
Embryonic Origin and Subclonal Evolution of Tumor-Associated Macrophages Imply Preventive Care for Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Facts
- TAMs mainly manifest protumoral activities, including tumor initiation, angiogenesis, metastasis, drug resistance, and antitumor immunosuppression.
- TAMs predominantly derive from circulating monocytes with bone marrow hematopoietic stem cell (HSC) origin. This concept was not questioned until the embryonic origin of tissue-resident macrophages (TRMs) was discovered.
- CCL2/C-C chemokine receptor type 2 (CCR2) signal axis is the most crucial chemokine signaling pathway on which monocyte recruitment depends.
- The local proliferation of macrophages is supported by cytokines, such as CSF-1, IL-34, GM-CSF, and Th2 cytokines, including interleukin (IL) 4 and IL-13, and regulated by transcription factors, such as MafB, cMaf, Gata6, and retinoid X receptors (RXRs). These biomarkers help track down TAM subclonal evolution.
- Metabolism and metabolites play crucial roles in shaping the phenotype and function of TAMs, especially under the framework of M1/M2 TAM activation. M1-TAM-like TAMs have antitumor activity, whereas M2-TAM-like phenotypes disrupt antitumor immunity and promote tumor growth. Therapeutics can be designed to balance the ratio of M1/M2 TAMs in a tumor.
- Spatiotemporal single-cell transcriptomics and single-cell tracing technology offer the potential to target the subclonal evolution of cancer development in patients.
2. Open Questions
- How to tear off the heterogeneous “camouflage” in TAMs by single-cell transcriptome to track down subclonal evolution to conduce to target de facto protumoral TAMs efficiently?
- How to identify in vivo the molecular targets and determinants of driver mutations for each subclone of TAMs?
- How to determine potential therapeutic manipulation related to diagnosis and prognosis of single-cell-based spatiotemporal subclonal evolution?
3. Background and Introduction
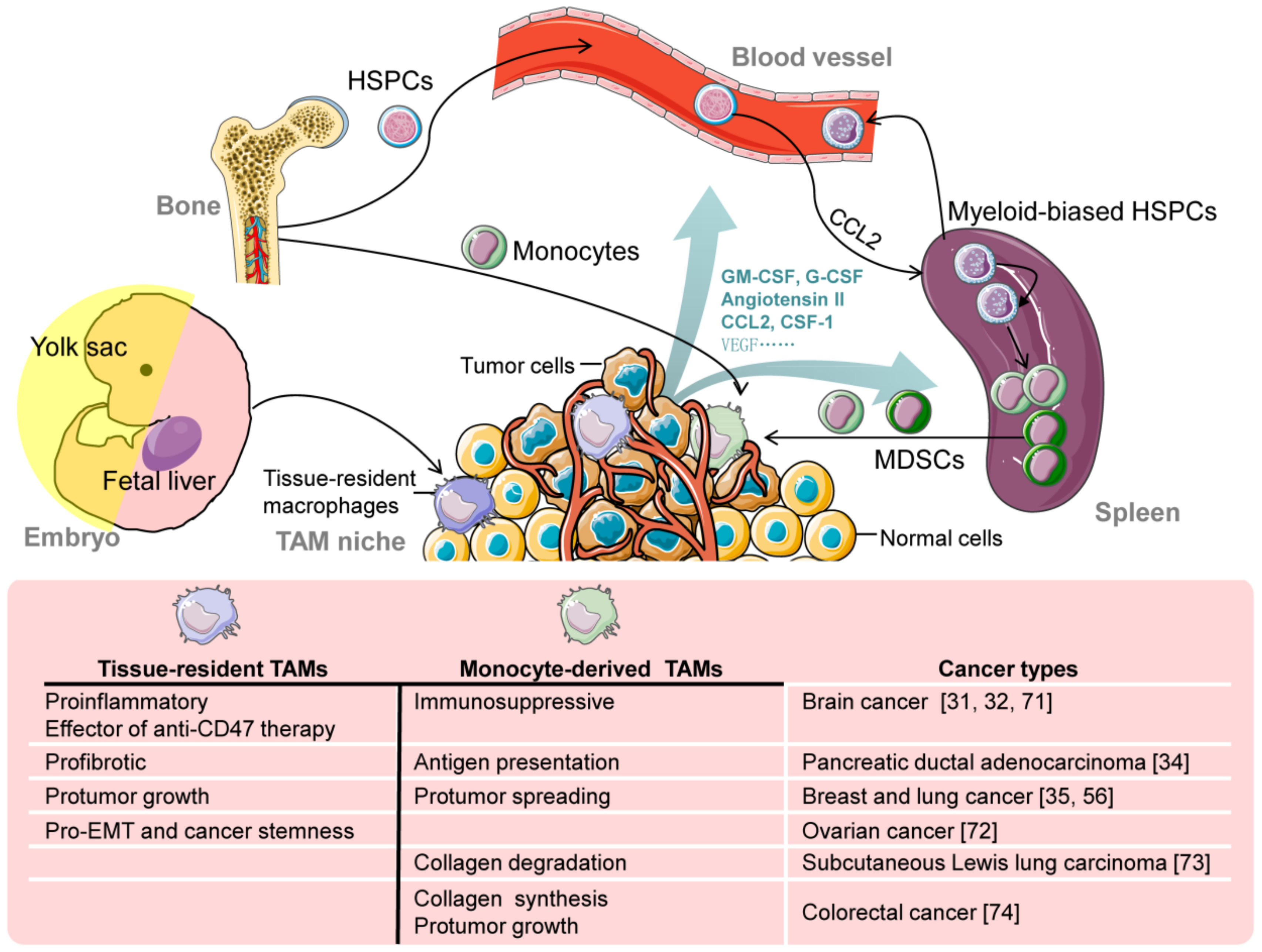
4. TAM Origins
5. Contribution of Different Origins to TAM Population
6. Functional Heterogeneity of TAMs from Different Origins
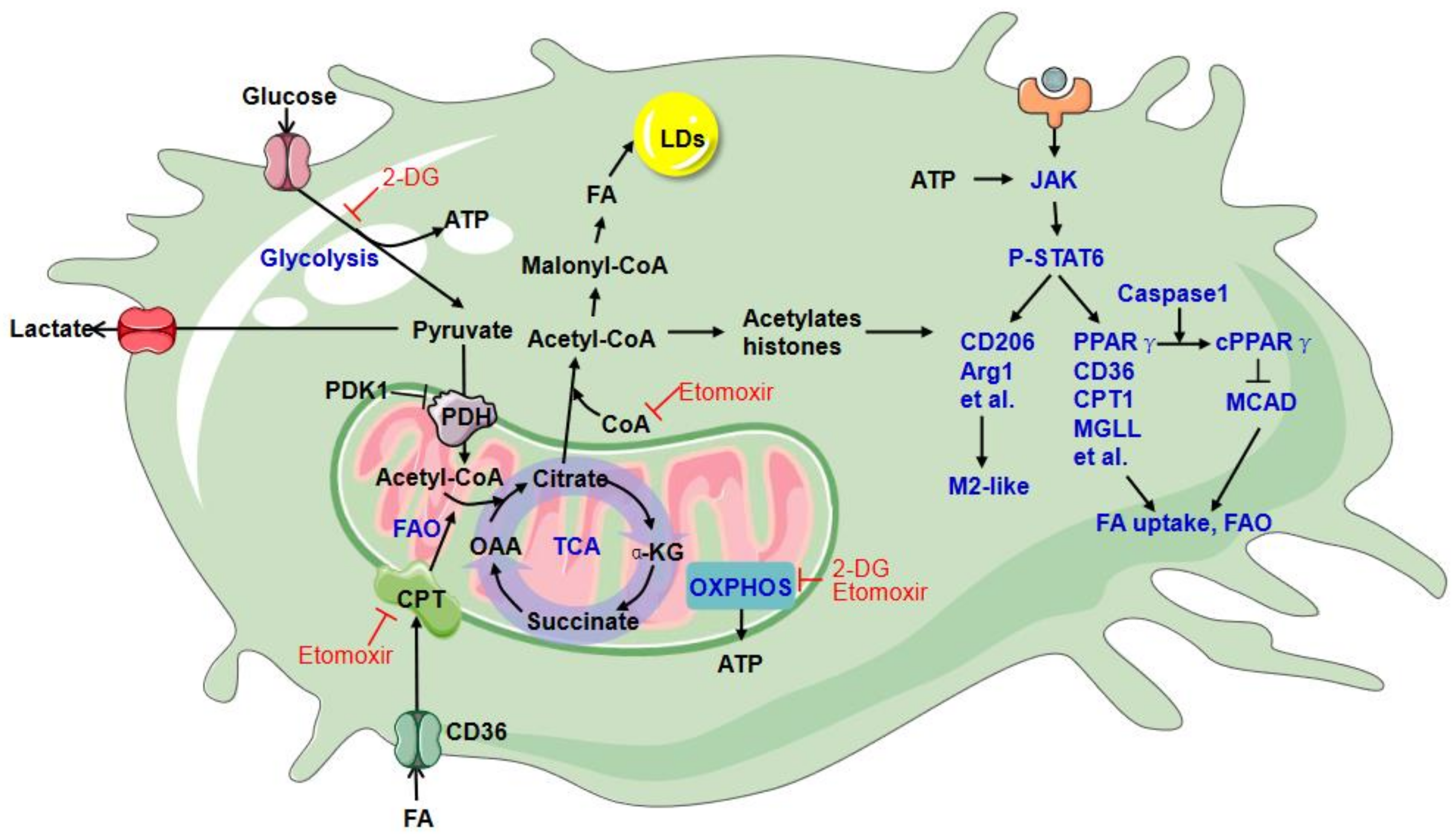
7. TAM Metabolic Reprogramming
8. Arginine Metabolism
9. Glucose Metabolism
10. Lipid Metabolism
11. Metabolic Regulation of TAM Ontogeny
12. Targeting Subclonal TAMs for Cancer Therapy
13. Spatiotemporal Single-Cell Transcriptomics and Single-Cell Tracking Technology Offer Subclonal Evolution of Cancer Development in Patients
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Gordon, S. Phagocytosis: An Immunobiologic Process. Immunity 2016, 44, 463–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamoutounour, S.; Guilliams, M.; Sanchis, F.M.; Liu, H.; Terhorst, D.; Malosse, C.; Pollet, E.; Ardouin, L.; Luche, H.; Sanchez, C.; et al. Origins and Functional Specialization of Macrophages and of Conventional and Monocyte-Derived Dendritic Cells in Mouse Skin. Immunity 2013, 39, 925–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353, aaf4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edholm, E.