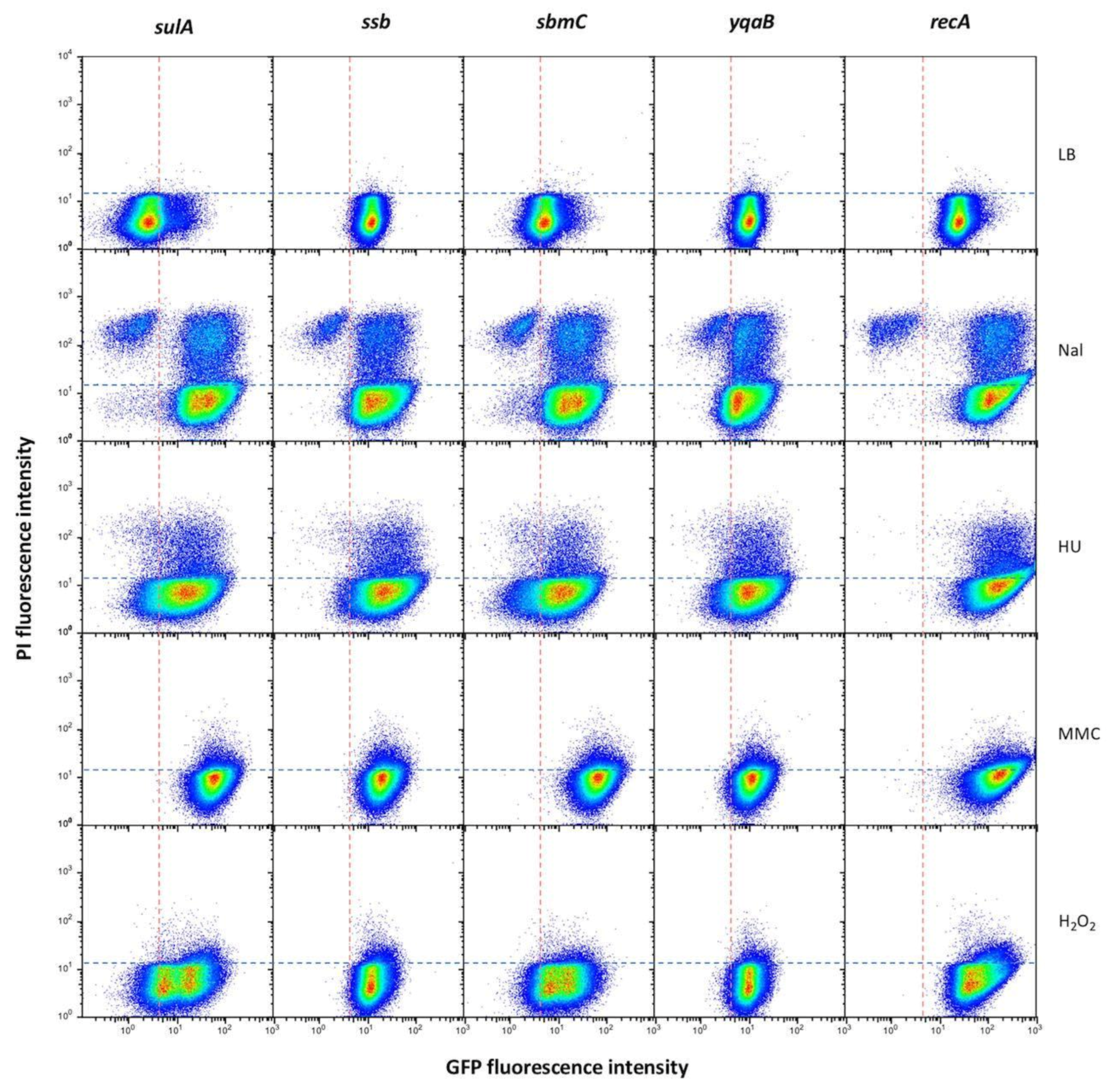
3.5. Gene Expression Patterns of S. enterica SOS Genes upon Activation of the SOS Response
- (i).
