
Clusterin Deficiency Exacerbates Hyperoxia-Induced Acute Lung Injury

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Oxygen Exposure
2.3. RNA Isolation and Quantitative Real-Time PCR
2.4. Statistical Analysis
3. Results
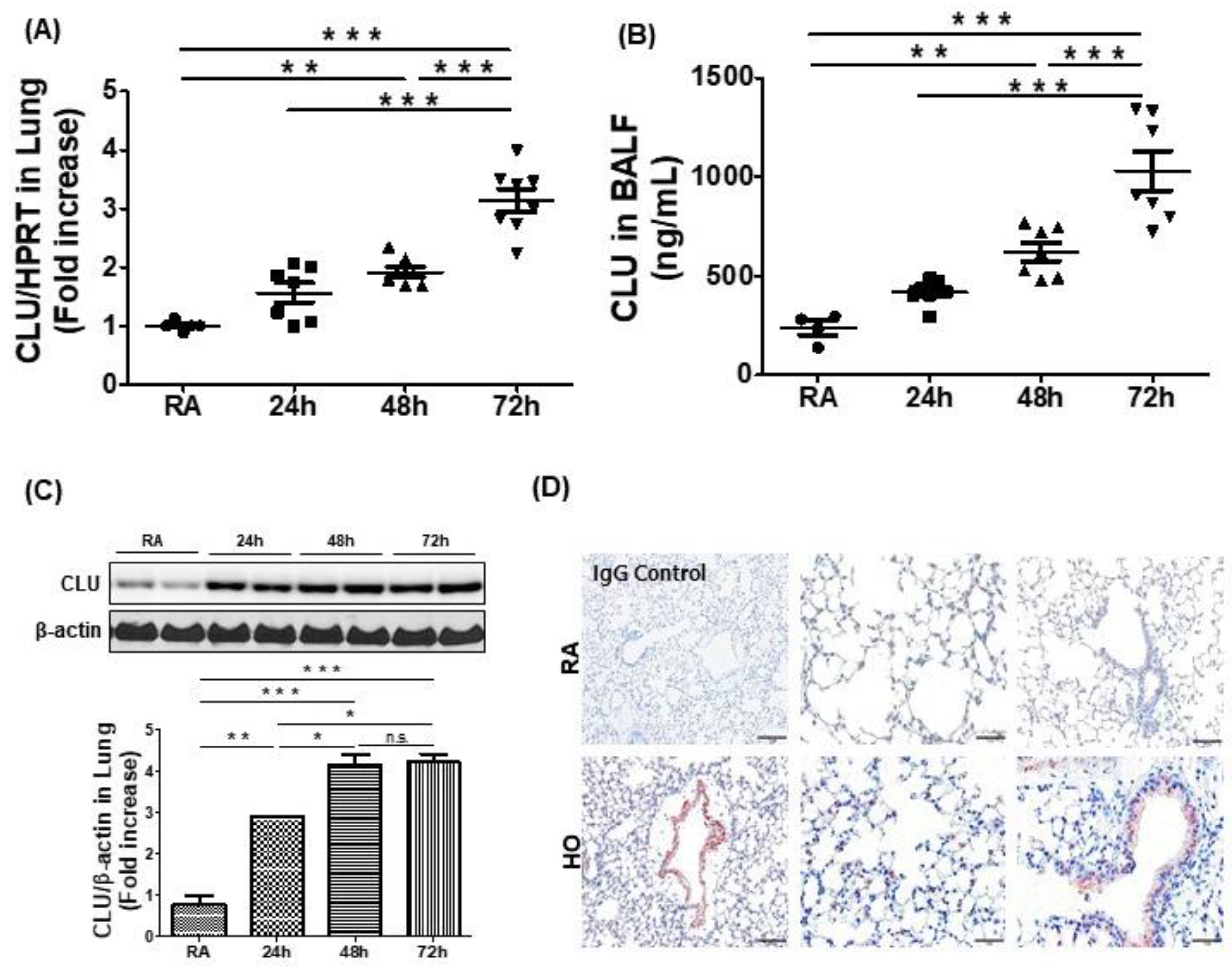
3.1. Hyperoxia-Induced Acute Lung Injury Is Associated with Elevated Levels of CLU
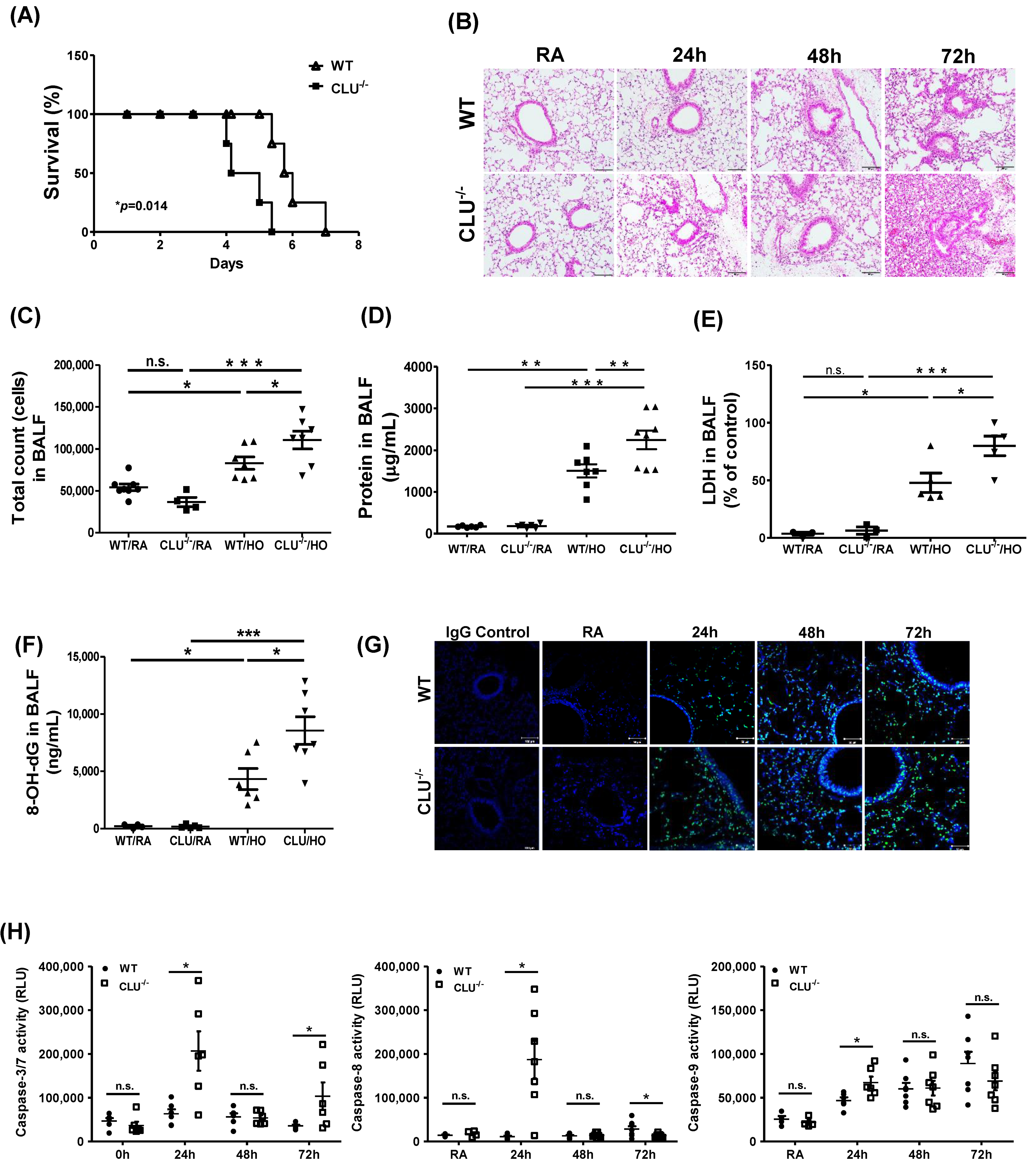
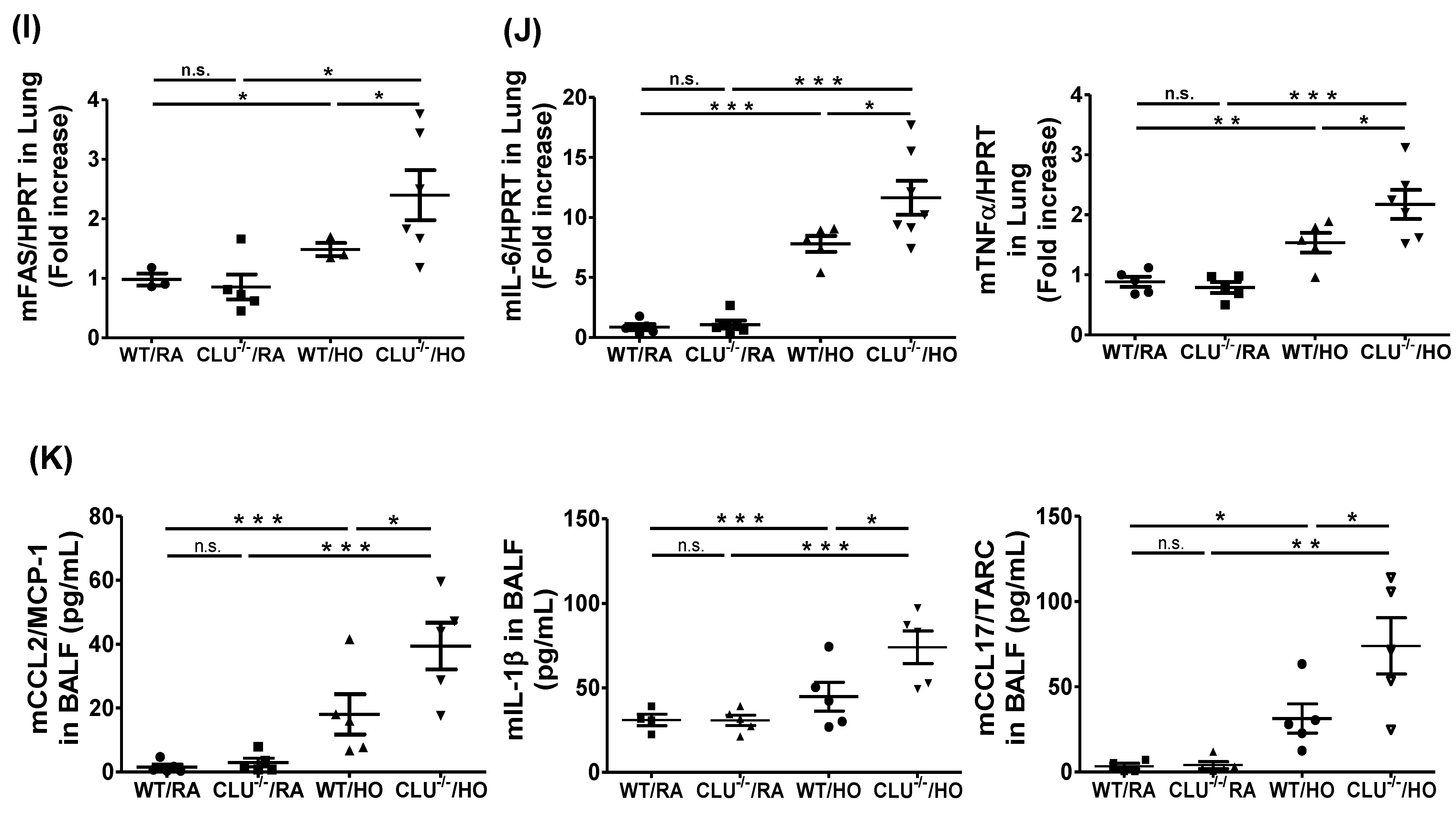
3.2. CLU Protects Against Hyperoxia-Induced Lung Injury and Apoptosis in Mice
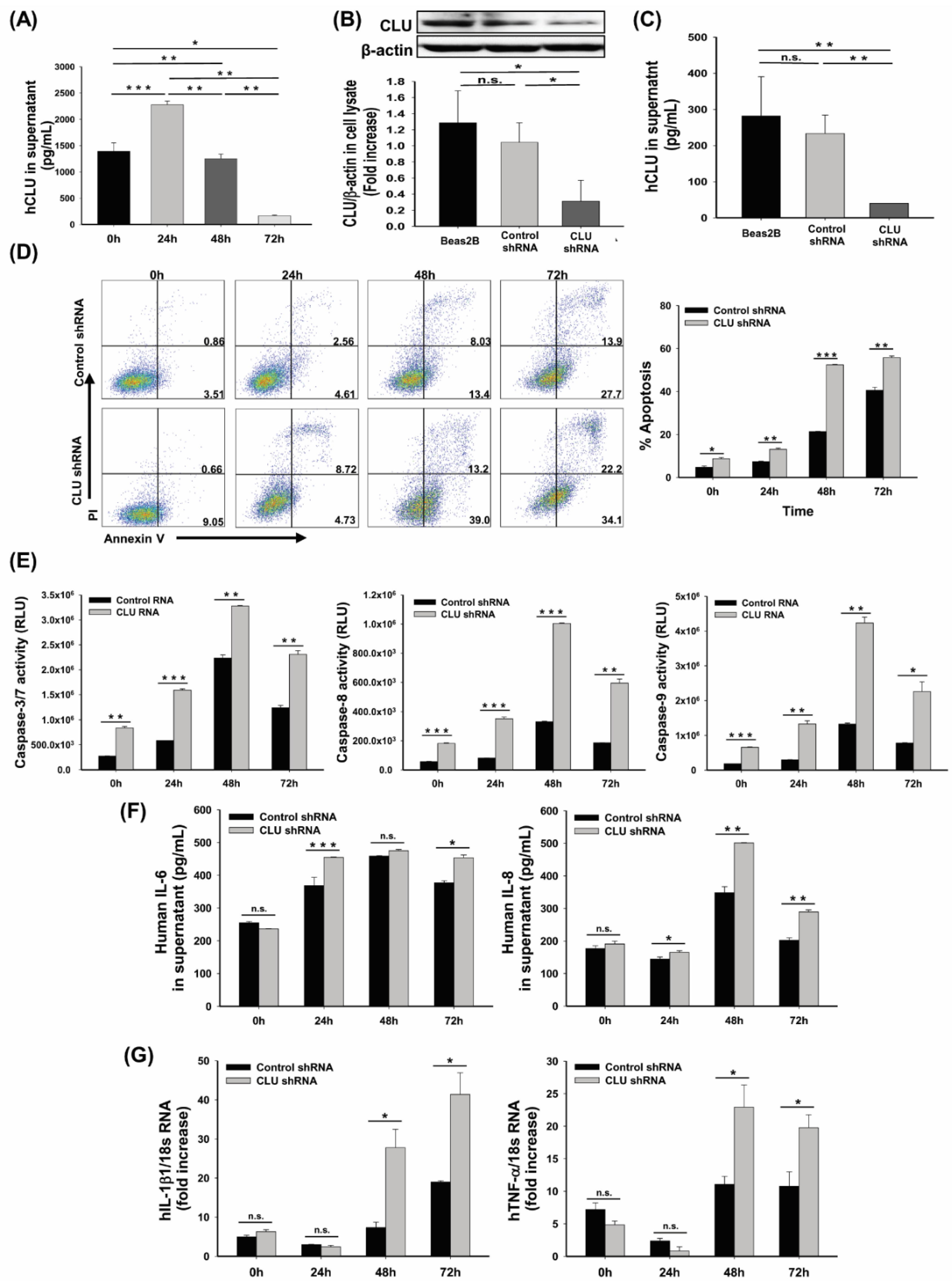
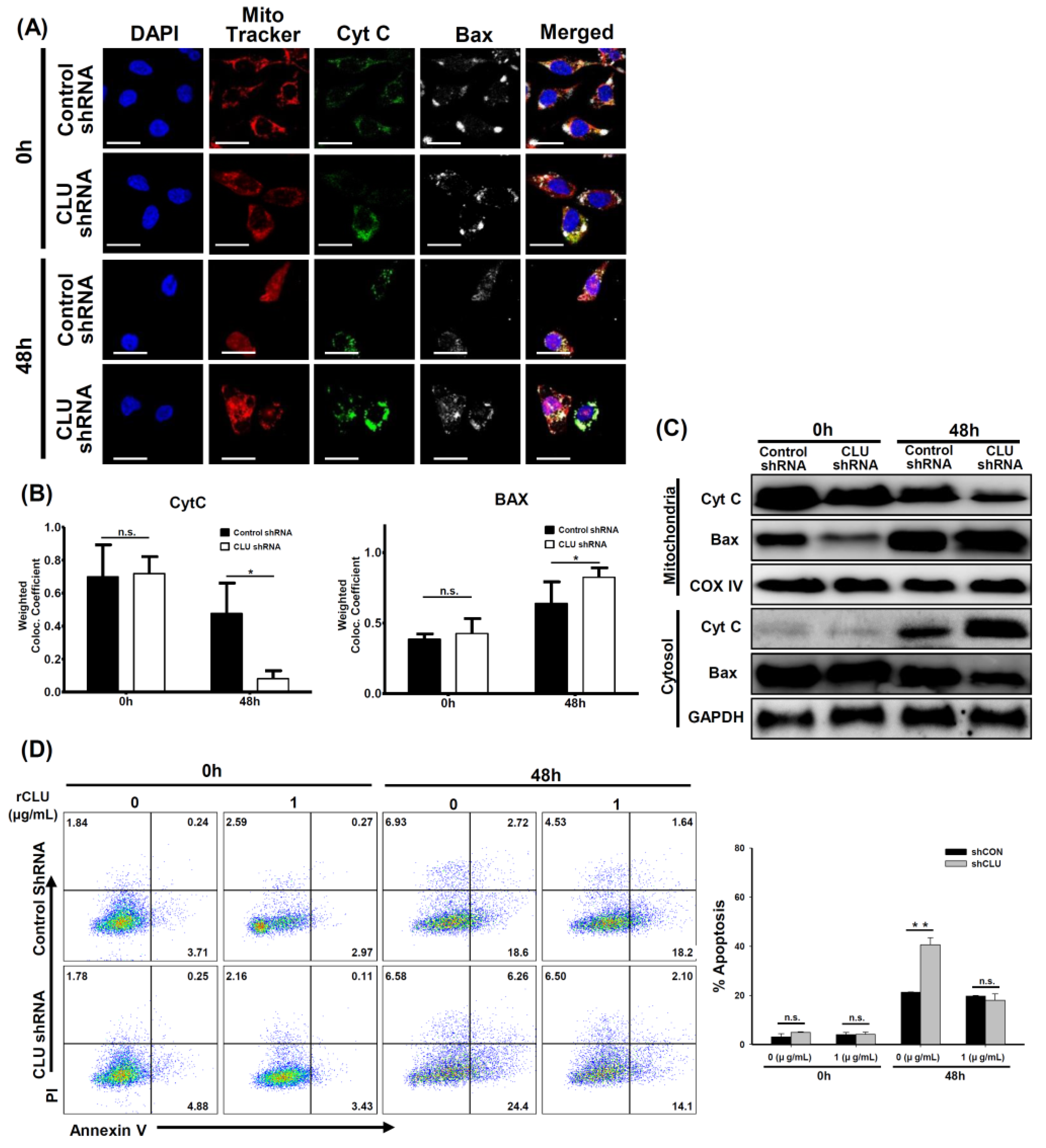
3.3. CLU Deficiency Increases Hyperoxia-Induced Apoptosis in Human Airway Epithelial Cells
3.4. CLU Regulates Mitochondrial Potential and Apoptosis in Hyperoxia-Exposed Mice and Human Airway Epithelial Cells
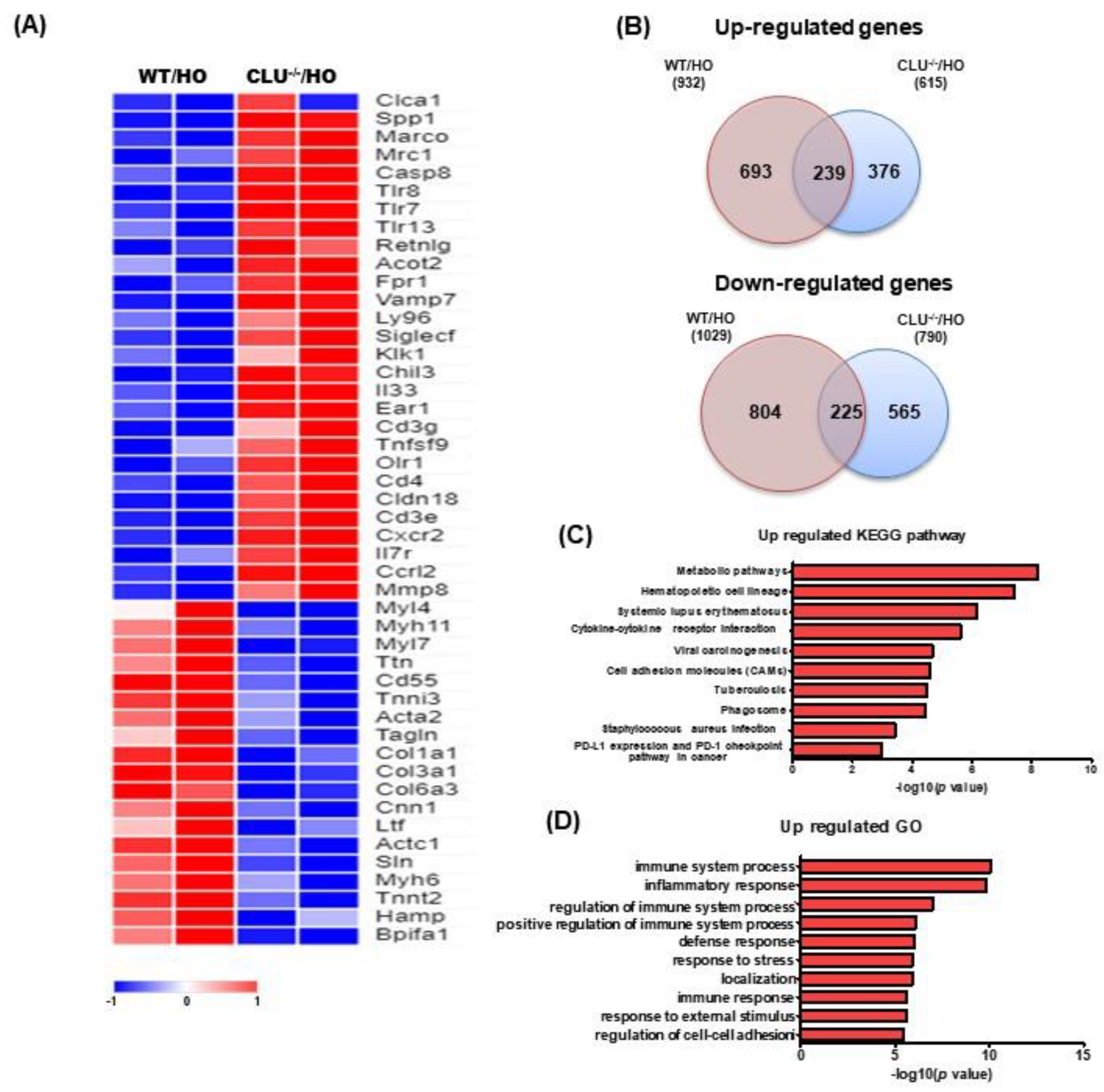
3.5. Analysis of DEGs and Pathways Based on Exposure to Hyperoxia
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sohn, M.H.; Kang, M.; Matsuura, H.; Bhandari, V.; Chen, N.; Lee, C.G.; Elias, J.A. The chitinase-like proteins breast regression protein-39 and YKL-40 regulate hyperoxia-induced acute lung injury. Am. J. Respir. Crit. Care Med. 2010, 182, 918–928. [Google Scholar] [CrossRef] [Green Version]
- Laube, M.; Amann, E.; Uhlig, U.; Yang, Y.; Fuchs, H.W.; Zemlin, M.; Mercier, J.-C.; Maier, R.F.; Hummler, H.D.; Uhlig, S.; et al. Inflammatory Mediators in Tracheal Aspirates of Preterm Infants Participating in a Randomized Trial of Inhaled Nitric Oxide. PLoS ONE 2017, 12, e0169352. [Google Scholar] [CrossRef] [Green Version]