-S.; Rhoo, K.H.; Robert, J. Evolutionary Aspects of Macrophages Polarization. In Tissue Engineering; J.B. Metzler: Stuttgart, Germany, 2017; Volume 62, pp. 3–22. [Google Scholar]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Gong, Y.; Huang, T.; Lee, C.Z.W.; Bian, L.; Bai, Z.; Shi, H.; Zeng, Y.; Liu, C.; He, J.; et al. Deciphering human macrophage development at single-cell resolution. Nat. Cell Biol. 2020, 582, 571–576. [Google Scholar] [CrossRef]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Jäppinen, N.; Félix, I.; Lokka, E.; Tyystjärvi, S.; Pynttäri, A.; Lahtela, T.; Gerke, H.; Elima, K.; Rantakari, P.; Salmi, M. Fetal-derived macrophages dominate in adult mammary glands. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Aegerter, H.; Kulikauskaite, J.; Crotta, S.; Patel, H.; Kelly, G.; Hessel, E.M.; Mack, M.; Beinke, S.; Wack, A. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat. Immunol. 2020, 21, 145–157. [Google Scholar] [CrossRef]
- Ydens, E.; Amann, L.; Asselbergh, B.; Scott, C.L.; Martens, L.; Sichien, D.; Mossad, O.; Blank, T.; De Prijck, S.; Low, D.; et al. Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat. Neurosci. 2020, 23, 676–689. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Macciò, A.; Gramignano, G.; Cherchi, M.C.; Tanca, L.; Melis, L.; Madeddu, C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 1–15. [Google Scholar]
- Wanderley, C.W.; Colon, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; A Leite, C.; A Pereira, J.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1- profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, R.A.; Li, M.O. Ontogeny of Tumor-Associated Macrophages and Its Implication in Cancer Regulation. Trends Cancer 2016, 2, 20–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputa, G.; Castoldi, A.; Pearce, E.J. Metabolic adaptations of tissue-resident immune cells. Nat. Immunol. 2019, 20, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Fernando, V.; Sharma, V.; Walia, Y.; Letson, J.; Furuta, S. Correction of arginine metabolism with sepiapterin—the precursor of nitric oxide synthase cofactor BH4—induces immunostimulatory-shift of breast cancer. Biochem. Pharmacol. 2020, 176, 113887. [Google Scholar] [CrossRef] [PubMed]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brüne, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from em-bryonic hematopoiesis and promote tumor progression. Immunity 2017, 47, 323–338.e326. [Google Scholar] [CrossRef]
- Loyher, P.-L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; De Chanville, C.B.; Combadière, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.-C.; Zhao, J.-L.; Lu, Y.-T.; Gao, C.-C.; Yang, Y.; Liang, S.-Q.; Lu, Y.-Y.; Wang, L.; Yue, S.-Q.; Dou, K.-F.; et al. NOTCH Signaling via WNT Regulates the Proliferation of Alternative, CCR2-Independent Tumor-Associated Macrophages in Hepatocellular Carcinoma. Cancer Res. 2019, 79, 4160–4172. [Google Scholar] [CrossRef]
- Soncin, I.; Sheng, J.; Chen, Q.; Foo, S.; Duan, K.; Lum, J.; Poidinger, M.; Zolezzi, F.; Karjalainen, K.; Ruedl, C. The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Camara, A.; Cordeiro, O.G.; Alloush, F.; Sponsel, J.; Chypre, M.; Onder, L.; Asano, K.; Tanaka, M.; Yagita, H.; Ludewig, B.; et al. Lymph Node Mesenchymal and Endothelial Stromal Cells Cooperate via the RANK-RANKL Cytokine Axis to Shape the Sinusoidal Macrophage Niche. Immunity 2019, 50, 1467–1481.e6. [Google Scholar] [CrossRef]