- The locus-specific expression patterns detected in non-inducing conditions largely disappeared upon SOS induction, with minor differences that did not correlate with the group classification (Figure 7). Differences were, however, seen depending on the DNA inducer, which seems to be a major determinant of the expression pattern.
- (ii).
- A large subpopulation of live cells (GFP+ PI−) with an active SOS response was detected in all loci under study.
- (iii).
- Subpopulations made of GFP+PI+ cells were also detected. The fact that such cells had an active SOS response but were a PI+ suggests that they may have a compromised cell membrane. Hence, they may be tentatively considered dead or bound to die.
- (iv).
- Subpopulations of dead cells that did not show SOS induction (GFP− PI+) were also detected. A subpopulation of this kind was especially conspicuous upon treatment with nalidixic acid. A tentative interpretation is either that SOS induction did not take place in such cells or that SOS induction failed to tolerate DNA damage.
- (v).
- Detection of subpopulations that did not show GFP nor PI fluorescence admits more than one explanation. One is that repair of DNA damage is highly efficient in such cells, thus permitting that the SOS response is rapidly turned off. This explanation seems, however, unlikely, as a relatively stable GFP variant has been used [39]. An alternative possibility is that the non-fluorescent subpopulation is made of cells that survive DNA damage without inducing the SOS response, and an attractive speculation is that they might be in a dormant state. However, we cannot ignore the limitations of using PI as indicator of cell death: as a DNA binding agent, if the cell has lost its genetic material due to severe damage it will not be stained and will appear in the “live” fraction (GFP− PI−).
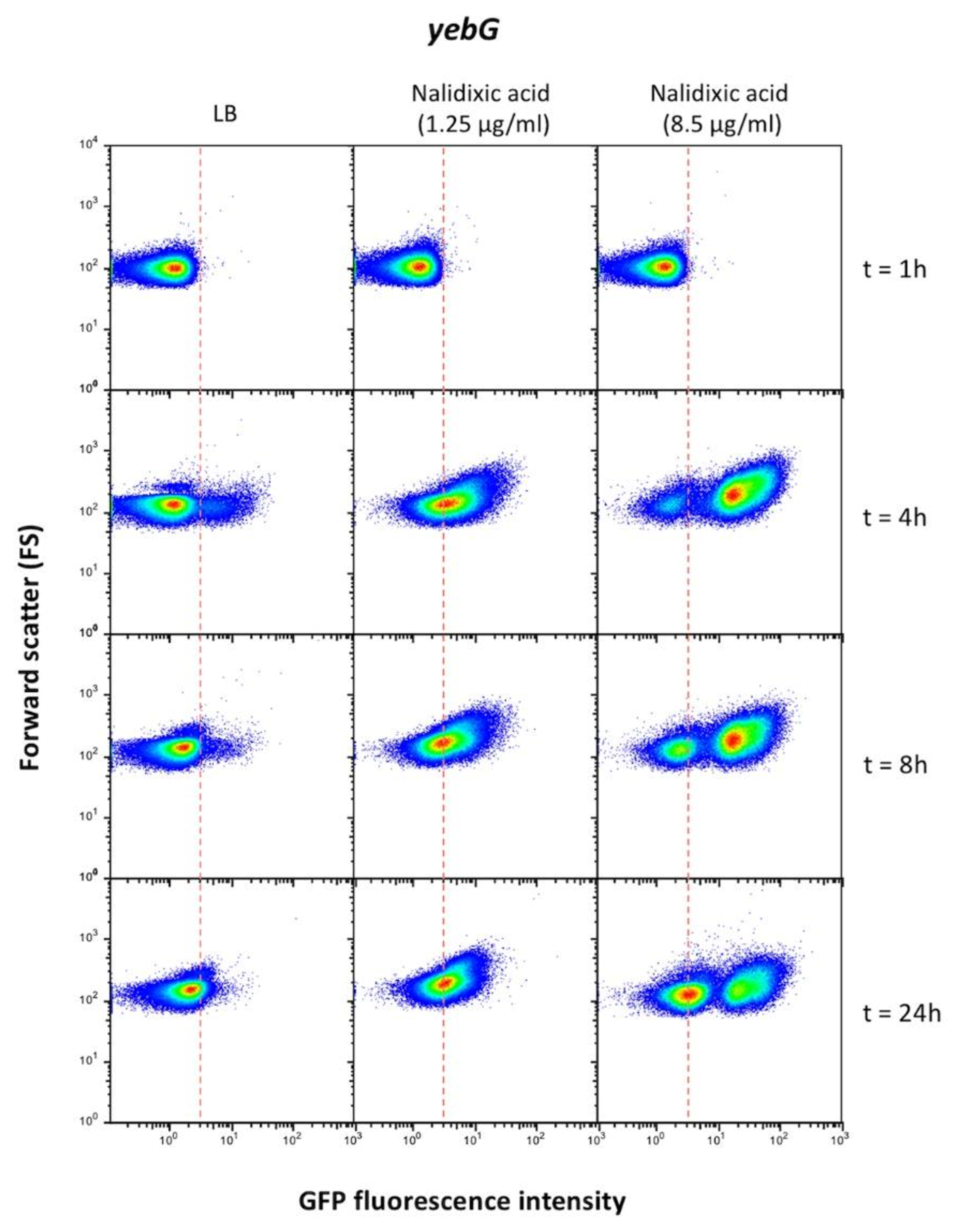
- (vi).
- When flow cytometry data were collected to represent cell sizes in the y-axis (instead of PI fluorescence as in Figure 7), filamentation was detected at various extents, especially when hydroxyurea or nalidixic acid were used as inducers (Supplementary Figure S7).
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radman, M. SOS repair hypothesis: Phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci. 1975, 5, 355–367. [Google Scholar]
- Erill, I.; Escribano, M.; Campoy, S.; Barbe, J. In silico analysis reveals substantial variability in the gene contents of the gamma proteobacteria LexA-regulon. Bioinformatics 2003, 19, 2225–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erill, I.; Campoy, S.; Barbe, J. Aeons of distress: An evolutionary perspective on the bacterial SOS response. FEMS Microbiol. Rev. 2007, 31, 637–656. [Google Scholar] [CrossRef] [Green Version]
- Simmons, L.A.; Foti, J.J.; Cohen, S.E.; Walker, G.C. The SOS regulatory network. EcoSal Plus 2008, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baharoglu, Z.; Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslowska, K.H.; Makiela-Dzbenska, K.; Fijalkowska, I.J. The SOS system: A complex and tightly regulated response to DNA damage. Environ. Mol. Mutagen. 2019, 60, 368–384. [Google Scholar] [CrossRef] [Green Version]