- Mizushina, Y.; Shirasuna, K.; Usui, F.; Karasawa, T.; Kawashima, A.; Kimura, H.; Kobayashi, M.; Komada, T.; Inoue, Y.; Mato, N.; et al. NLRP3 Protein Deficiency Exacerbates Hyperoxia-induced Lethality through Stat3 Protein Signaling Independent of Interleukin-1β. J. Biol. Chem. 2015, 290, 5065–5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, V.; Choo-Wing, R.; Homer, R.J.; Elias, J.A. Increased Hyperoxia-Induced Mortality and Acute Lung Injury in IL-13 Null Mice. J. Immunol. 2007, 178, 4993–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budinger, G.R.S.; Mutlu, G.M.; Urich, D.; Soberanes, S.; Buccellato, L.J.; Hawkins, K.; Chiarella, S.E.; Radigan, K.A.; Eisenbart, J.; Agrawal, H.; et al. Epithelial Cell Death Is an Important Contributor to Oxidant-mediated Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2011, 183, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Chen, M.; Ji, H.; Liu, G.; Mei, H.; Li, K.; Chen, T. miR-21-5p regulates type II alveolar epithelial cell apoptosis in hyperoxic acute lung injury. Mol. Med. Rep. 2018, 17, 5796–5804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, D.M.L.; Mekary, R.A.; Rodrigues, K.C.D.C.; Anaruma, C.P.; Ropelle, E.R.; Da Silva, A.S.R.; Cintra, D.E.; Pauli, J.R.; De Moura, L.P. Protective molecular mechanisms of clusterin against apoptosis in cardiomyocytes. Heart Fail. Rev. 2018, 23, 123–129. [Google Scholar] [CrossRef]
- Rohne, P.; Prochnow, H.; Koch-Brandt, C. The CLU-files: Disentanglement of a mystery. Biomol. Concepts 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Hong, G.H.; Kwon, H.-S.; Moon, K.-A.; Park, S.Y.; Park, S.; Lee, K.Y.; Ha, E.H.; Kim, T.-B.; Moon, H.-B.; Lee, H.K.; et al. Clusterin Modulates Allergic Airway Inflammation by Attenuating CCL20-Mediated Dendritic Cell Recruitment. J. Immunol. 2016, 196, 2021–2030. [Google Scholar] [CrossRef] [Green Version]