- Mondor, I.; Baratin, M.; Lagueyrie, M.; Saro, L.; Henri, S.; Gentek, R.; Suerinck, D.; Kastenmuller, W.; Jiang, J.X.; Bajénoff, M. Lymphatic Endothelial Cells Are Essential Components of the Subcapsular Sinus Macrophage Niche. Immunity 2019, 50, 1453–1466.e4. [Google Scholar] [CrossRef]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; Van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local Macrophage Proliferation, Rather than Recruitment from the Blood, Is a Signature of TH2 Inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braune, J.; Weyer, U.; Hobusch, C.; Mauer, J.; Brüning, J.C.; Bechmann, I.; Gericke, M. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J. Immunol. 2017, 198, 2927–2934. [Google Scholar] [CrossRef] [PubMed]
- Soucie, E.L.; Weng, Z.; Geirsdóttir, L.; Molawi, K.; Maurizio, J.; Fenouil, R.; Mossadegh-Keller, N.; Gimenez, G.; Vanhille, L.; Beniazza, M.; et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 2016, 351, aad5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas, M.; Davies, L.C.; Giles, P.J.; Liao, C.-T.; Kharfan, B.; Stone, T.C.; O’Donnell, V.B.; Fraser, D.J.; Jones, S.A.; Taylor, P.R. The Transcription Factor Gata6 Links Tissue Macrophage Phenotype and Proliferative Renewal. Science 2014, 344, 645–648. [Google Scholar] [CrossRef] [Green Version]
- Casanova-Acebes, M.; Menéndez-Gutiérrez, M.P.; Porcuna, J.; Álvarez-Errico, D.; Lavin, Y.; García, A.; Kobayashi, S.; Le Berichel, J.; Núñez, V.; Were, F.; et al. RXRs control serous macrophage neonatal expansion and identity and contribute to ovarian cancer progression. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Sieweke, M.H.; Allen, J.E. Beyond Stem Cells: Self-Renewal of Differentiated Macrophages. Science 2013, 342, 1242974. [Google Scholar] [CrossRef]
- Van Overmeire, E.; Stijlemans, B.; Heymann, F.; Keirsse, J.; Morias, Y.; Elkrim, Y.; Brys, L.; Abels, C.; Lahmar, Q.; Ergen, C.; et al. M-CSF and GM-CSF Receptor Signaling Differentially Regulate Monocyte Maturation and Macrophage Polarization in the Tumor Microenvironment. Cancer Res. 2016, 76, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Ao, J.-Y.; Zhu, X.-D.; Chai, Z.-T.; Cai, H.; Zhang, Y.-Y.; Zhang, K.-Z.; Kong, L.-Q.; Zhang, N.; Ye, B.-G.; Ma, D.-N.; et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Sidibe, A.; Ropraz, P.; Jemelin, S.; Emre, Y.; Poittevin, M.; Pocard, M.; Bradfield, P.F.; Imhof, B.A. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat. Commun. 2018, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Bu, W.; Meng, L.; Liu, X.; Wang, S.; Jiang, L.; Ren, M.; Fan, Y.; Sun, H. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp. Cell Res. 2019, 378, 131–138. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Tymoszuk, P.; Evens, H.; Marzola, V.; Wachowicz, K.; Wasmer, M.-H.; Datta, S.; Müller-Holzner, E.; Fiegl, H.; Böck, G.; Van Rooijen, N.; et al. In situ proliferation contributes to accumulation of tumor-associated macrophages in spontaneous mammary tumors. Eur. J. Immunol. 2014, 44, 2247–2262. [Google Scholar] [CrossRef]