- d’Ari, R. The SOS system. Biochimie 1985, 67, 343–347. [Google Scholar] [CrossRef]
- Courcelle, J.; Khodursky, A.; Peter, B.; Brown, P.O.; Hanawalt, P.C. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001, 158, 41–64. [Google Scholar]
- Wade, J.T.; Reppas, N.B.; Church, G.M.; Struhl, K. Genomic analysis of LexA binding reveals the permissive nature of the Escherichia coli genome and identifies unconventional target sites. Genes Dev. 2005, 19, 2619–2630. [Google Scholar] [CrossRef] [Green Version]
- Au, N.; Kuester-Schoeck, E.; Mandava, V.; Bothwell, L.E.; Canny, S.P.; Chachu, K.; Colavito, S.A.; Fuller, S.N.; Groban, E.S.; Hensley, L.A.; et al. Genetic composition of the Bacillus subtilis SOS system. J. Bacteriol. 2005, 187, 7655–7666. [Google Scholar] [CrossRef] [Green Version]
- Cirz, R.T.; O’Neill, B.M.; Hammond, J.A.; Head, S.R.; Romesberg, F.E. Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J. Bacteriol. 2006, 188, 7101–7110. [Google Scholar] [CrossRef] [Green Version]
- Pang, P.P.; Walker, G.C. The Salmonella typhimurium LT2 uvrD gene is regulated by the lexA gene product. J. Bacteriol. 1983, 154, 1502–1504. [Google Scholar] [CrossRef] [Green Version]
- Pierre, A.; Paoletti, C. Purification and characterization of RecA protein from Salmonella typhimurium. J. Biol. Chem. 1983, 258, 2870–2874. [Google Scholar] [CrossRef]
- Freudl, R.; Braun, G.; Honore, N.; Cole, S.T. Evolution of the enterobacterial sulA gene: A component of the SOS system encoding an inhibitor of cell division. Gene 1987, 52, 31–40. [Google Scholar] [CrossRef]
- Smith, C.M.; Arany, Z.; Orrego, C.; Eisenstadt, E. DNA damage-inducible loci in Salmonella typhimurium. J. Bacteriol. 1991, 173, 3587–3590. [Google Scholar] [CrossRef] [Green Version]
- Benson, N.R.; Wong, R.M.; McClelland, M. Analysis of the SOS response in Salmonella enterica serovar Typhimurium using RNA fingerprinting by arbitrarily primed PCR. J. Bacteriol. 2000, 182, 3490–3497. [Google Scholar] [CrossRef] [Green Version]
- Clerch, B.; Garriga, X.; Torrents, E.; Rosales, C.M.; Llagostera, M. Construction and characterization of two lexA mutants of Salmonella typhimurium with different UV sensitivities and UV mutabilities. J. Bacteriol. 1996, 178, 2890–2896. [Google Scholar] [CrossRef] [Green Version]
- Bunny, K.; Liu, J.; Roth, J. Phenotypes of lexA mutations in Salmonella enterica: Evidence for a lethal lexA null phenotype due to the Fels-2 prophage. J. Bacteriol. 2002, 184, 6235–6249. [Google Scholar] [CrossRef] [Green Version]
- Buchmeier, N.A.; Lipps, C.J.; So, M.Y.; Heffron, F. Recombination-deficient mutants of Salmonella typhimurium are avirulent and sensitive to the oxidative burst of macrophages. Mol. Microbiol. 1993, 7, 933–936. [Google Scholar] [CrossRef]