- Bae, C.H.; Na, H.G.; Choi, Y.S.; Song, S.; Kim, Y. Clusterin Induces MUC5AC Expression via Activation of NF-κB in Human Airway Epithelial. Cells Clin. Exp. Otorhinolaryngol. 2018, 11, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trougakos, I.P.; So, A.; Jansen, B.; Gleave, M.E.; Gonos, E.S. Silencing expression of the clusterin/apolipoprotein j gene in human cancer cells using small interfering RNA induces spontaneous apoptosis, reduced growth ability, and cell sensitization to genotoxic and oxidative stress. Cancer Res. 2004, 64, 1834–1842. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.; Kim, D.; Lee, S.; Lee, H.S.; Seo, J.H.; Kim, J.H.; Yu, Y.S.; Min, B.H.; Kim, K. Clusterin protects H9c2 cardiomyocytes from oxidative stress-induced apoptosis via Akt/GSK-3β signaling pathway. Exp. Mol. Med. 2011, 43, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Habiel, D.M.; Camelo, A.; Espindola, M.; Burwell, T.; Hanna, R.; Miranda, E.; Carruthers, A.; Bell, M.; Coelho, A.L.; Liu, H.; et al. Divergent roles for Clusterin in Lung Injury and Repair. Sci. Rep. 2017, 7, 15444. [Google Scholar] [CrossRef] [Green Version]
- Takase, O.; Minto, A.W.; Puri, T.S.; Cunningham, P.N.; Jacob, A.; Hayashi, M.; Quigg, R.J. Inhibition of NF-kappaB-dependent Bcl-xL expression by clusterin promotes albumin-induced tubular cell apoptosis. Kidney Int. 2008, 73, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnevali, S.; Luppi, F.; D’Arca, D.; Caporali, A.; Ruggieri, M.P.; Vettori, M.V.; Caglieri, A.; Astancolle, S.; Panico, F.; Davalli, P.; et