- Campbell, M.J.; Tonlaar, N.Y.; Garwood, E.R.; Huo, D.; Moore, D.H.; Khramtsov, A.I.; Au, A.; Baehner, F.; Chen, Y.; Malaka, D.O.; et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res. Treat. 2010, 128, 703–711. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.-L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; He, R.; Long, H.; Guo, B.; Jia, Q.; Qin, D.; Liu, S.-Q.; Wang, Z.; Xiang, T.; Zhang, J.; et al. Late-stage tumors induce anemia and immunosuppressive extramedullary erythroid progenitor cells. Nat. Med. 2018, 24, 1536–1544. [Google Scholar] [CrossRef]
- Yang, X.; Chen, D.; Long, H.; Zhu, B. The mechanisms of pathological extramedullary hematopoiesis in diseases. Cell. Mol. Life Sci. 2020, 77, 2723–2738. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Rauch, P.J.; Chudnovskiy, A.; Berger, C.; Ryan, R.J.H.; Iwamoto, Y.; Marinelli, B.; Gorbatov, R.; et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 2012, 109, 2491–2496. [Google Scholar]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Wu, C.; Ning, H.; Liu, M.; Lin, J.; Luo, S.; Zhu, W.; Xu, J.; Wu, W.-C.; Liang, J.; Shao, C.-K.; et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J. Clin. Investig. 2018, 128, 3425–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.-C.; Sun, H.-W.; Chen, H.-T.; Liang, J.; Yu, X.-J.; Wu, C.; Wang, Z.; Zheng, L. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc. Natl. Acad. Sci. USA 2014, 111, 4221–4226. [Google Scholar]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Ryan, R.; Pucci, F.; Sio, S.W.; Kuswanto, W.; Rauch, P.J.; Chudnovskiy, A.; Iwamoto, Y.; et al. Angiotensin II Drives the Production of Tumor-Promoting Macrophages. Immunity 2013, 38, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Li, X.; Li, J.; Lu, Y.; Zhao, S.; Tang, X.; Chen, X.; Li, J.; Zheng, Y.; Li, S.; et al. APOBEC3B interaction with PRC2 modulates microenvironment to promote HCC progression. Gut 2019, 68, 1846–1857. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Ginhoux, F.; Schultze, J.L.; Murray, P.J.; Ochando, J.; Biswas, S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2015, 17, 34–40. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar]
- Etzerodt, A.; Moulin, M.; Doktor, T.K.; Delfini, M.; Mossadegh-Keller, N.; Bajenoff, M.; Sieweke, M.H.; Moestrup, S.K.; Auphan-Anezin, N.; Lawrence, T. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Madsen, D.H.; Jürgensen, H.J.; Siersbæk, M.S.; Kuczek, D.E.; Cloud, L.G.; Liu, S.; Behrendt, N.; Grøntved, L.; Weigert, R.; Bugge, T.H. Tumor-Associated Macrophages Derived from Circulating Inflammatory Monocytes Degrade Collagen through Cellular Uptake. Cell Rep. 2017, 21, 3662–3671. [Google Scholar] [CrossRef] [Green Version]
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T.; et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 2016, 213, 2315–2331. [Google Scholar] [CrossRef]
- Gadiyar, V.; Patel, G.; Davra, V. Immunological role of TAM receptors in the cancer microenvironment. Int. Rev. Cell Mol. Biol. Vol. 298 2020, 357, 57–79. [Google Scholar] [CrossRef]