- Cano, D.A.; Pucciarelli, M.G.; Garcia-del Portillo, F.; Casadesus, J. Role of the RecBCD recombination pathway in Salmonella virulence. J. Bacteriol. 2002, 184, 592–595. [Google Scholar] [CrossRef] [Green Version]
- Suvarnapunya, A.E.; Lagasse, H.A.; Stein, M.A. The role of DNA base excision repair in the pathogenesis of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2003, 48, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Prieto, A.I.; Ramos-Morales, F.; Casadesus, J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics 2006, 174, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrande, M.; Andrews-Polymenis, H.; Twedt, D.J.; Steele-Mortimer, O.; Porwollik, S.; McClelland, M.; Knodler, L.A. Genetic determinants of Salmonella enterica serovar Typhimurium proliferation in the cytosol of epithelial cells. Infect. Immun. 2016, 84, 3517–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culyba, M.J.; Mo, C.Y.; Kohli, R.M. Targets for combating the evolution of acquired antibiotic resistance. Biochemistry 2015, 54, 3573–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, J.D.; Taylor, R.J. The estimation of doses of Salmonella typhimurium suitable for the experimental production of disease in calves. Vet. Rec. 1966, 78, 706–707. [Google Scholar] [CrossRef]
- Schmieger, H. Phage P22-mutants with increased or decreased transduction abilities. Mol Gen Genet 1972, 119, 75–88. [Google Scholar] [CrossRef]
- Torreblanca, J.; Marques, S.; Casadesus, J. Synthesis of FinP RNA by plasmids F and pSLT is regulated by DNA adenine methylation. Genetics 1999, 152, 31–45. [Google Scholar]
- Chan, R.K.; Botstein, D.; Watanabe, T.; Ogata, Y. Specialized transduction of tetracycline resistance by phage P22 in Salmonella typhimurium. II. Properties of a high-frequency-transducing lysate. Virology 1972, 50, 883–898. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The sequence alignment/map format and SAM tools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. A model based criterion for gene expression calls using RNA-seq data. Theory Biosci. 2013, 132, 159–164. [Google Scholar] [CrossRef]
- Kroger, C.; Colgan, A.; Srikumar, S.; Handler, K.; Sivasankaran, S.K.; Hammarlof, D.L.; Canals, R.; Grissom, J.E.; Conway, T.; Hokamp, K.; et al. An infection-relevant transcriptomic compendium for Salmonella enterica serovar Typhimurium. Cell Host Microbe 2013, 14, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Berg, O.G. Selection of DNA binding sites by regulatory proteins: The LexA protein and the arginine repressor use different strategies for functional specificity. Nucleic Acids Res. 1988, 16, 5089–5105. [Google Scholar] [CrossRef] [Green Version]
- Hautefort, I.; Proenca, M.J.; Hinton, J.C. Single-copy green fluorescent protein gene fusions allow accurate measurement of Salmonella gene expression in vitro and during infection of mammalian cells. Appl. Environ. Microbiol. 2003, 69, 7480–7491. [Google Scholar] [CrossRef] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 90, 6640–6645. [Google Scholar] [CrossRef] [Green Version]