al. Clusterin Decreases Oxidative Stress in Lung Fibroblasts Exposed to Cigarette Smoke. Am. J. Respir. Crit. Care Med. 2006, 174, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.Y.; Na Kim, M.; Kwak, E.J.; Kim, E.G.; Kim, K.W.; Sohn, M.H. Role of Clusterin in Protection Against Hyperoxic Lung Injury in Mice. Mech. Lung Inj. Repair 2019, 54, 4108. [Google Scholar] [CrossRef]
- Hong, J.Y.; Kim, M.; Sol, I.S.; Kim, K.W.; Lee, C.-M.; Elias, J.A.; Sohn, M.H.; Lee, C.-M.; Lee, C.G. Chitotriosidase inhibits allergic asthmatic airways via regulation of TGF-β expression and Foxp3+ Treg cells. Allergy 2018, 73, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Hong, J.Y.; Lee, K.E.; Kim, K.W.; Sohn, M.H.; Kim, K.-E. Effect of Cholesterol Depletion on Interleukin-8 Production in Human Respiratory Epithelial Cells. Allergy Asthma Immunol. Res. 2013, 5, 402–408. [Google Scholar] [CrossRef] [Green Version]
- De Paepe, M.E.; Mao, Q.; Chao, Y.; Powell, J.L.; Rubin, L.P.; Sharma, S. Hyperoxia-induced apoptosis and Fas/FasL expression in lung epithelial cells. AJP-Lung Cell. Mol. Physiol. 2005, 289, L647–L659. [Google Scholar] [CrossRef] [Green Version]
- Albertine, K.H.; Soulier, M.F.; Wang, Z.; Ishizaka, A.; Hashimoto, S.; Zimmerman, G.A.; Matthay, M.A.; Ware, L.B. Fas and Fas Ligand Are Up-Regulated in Pulmonary Edema Fluid and Lung Tissue of Patients with Acute Lung Injury and the Acute Respiratory Distress Syndrome. Am. J. Pathol. 2002, 161, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.E.; Jee, H.M.; Hong, J.Y.; Na Kim, M.; Oh, M.S.; Kim, Y.S.; Kim, K.W.; Kim, K.E.; Sohn, M.H. German Cockroach Extract Induces Matrix Metalloproteinase-1 Expression, Leading to Tight Junction Disruption in Human Airway Epithelial Cells. Yonsei Med. J. 2018, 59, 1222–1231. [Google Scholar] [CrossRef]