- Saha, S.; Shalova, I.N.; Biswas, S.K. Metabolic regulation of macrophage phenotype and function. Immunol. Rev. 2017, 280, 102–111. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D. Macrophage Arginine Metabolism to Ornithine/Urea or Nitric Oxide/Citrulline: A Life or Death Issue. Crit. Rev. Immunol. 2001, 21, 28–425. [Google Scholar] [CrossRef]
- Carmona-Fontaine, C.; Deforet, M.; Akkari, L.; Thompson, C.B.; Joyce, J.A.; Xavier, J.B. Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2017, 114, 2934–2939. [Google Scholar]
- Ryan, D.G.; O’Neill, L.A. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu. Rev. Immunol. 2020, 38, 289–313. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.C.-C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 2016, 5, e11612. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475.e4. [Google Scholar] [CrossRef] [Green Version]
- Galdieri, L.; Vancura, A. Acetyl-CoA Carboxylase Regulates Global Histone Acetylation. J. Biol. Chem. 2012, 287, 23865–23876. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Xie, N.; Cui, H.; Moellering, D.R.; Abraham, E.; Thannickal, V.J.; Liu, G. Pyruvate Dehydrogenase Kinase 1 Participates in Macrophage Polarization via Regulating Glucose Metabolism. J. Immunol. 2015, 194, 6082–6089. [Google Scholar] [CrossRef]
- De-Brito, N.M.; Duncan-Moretti, J.; Da-Costa, H.C.; Saldanha-Gama, R.; Paula-Neto, H.A.; Dorighello, G.G.; Simões, R.L.; Barja-Fidalgo, C. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim. Biophys. Acta (BBA) Bioenerg. 2020, 1867, 118604. [Google Scholar] [CrossRef]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; Ee, P.S.C.; Luis, B.S.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. OncoImmunology 2016, 5, e1191731. [Google Scholar] [CrossRef]
- Chen, D.-P.; Ning, W.-R.; Jiang, Z.-Z.; Peng, Z.-P.; Zhu, L.-Y.; Zhuang, S.-M.; Kuang, D.-M.; Zheng, L.; Wu, Y. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J. Hepatol. 2019, 71, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houten, S.M.; Wanders, R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef]
- Yao, C.-H.; Liu, G.-Y.; Wang, R.; Moon, S.H.; Gross, R.W.; Patti, G.J. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of β-oxidation. PLoS Biol. 2018, 16, e2003782. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, R.S.; Guo, L.; Ghassemi, S.; Snyder, N.W.; Worth, A.J.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef] [Green Version]
- Nomura, M.; Liu, J.; I Rovira, I.; Gonzalez-Hurtado, E.; Lee, J.; Wolfgang, M.J.; Finkel, T. Fatty acid oxidation in macrophage polarization. Nat. Immunol. 2016, 17, 216–217. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Hsieh, W.Y.; Minarrieta, L.; Duong, T.N.; Kim, K.K.; Desousa, B.R.; Andreyev, A.Y.; Bowman, C.E.; Caradonna, K.; Dranka, B.P.; et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018, 28, 490–503.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Weinberg, S.; Deberge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Charvet, L.Y.-; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456.e5. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, N.-A.; Quintana, J.A.; García-Silva, S.; Mazariegos, M.; De La Aleja, A.G.; Nicolás-Ávila, J.A.; Walter, W.; Adrover, J.M.; Crainiciuc, G.; Kuchroo, V.K.; et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J. Exp. Med. 2017, 214, 1281–1296. [Google Scholar] [CrossRef] [Green Version]