- Lehtinen, J.; Nuutila, J.; Lilius, E.M. Green fluorescent protein-propidium iodide (GFP-PI) based assay for flow cytometric measurement of bacterial viability. Cytom. A 2004, 60, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Fernandez De Henestrosa, A.R.; Ogi, T.; Aoyagi, S.; Chafin, D.; Hayes, J.J.; Ohmori, H.; Woodgate, R. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 2000, 35, 1560–1572. [Google Scholar] [CrossRef] [PubMed]
- Ysern, P.; Clerch, B.; Castano, M.; Gibert, I.; Barbe, J.; Llagostera, M. Induction of SOS genes in Escherichia coli and mutagenesis in Salmonella typhimurium by fluoroquinolones. Mutagenesis 1990, 5, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Newmark, K.G.; O’Reilly, E.K.; Pohlhaus, J.R.; Kreuzer, K.N. Genetic analysis of the requirements for SOS induction by nalidixic acid in Escherichia coli. Gene 2005, 356, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.; Argaman, L.; Wagner, E.G.; Altuvia, S. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr. Biol. 2004, 14, 2271–2276. [Google Scholar] [CrossRef] [Green Version]
- Lemire, S.; Figueroa-Bossi, N.; Bossi, L. Bacteriophage crosstalk: Coordination of prophage induction by trans-acting antirepressors. PLoS Genet 2011, 7, e1002149. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-Bossi, N.; Coissac, E.; Netter, P.; Bossi, L. Unsuspected prophage-like elements in Salmonella typhimurium. Mol. Microbiol. 1997, 25, 161–173. [Google Scholar] [CrossRef]
- Hryniewicz, M.; Sirko, A.; Palucha, A.; Bock, A.; Hulanicka, D. Sulfate and thiosulfate transport in Escherichia coli K-12: Identification of a gene encoding a novel protein involved in thiosulfate binding. J. Bacteriol. 1990, 172, 3358–3366. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, Q.; Guan, S.; Shi, H. Quantitative connection between cell size and growth rate by phospholipid metabolism. Cells 2020, 9, 391. [Google Scholar] [CrossRef] [Green Version]
- Hillyard, D.R.; Edlund, M.; Hughes, K.T.; Marsh, M.; Higgins, N.P. Subunit-specific phenotypes of Salmonella typhimurium HU mutants. J. Bacteriol. 1990, 172, 5402–5407. [Google Scholar] [CrossRef] [Green Version]
- Krin, E.; Pierle, S.A.; Sismeiro, O.; Jagla, B.; Dillies, M.A.; Varet, H.; Irazoki, O.; Campoy, S.; Rouy, Z.; Cruveiller, S.; et al. Expansion of the SOS regulon of Vibrio cholerae through extensive transcriptome analysis and experimental validation. BMC Genom. 2018, 19, 373. [Google Scholar] [CrossRef] [PubMed]
- McCool, J.D.; Long, E.; Petrosino, J.F.; Sandler, H.A.; Rosenberg, S.M.; Sandler, S.J. Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol. Microbiol. 2004, 53, 1343–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, J.M.; Rosenberg, S.M. Spontaneous DNA breakage in single living Escherichia coli cells. Nat. Genet 2007, 39, 797–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamensek, S.; Podlesek, Z.; Gillor, O.; Zgur-Bertok, D. Genes regulated by the Escherichia coli SOS repressor LexA exhibit heterogeneous expression. BMC Microbiol. 2010, 10, 283. [Google Scholar] [CrossRef] [Green Version]