- Wang, Y.; Lee, C.G.L. MicroRNA and cancer-focus on apoptosis. J. Cell. Mol. Med. 2008, 13, 12–23. [Google Scholar] [CrossRef]
- Buccellato, L.J.; Tso, M.; Akinci, O.I.; Chandel, N.S.; Budinger, G.R.S. Reactive Oxygen Species Are Required for Hyperoxia-induced Bax Activation and Cell Death in Alveolar Epithelial Cells. J. Biol. Chem. 2004, 279, 6753–6760. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.R.; Sengupta, S.; Biswas, S.; Sinha, T.K.; Sen, R.; Basak, R.K.; Adhikari, B.; Bhattacharyya, A. Bacterial fucose-rich polysaccharide stabilizes MAPK-mediated Nrf2/Keap1 signaling by directly scavenging reactive oxygen species during hydrogen peroxide-induced apoptosis of human lung fibroblast cells. PLoS ONE 2014, 9, e113663. [Google Scholar]
- Pagano, A.; Donati, Y.; Métrailler, I.; Argiroffo, C.B. Mitochondrial cytochrome c release is a key event in hyperoxia-induced lung injury: Protection by cyclosporin A. Am. J. Physiol. Cell. Mol. Physiol. 2004, 286, L275–L283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koltai, T. Clusterin: A key player in cancer chemoresistance and its inhibition. OncoTargets Ther. 2014, 7, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Kim, J.K.; Edwards, C.A.; Xu, Z.; Taichman, R.; Wang, C.-Y. Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 2005, 7, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Lourda, M.; Antonelou, M.H.; Kletsas, D.; Gorgoulis, V.G.; Papassideri, I.S.; Zou, Y.; Margaritis, L.H.; Boothman, D.A.; Gonos, E.S. Intracellular Clusterin Inhibits Mitochondrial Apoptosis by Suppressing p53-Activating Stress Signals and Stabilizing the Cytosolic Ku70-Bax Protein Complex. Clin. Cancer Res. 2009, 15, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klock, G.; Baiersdörfer, M.; Koch-Brandt, C. Chapter 7: Cell protective functions of secretory Clusterin (sCLU). Adv. Cancer Res. 2009, 204, 115–138. [Google Scholar]
- Patel, V.A.; Longacre, A.; Hsiao, K.; Fan, H.; Meng, F.; Mitchell, J.E.; Rauch, J.; Ucker, D.S.; Levine, J.S. Apoptotic cells, at all stages of the death process, trigger characteristic signaling events that are divergent from and dominant over those triggered by necrotic cells: Implications for the delayed clearance model of autoimmunity. J. Biol. Chem. 2006, 281, 4663–4670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, E.M.; Aquilina, J.A.; Easterbrook-Smith, S.B.; Murphy-Durland, D.; Jacobsen, C.; Moestrup, S.K.; Wilson, M.R. Effects of Glycosylation on the Structure and Function of the Extracellular Chaperone Clusterin. Biochemistry 2007, 46, 1412–1422. [Google Scholar] [CrossRef]
- Cunin, P.; Beauvillain, C.; Miot, C.; Augusto, J.-F.; Preisser, L.; Blanchard, S.; Pignon, P.; Scotet, M.; Garo, E.; Fremaux, I.; et al. Clusterin facilitates apoptotic cell clearance and prevents apoptotic cell-induced autoimmune responses. Cell Death Dis. 2016, 7, e2215. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, V.; Choo-Wing, R.; Lee, C.G.; Zhu, Z.; Nedrelow, J.H.; Chupp, G.L.; Zhang, X.; Matthay, M.A.; Ware, L.B.; Homer, R.J.; et al. Hyperoxia causes angiopoietin 2–mediated acute lung injury and necrotic cell death. Nat. Med. 2006, 12, 1286–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, V.; Elias, J.A. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radic. Biol. Med. 2006, 41, 4–18. [Google Scholar] [CrossRef] [PubMed]