- Cunha, L.D.; Yang, M.; Carter, R.; Guy, C.; Harris, L.; Crawford, J.C.; Quarato, G.; Boada-Romero, E.; Kalkavan, H.; Johnson, M.D.; et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 2018, 175, 429–441.e16. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Han, Y.; Sillke, Y.R.; Deng, H.; Siddiqui, S.; Treese, C.; Schmidt, F.; Friedrich, M.; Keye, J.; Wan, J.; et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol. Med. 2019, 11, e10698. [Google Scholar] [CrossRef]
- Li, F.; Okreglicka, K.M.; Pohlmeier, L.M.; Schneider, C.; Kopf, M. Fetal monocytes possess increased metabolic capacity and replace primitive macrophages in tissue macrophage development. EMBO J. 2020, 39, e103205. [Google Scholar] [CrossRef]
- Dreschers, S.; Ohl, K.; Lehrke, M.; Möllmann, J.; Denecke, B.; Costa, I.; Vogl, T.; Viemann, D.; Roth, J.; Orlikowsky, T.; et al. Impaired cellular energy metabolism in cord blood macrophages contributes to abortive response toward inflammatory threats. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.-H.; Collins, S.L.; Sun, I.-H.; Tam, A.J.; Patel, C.H.; Arwood, M.L.; Chan-Li, Y.; Powell, J.D.; Horton, M.R. mTORC2 Signaling Selectively Regulates the Generation and Function of Tissue-Resident Peritoneal Macrophages. Cell Rep. 2017, 20, 2439–2454. [Google Scholar] [CrossRef] [Green Version]
- Woods, P.S.; Kimmig, L.M.; Meliton, A.Y.; Sun, K.A.; Tian, Y.; O’Leary, E.M.; Gökalp, G.A.; Hamanaka, R.B.; Mutlu, G.M. Tissue-Resident Alveolar Macrophages Do Not Rely on Glycolysis for LPS-induced Inflammation. Am. J. Respir. Cell Mol. Biol. 2020, 62, 243–255. [Google Scholar] [CrossRef]
- Svedberg, F.R.; Brown, S.L.; Krauss, M.Z.; Campbell, L.; Sharpe, C.; Clausen, M.; Howell, G.J.; Clark, H.; Madsen, J.; Evans, C.M.; et al. The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nat. Immunol. 2019, 20, 571–580. [Google Scholar] [CrossRef]
- Huang, L.; Nazarova, E.V.; Tan, S.; Liu, Y.; Russell, D.G. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med. 2018, 215, 1135–1152. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.H.; Clark, N.; Brennan, A.L.; Fisher, D.A.; Gyi, K.M.; Hodson, M.E.; Philips, B.J.; Baines, D.L.; Wood, D.M. Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J. Appl. Physiol. 2007, 102, 1969–1975. [Google Scholar] [CrossRef]
- Tan, S.Y.S.; Krasnow, M.A. Developmental origin of lung macrophage diversity. Dev. 2016, 143, 1318–1327. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.-Y.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.W.; et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Lamb, Y.N. Pexidartinib: First Approval. Drugs 2019, 79, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, P.P.; Surriga, O.; Beckman, M.J.; De Stanchina, E.; DeMatteo, R.P.; Tap, W.D.; Schwartz, G.K. Sustained Inhibition of Receptor Tyrosine Kinases and Macrophage Depletion by PLX3397 and Rapamycin as a Potential New Approach for the Treatment of MPNSTs. Clin. Cancer Res. 2014, 20, 3146–3158. [Google Scholar] [CrossRef] [Green Version]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar]