- Kuzminov, A.; Stahl, F.W. Double-strand end repair via the RecBC pathway in Escherichia coli primes DNA replication. Genes Dev. 1999, 13, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Kowalczykowski, S.C. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 2000, 25, 156–165. [Google Scholar] [CrossRef]
- D’Ari, R.; Casadesus, J. Underground metabolism. Bioessays 1998, 20, 181–186. [Google Scholar] [CrossRef]
- Butala, M.; Zgur-Bertok, D.; Busby, S.J. The bacterial LexA transcriptional repressor. Cell Mol. Life Sci. 2009, 66, 82–93. [Google Scholar] [CrossRef]
- von Hippel, P.H.; Berg, O.G. Facilitated target location in biological systems. J. Biol. Chem. 1989, 264, 675–678. [Google Scholar] [CrossRef]
- Lewis, L.K.; Harlow, G.R.; Gregg-Jolly, L.A.; Mount, D.W. Identification of high affinity binding sites for LexA which define new DNA damage-inducible genes in Escherichia coli. J. Mol. Biol. 1994, 241, 507–523. [Google Scholar] [CrossRef]
- Barbe, J.; Villaverde, A.; Guerrero, R. Induction of the SOS response by hydroxyurea in Escherichia coli K12. Mutat. Res. 1987, 192, 105–108. [Google Scholar] [CrossRef]
- Otsuji, N.; Sekiguchi, M.; Iijima, T.; Takagi, Y. Induction of phage formation in the lysogenic Escherichia coli K-12 by mitomycin C. Nature 1959, 184, 1079–1080. [Google Scholar] [CrossRef]
- Imlay, J.A.; Linn, S. Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J. Bacteriol. 1987, 169, 2967–2976. [Google Scholar] [CrossRef] [Green Version]
- Vila, J.; Fabrega, A.; Roca, I.; Hernandez, A.; Martinez, J.L. Efflux pumps as an important mechanism for quinolone resistance. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 77, 167–235. [Google Scholar]
- Yang, L.; Mih, N.; Anand, A.; Park, J.H.; Tan, J.; Yurkovich, J.T.; Monk, J.M.; Lloyd, C.J.; Sandberg, T.E.; Seo, S.W.; et al. Cellular responses to reactive oxygen species are predicted from molecular mechanisms. Proc. Natl. Acad. Sci. USA 2019, 116, 14368–14373. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Chiu, L.Y.; Nehring, R.B.; Bravo Nunez, M.A.; Mei, Q.; Perez, M.; Zhai, Y.; Fitzgerald, D.M.; Pribis, J.P.; Wang, Y.; et al. Bacteria-to-human protein networks reveal origins of endogenous DNA damage. Cell 2019, 176, 127–143 e24. [Google Scholar] [CrossRef] [Green Version]
- Sigal, N.; Delius, H.; Kornberg, T.; Gefter, M.L.; Alberts, B. A DNA-unwinding protein isolated from Escherichia coli: Its interaction with DNA and with DNA polymerases. Proc. Natl. Acad. Sci. USA 1972, 69, 3537–3541. [Google Scholar] [CrossRef] [Green Version]
- Shinagawa, H.; Iwasaki, H. Processing the holliday junction in homologous recombination. Trends Biochem. Sci. 1996, 21, 107–111. [Google Scholar] [CrossRef]
- Seeberg, E.; Nissen-Meyer, J.; Strike, P. Incision of ultraviolet-irradiated DNA by extracts of E. coli requires three different gene products. Nature 1976, 263, 524–526. [Google Scholar] [CrossRef]