- D’Angio, C.T.; Lomonaco, M.B.; Chaudhry, S.A.; Paxhia, A.; Ryan, R.M. Discordant pulmonary proinflammatory cytokine expression during acute hyperoxia in the newborn rabbit. Exp. Lung Res. 1999, 25, 443–465. [Google Scholar] [PubMed]
- Higgins, R.D.; Jobe, A.H.; Koso-Thomas, M.; Bancalari, E.; Viscardi, R.M.; Hartert, T.V.; Ryan, R.M.; Kallapur, S.G.; Steinhorn, R.H.; Konduri, G.G.; et al. Bronchopulmonary Dysplasia: Executive Summary of a Workshop. J. Pediatr. 2018, 197, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Buczynski, B.W.; Maduekwe, E.T.; O’Reilly, M.A. The Role of Hyperoxia in the Pathogenesis of Experimental BPD. Semin. Perinatol. 2013, 37, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dong, W. Oxidative stress and bronchopulmonary dysplasia. Gene 2018, 678, 177–183. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | p-Value | Number of DEGs | DEGs |
|---|---|---|---|
| Metabolic pathways | 6.2063 × 10−9 | 28 | Cth, Prps2, Alas2, Aldoc, Alox5, Bst1, Bpgm, Cat, Cyp3a13, Cyp51, Hnmt, Gcnt2, Lss, Acot2, Rdh11, Pla2g1b, Sephs2, Man1c1, Nt5e, Papss2, Atp6v0d2, Uprt, Gatc, Sgpp2, Coq5, Mocs1, Lipf, Chia1 |
| Hematopoietic cell lineage | 3.7106 × 10−8 | 9 | Ms4a1, Cd33, Cd3e, Cd3g, Cd4, Csf2ra, Gypa, Il7r, Gp9 |
| Systemic lupus erythematosus | 6.7975 × 10−7 | 9 | C6, Fcgr4, Hist1h4c, Hist1h4d, Hist1h4i, Hist1h4n, Hist1h2bg, Hist4h4, Hist1h4h |