- Dammeijer, F.; Lievense, L.A.; Kaijen-Lambers, M.E.; Van Nimwegen, M.; Bezemer, K.; Hegmans, J.P.; Van Hall, T.; Hendriks, R.W.; Aerts, J.G. Depletion of Tumor-Associated Macrophages with a CSF-1R Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by DC Immunotherapy. Cancer Immunol. Res. 2017, 5, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Fein, M.R.; He, X.-Y.; Almeida, A.S.; Bružas, E.; Pommier, A.; Yan, R.; Eberhardt, A.; Fearon, D.T.; Van Aelst, L.; Wilkinson, J.E.; et al. Cancer cell CCR2 orchestrates suppression of the adaptive immune response. J. Exp. Med. 2020, 217, e20181551. [Google Scholar] [CrossRef]
- Terashima, Y.; Toda, E.; Itakura, M.; Otsuji, M.; Yoshinaga, S.; Okumura, K.; Shand, F.H.W.; Komohara, Y.; Takeda, M.; Kokubo, K.; et al. Targeting FROUNT with disulfiram suppresses macrophage accumulation and its tumor-promoting properties. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.; Huang, H.; Guo, M.; Chen, J.; Wu, W.; Li, W.; Xu, X.; Lin, X.; Fu, W.; Yao, Y.-D.; et al. Breast Phyllodes Tumors Recruit and Repolarize Tumor-Associated Macrophages via Secreting CCL5 to Promote Malignant Progression, Which Can Be Inhibited by CCR5 Inhibition Therapy. Clin. Cancer Res. 2019, 25, 3873–3886. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xavy, S.; Mihardja, S.; Chen, S.; Sompalli, K.; Feng, D.; Choi, T.; Agoram, B.; Majeti, R.; Weissman, I.L.; et al. Targeting macrophage checkpoint inhibitor SIRPα for anticancer therapy. JCI Insight 2020, 5, 134728. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [Green Version]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.Y.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPαFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Puro, R.J.; Bouchlaka, M.N.; Hiebsch, R.R.; Capoccia, B.J.; Donio, M.J.; Manning, P.T.; Frazier, W.A.; Karr, R.W.; Pereira, D.S. Development of AO-176, a Next-Generation Humanized Anti-CD47 Antibody with Novel Anticancer Properties and Negligible Red Blood Cell Binding. Mol. Cancer Ther. 2019, 19, 835–846. [Google Scholar] [CrossRef] [Green Version]
- Cabrales, P. RRx-001 Acts as a Dual Small Molecule Checkpoint Inhibitor by Downregulating CD47 on Cancer Cells and SIRP-α on Monocytes/Macrophages. Transl. Oncol. 2019, 12, 626–632. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Lossos, C.; Liu, Y.; Kolb, K.E.; Christie, A.L.; Van Scoyk, A.; Prakadan, S.M.; Shigemori, K.; Stevenson, K.E.; Morrow, S.; Plana, O.D.; et al. Mechanisms of Lymphoma Clearance Induced by High-Dose Alkylating Agents. Cancer Discov. 2019, 9, 944–961. [Google Scholar] [CrossRef]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.-C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373.e9. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Qorraj, M.; Bruns, H.; Böttcher, M.; Weigand, L.; Saul, D.; Mackensen, A.; Jitschin, R.; Mougiakakos, D. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia 2016, 31, 470–478. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef]