- Culyba, M.J.; Kubiak, J.M.; Mo, C.Y.; Goulian, M.; Kohli, R.M. Non-equilibrium repressor binding kinetics link DNA damage dose to transcriptional timing within the SOS gene network. PLoS Genet 2018, 14, e1007405. [Google Scholar] [CrossRef] [Green Version]
- Davidson, C.J.; Surette, M.G. Individuality in bacteria. Annu. Rev. Genet 2008, 42, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Silva-Rocha, R.; de Lorenzo, V. Noise and robustness in prokaryotic regulatory networks. Annu. Rev. Microbiol. 2010, 64, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Goni-Moreno, A.; Benedetti, I.; Kim, J.; de Lorenzo, V. Deconvolution of gene expression noise into spatial dynamics of transcription factor-promoter Interplay. ACS Synth. Biol. 2017, 6, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Kaern, M.; Elston, T.C.; Blake, W.J.; Collins, J.J. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genet 2005, 6, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Silander, O.K.; Nikolic, N.; Zaslaver, A.; Bren, A.; Kikoin, I.; Alon, U.; Ackermann, M. A genome-wide analysis of promoter-mediated phenotypic noise in Escherichia coli. PLoS Genet 2012, 8, e1002443. [Google Scholar] [CrossRef]
- Woo, A.C.; Faure, L.; Dapa, T.; Matic, I. Heterogeneity of spontaneous DNA replication errors in single isogenic Escherichia coli cells. Sci. Adv. 2018, 4, eaat1608. [Google Scholar] [CrossRef] [Green Version]
- Pribis, J.P.; Garcia-Villada, L.; Zhai, Y.; Lewin-Epstein, O.; Wang, A.Z.; Liu, J.; Xia, J.; Mei, Q.; Fitzgerald, D.M.; Bos, J.; et al. Gamblers: An antibiotic-induced evolvable cell subpopulation differentiated by reactive-oxygen-induced general stress response. Mol. Cell 2019, 74, 785–800 e7. [Google Scholar] [CrossRef]
- Uphoff, S.; Lord, N.D.; Okumus, B.; Potvin-Trottier, L.; Sherratt, D.J.; Paulsson, J. Stochastic activation of a DNA damage response causes cell-to-cell mutation rate variation. Science 2016, 351, 1094–1097. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.S.; Uphoff, S. Bacterial phenotypic heterogeneity in DNA repair and mutagenesis. Biochem. Soc. Trans. 2020, 48, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol. 2015, 13, 497–508. [Google Scholar] [CrossRef]
- van Boxtel, C.; van Heerden, J.H.; Nordholt, N.; Schmidt, P.; Bruggeman, F.J. Taking chances and making mistakes: Non-genetic phenotypic heterogeneity and its consequences for surviving in dynamic environments. J. R. Soc. Interface 2017, 14, 20170141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroter, L.; Dersch, P. Phenotypic diversification of microbial p athogens—cooperating and preparing for the future. J. Mol. Biol. 2019, 431, 4645–4655. [Google Scholar] [CrossRef] [PubMed]
- Zgur-Bertok, D. Phenotypic heterogeneity in bacterial populations. Acta Agric. Slov. 2007, 90, 17–24. [Google Scholar]
- Hernandez, S.B.; Cota, I.; Ducret, A.; Aussel, L.; Casadesus, J. Adaptation and preadaptation of Salmonella enterica to bile. PLoS Genet 2012, 8, e1002459. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | Fold Change | Number of SOS Boxes | Distance to ATG (bp) | Heterology Index * |