| Cytokine-cytokine receptor interaction | 2.3806 × 10−6 | 11 | LOC100041504, Gm13304, Gm1987, Bmp4, Cd4, Cxcr2, Csf2ra, Il1rn, Il7r, Tnfsf9, Il33 |
| Viral carcinogenesis | 2.1249 × 10−5 | 9 | Casp8, Atp6v0d2, Hist1h4c, Hist1h4d, Hist1h4i, Hist1h4n, Hist1h2bg, Hist4h4, Hist1h4h |
| Cell adhesion molecules (CAMs) | 2.5415 × 10−5 | 8 | Cd4, Itgb2, Selp, Selplg, Siglec1, Cd226, Cldn18, Cd274 |
| Tuberculosis | 3.2532 × 10−5 | 8 | Casp8, Itgax, Itgb2, Mrc1, Atp6v0d2, Fcgr4, Clec4e, Clec7a |
| Phagosome | 3.4842 × 10−5 | 8 | Olr1, Cybb, Itgb2, Marco, Mrc1, Atp6v0d2, Fcgr4, Clec7a |
| Staphylococcus aureus infection | 0.0003783 | 6 | Fpr2, Fpr1, Itgb2, Selp, Selplg, Fcgr4 |
| Alcoholism | 0.00050774 | 7 | Hist1h4c, Hist1h4d, Hist1h4i, Hist1h4n, Hist1h2bg, Hist4h4, Hist1h4h |
| PD-L1 expression and PD-1 checkpoint pathway in cancer | 0.00102848 | 5 | Cd3e, Cd3g, Cd4, Ptpn6, Cd274 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, J.Y.; Kim, M.N.; Kim, E.G.; Lee, J.W.; Kim, H.R.; Kim, S.Y.; Lee, S.M.; Kim, Y.H.; Kim, K.W.; Sohn, M.H. Clusterin Deficiency Exacerbates Hyperoxia-Induced Acute Lung Injury. Cells 2021, 10, 944. https://doi.org/10.3390/cells10040944
Hong JY, Kim MN, Kim EG, Lee JW, Kim HR, Kim SY, Lee SM, Kim YH, Kim KW, Sohn MH. Clusterin Deficiency Exacerbates Hyperoxia-Induced Acute Lung Injury. Cells. 2021; 10(4):944. https://doi.org/10.3390/cells10040944
Chicago/Turabian StyleHong, Jung Yeon, Mi Na Kim, Eun Gyul Kim, Jae Woo Lee, Hye Rin Kim, Soo Yeon Kim, Soon Min Lee, Yoon Hee Kim, Kyung Won Kim, and Myung Hyun Sohn. 2021. "Clusterin Deficiency Exacerbates Hyperoxia-Induced Acute Lung Injury" Cells 10, no. 4: 944. https://doi.org/10.3390/cells10040944