- Liu, M.; Tong, Z.; Ding, C.; Luo, F.; Wu, S.; Wu, C.; Albeituni, S.; He, L.; Hu, X.; Tieri, D.; et al. Transcription factor c-Maf is a checkpoint that programs macrophages in lung cancer. J. Clin. Investig. 2020, 130, 2081–2096. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Wu, L.; Hollinshead, K.E.; Hao, Y.; Au, C.; Kroehling, L.; Ng, C.; Lin, W.-Y.; Li, D.; Silva, H.M.; Shin, J.; et al. Niche-Selective Inhibition of Pathogenic Th17 Cells by Targeting Metabolic Redundancy. Cell 2020, 182, 641–654.e20. [Google Scholar] [CrossRef]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Li, S.-C.; Stucky, A.; Chen, X.; Kabeer, M.H.; Loudon, W.G.; Plant, A.S.; Torno, L.; Nangia, C.S.; Cai, J.; Zhang, G.; et al. Single-cell transcriptomes reveal the mechanism for a breast cancer prognostic gene panel. Oncotarget 2018, 9, 33290–33301. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.X.; Li, S.C. Hunting down the dominating subclone of cancer stem cells as a potential new therapeutic target in multiple myeloma: An artificial intelligence perspective. World J. Stem Cells 2020, 12, 706–720. [Google Scholar] [CrossRef]
- Li, S.C.; Tachiki, L.M.L.; Luo, J.; Dethlefs, B.A.; Chen, Z.; Loudon, W.G. A Biological Global Positioning System: Considerations for Tracking Stem Cell Behaviors in the Whole Body. Stem Cell Rev. Rep. 2010, 6, 317–333. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-C.; Lee, K.L.; Luo, J. Control Dominating Subclones for Managing Cancer Progression and Posttreatment Recurrence by Subclonal Switchboard Signal: Implication for New Therapies. Stem Cells Dev. 2012, 21, 503–506. [Google Scholar] [CrossRef]
- Zhou, M.-Y.; Xie, X.-L.; Peng, Y.-G.; Wu, M.-J.; Deng, X.-Z.; Wu, Y.; Xiong, L.-J.; Shang, L.-H. From SARS to COVID-19: What we have learned about children infected with COVID-19. Int. J. Infect. Dis. 2020, 96, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.-B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef] [PubMed]
- Diorio, C.; Henrickson, S.E.; Vella, L.A.; McNerney, K.O.; Chase, J.M.; Burudpakdee, C.; Lee, J.H.; Jasen, C.; Balamuth, F.; Barrett, D.M.; et al. Multisystem inflammatory syndrome in children and COVID-19 are distinct presentations of SARS–CoV-2. J. Clin. Investig. 2020, 130, 5967–5975. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wen, Q.; Stucky, A.; Zeng, Y.; Gao, S.; Loudon, W.G.; Ho, H.W.; Kabeer, M.H.; Li, S.C.; Zhang, X.; et al. Relapse pathway of glioblastoma revealed by single-cell molecular analysis. Carcinogenesis 2018, 39, 931–936. [Google Scholar] [CrossRef]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 1–10. [Google Scholar] [CrossRef]
- Quinn, J.J.; Jones, M.G.; Okimoto, R.A.; Nanjo, S.; Chan, M.M.; Yosef, N.; Bivona, T.G.; Weissman, J.S. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 2021, 371, eabc1944. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.-M.; Chen, D.-G.; Li, S.C.; Zhu, B.; Li, Z.-J. Embryonic Origin and Subclonal Evolution of Tumor-Associated Macrophages Imply Preventive Care for Cancer. Cells 2021, 10, 903. https://doi.org/10.3390/cells10040903
Zhang X-M, Chen D-G, Li SC, Zhu B, Li Z-J. Embryonic Origin and Subclonal Evolution of Tumor-Associated Macrophages Imply Preventive Care for Cancer. Cells. 2021; 10(4):903. https://doi.org/10.3390/cells10040903
Chicago/Turabian StyleZhang, Xiao-Mei, De-Gao Chen, Shengwen Calvin Li, Bo Zhu, and Zhong-Jun Li. 2021. "Embryonic Origin and Subclonal Evolution of Tumor-Associated Macrophages Imply Preventive Care for Cancer" Cells 10, no. 4: 903. https://doi.org/10.3390/cells10040903