|---|---|---|---|---|---|
| STM14_4584 | tisB | 610.09 | 1 | 236 | 6.13 |
| STM14_2287 | yebG | 41.34 | 1 | 17 | 7.03 |
| STM14_3289 | recN | 39.41 | 3 | 8, 26, 48 | 12.97, 7.72, 8.82 |
| STM14_3417 | recA | 35.71 | 1 | 64 | 5.92 |
| STM14_1215 | sulA | 26.70 | 1 | 21 | 1.49 |
| STM14_3210 | dinIGifsy-1 | 23.20 | 1 | 19 | 4.55 |
| STM14_1331 | dinI | 22.82 | 2 | 19, 42 | 4.00, 17.60 |
| STM14_2423 | umuD ** | 20.10 | 1 | 15 | 4.38 |
| STM14_4775 | yigN | 19.46 | 1 | 44 | 8.71 |
| STM14_3002 | cysP | 15.47 | 1 | 65 | 18.16 |
| STM14_4846 | nlhH | 15.11 | 1 | 13 | 7.63 |
| STM14_1156 | dinIGifsy-2 | 14.46 | 1 | 19 | 3.92 |
| STM14_0926 | uvrB | 14.13 | 1 | 73 | 5.66 |
| STM14_4752 | uvrD | 13.87 | 1 | 105 | 11.48 |
| STM14_0369 | dinP *** | 11.30 | 1 | 15 | 6.80 |
| STM14_5112 | uvrA | 11.16 | 1 | 80 | 6.99 |
| STM14_953 | dinG | 10.61 | 1 | 14 | 10.08 |
| IstR-1,2 | istR-1,2 | 10.26 | 1 | 40 | 6.13 |
| STM14_5114 | ssb | 9.63 | 1 | 24 | 6.99 |
| STM14_4847 | higB-2 | 8.05 | 1 | 184 | 7.63 |
| STM14_4236 | dinJ | 7.82 | 1 | 14 | 8.27 |
| STM14_3214 | -- | 7.70 | 1 | 6 | 12.33 |
| STM14_5094 | lexA | 6.86 | 2 | 6, 27 | 14.48, 7.94 |
| STM14_3568 | gudD | 5.43 | 1 | 0 | 16.97 |
| STM14_3405 | yqaB | 5.32 | 1 | 12 | 16.26 |
| STM14_1439 | dinIGifsy-3 | 4.56 | 1 | 19 | 5.26 |
| STM14_2752 | yejK | 4.54 | 1 | 102 | 20.89 |
| STM14_2422 | umuC ** | 3.91 | 1 | -- | 4.38 |
| STM14_2753 | yejL | 3.74 | 1 | 57 | 20.89 |
| STM14_2648 | thiM | 3.62 | 1 | 177 | 18.02 |
| STM14_3627 | mutH | 3.58 | 1 | 228 | 12.99 |
| STM14_2551 | sbmC | 3.55 | 1 | 21 | 6.32 |
| STM14_1492 | msgA | 3.44 | 1 | 14 | 11.88 |
| STM14_2650 | rcnB | 3.38 | 1 | 212 | 18.02 |
| STM14_1589 | ydjQ | 3.33 | 1 | 0 | 6.69 |
| STM14_0161 | yacA | 3.30 | 1 | 403 | 19.46 |
| STM14_4344 | yhjE | 3.05 | 1 | 202 | 14.01 |
| STM14_5011 | hupA | 2.74 | 1 | 190 | 15.31 |
| STM14_1432 | -- | 2.71 | 1 | 6 | 12.33 |
| STM14_4158 | slyX | 2.70 | 1 | 146 | 21.28 |
| STM14_3283 | corE | 2.54 | 1 | 98 | 20.11 |
| STM14_1385 | ndh | 2.37 | 1 | 157 | 12.04 |
| STM14_0374 | frsA | 2.33 | 1 | 360 | 13.12 |
| STM14_5490 | deoD | 2.21 | 1 | 210 | 18.78 |
| STM14_5518 | -- | 2.19 | 1 | 71 | 17.26 |
| STM14_1605 | ydjM | 2.11 | 2 | 14, 32 | 9.12, 12.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mérida-Floriano, A.; Rowe, W.P.M.; Casadesús, J. Genome-Wide Identification and Expression Analysis of SOS Response Genes in Salmonella enterica Serovar Typhimurium. Cells 2021, 10, 943. https://doi.org/10.3390/cells10040943
Mérida-Floriano A, Rowe WPM, Casadesús J. Genome-Wide Identification and Expression Analysis of SOS Response Genes in Salmonella enterica Serovar Typhimurium. Cells. 2021; 10(4):943. https://doi.org/10.3390/cells10040943
Chicago/Turabian StyleMérida-Floriano, Angela, Will P. M. Rowe, and Josep Casadesús. 2021. "Genome-Wide Identification and Expression Analysis of SOS Response Genes in Salmonella enterica Serovar Typhimurium" Cells 10, no. 4: 943. https://doi.org/10.3390/cells10040943