
Structural Alternation in Heat Shock Proteins of Activated Macrophages

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and LPS Stimulation
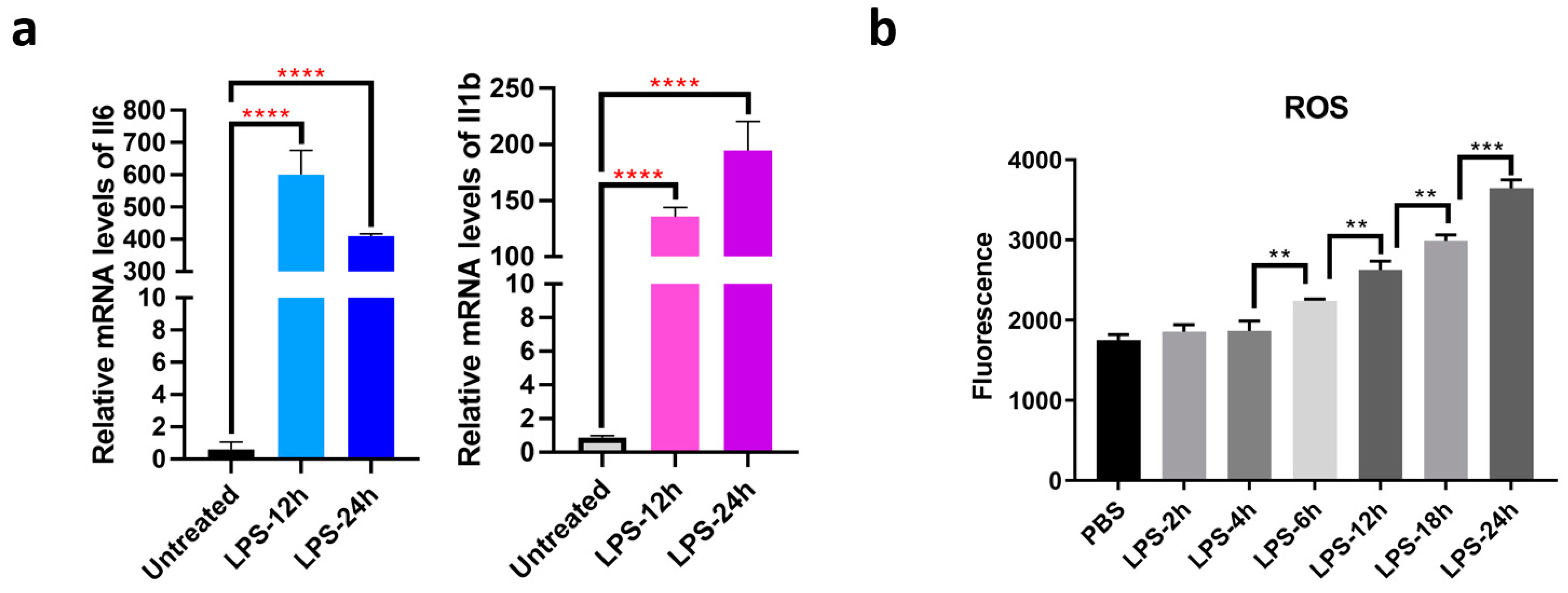
2.2. Quantitative RT-PCR Assay
2.3. Detection of Cellular Reactive Oxygen Species
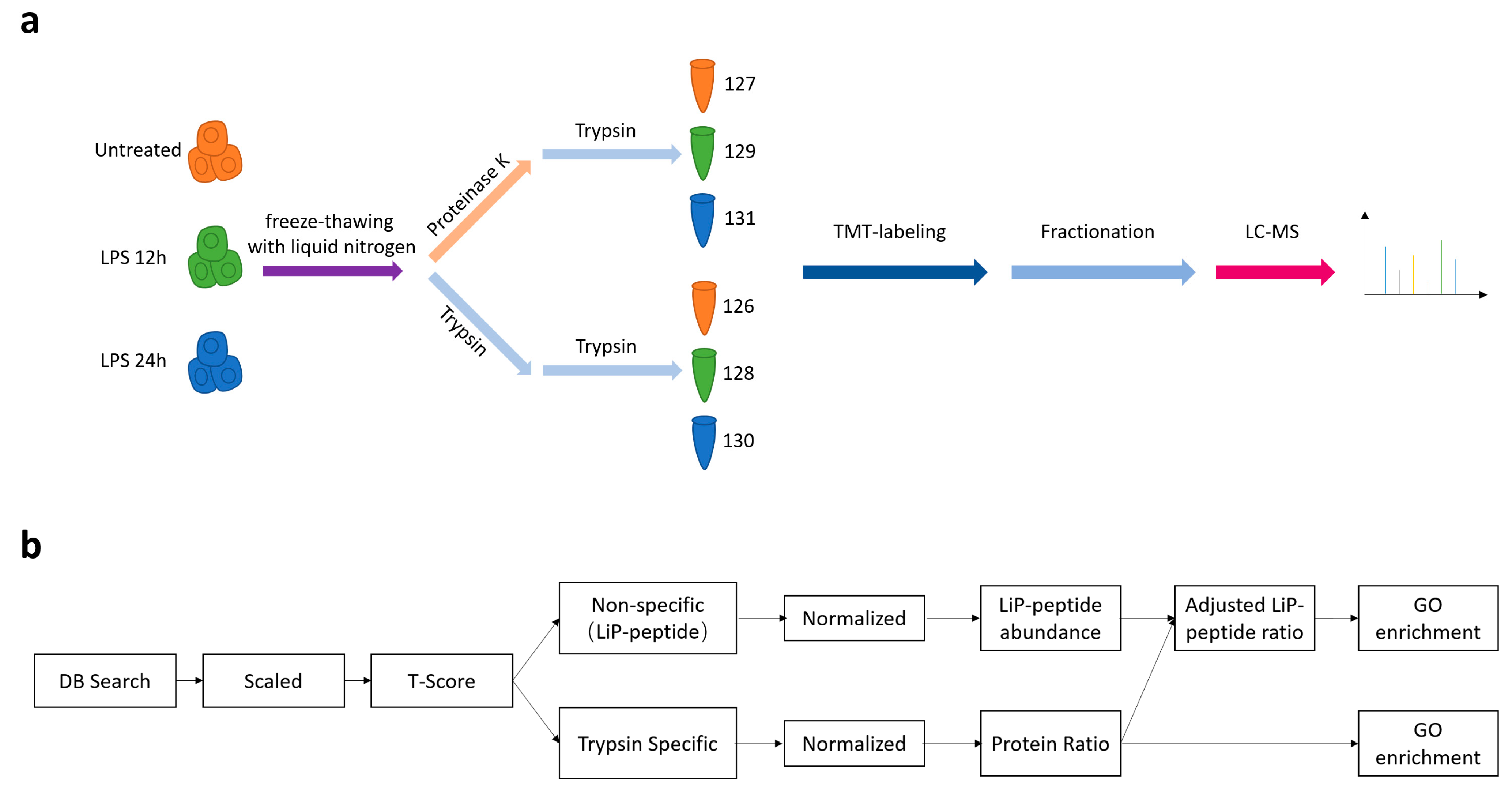
2.4. LiP-MS Analysis
2.5. LC-MS Analysis
2.6. LiP-MS Data Analysis
2.7. Cellular Thermal Shift Assay (CETSA)
2.8. Western Blot Analysis
3. Results
3.1. Modification of the LiP-MS Workflow for Characterization of the Structure-Ome
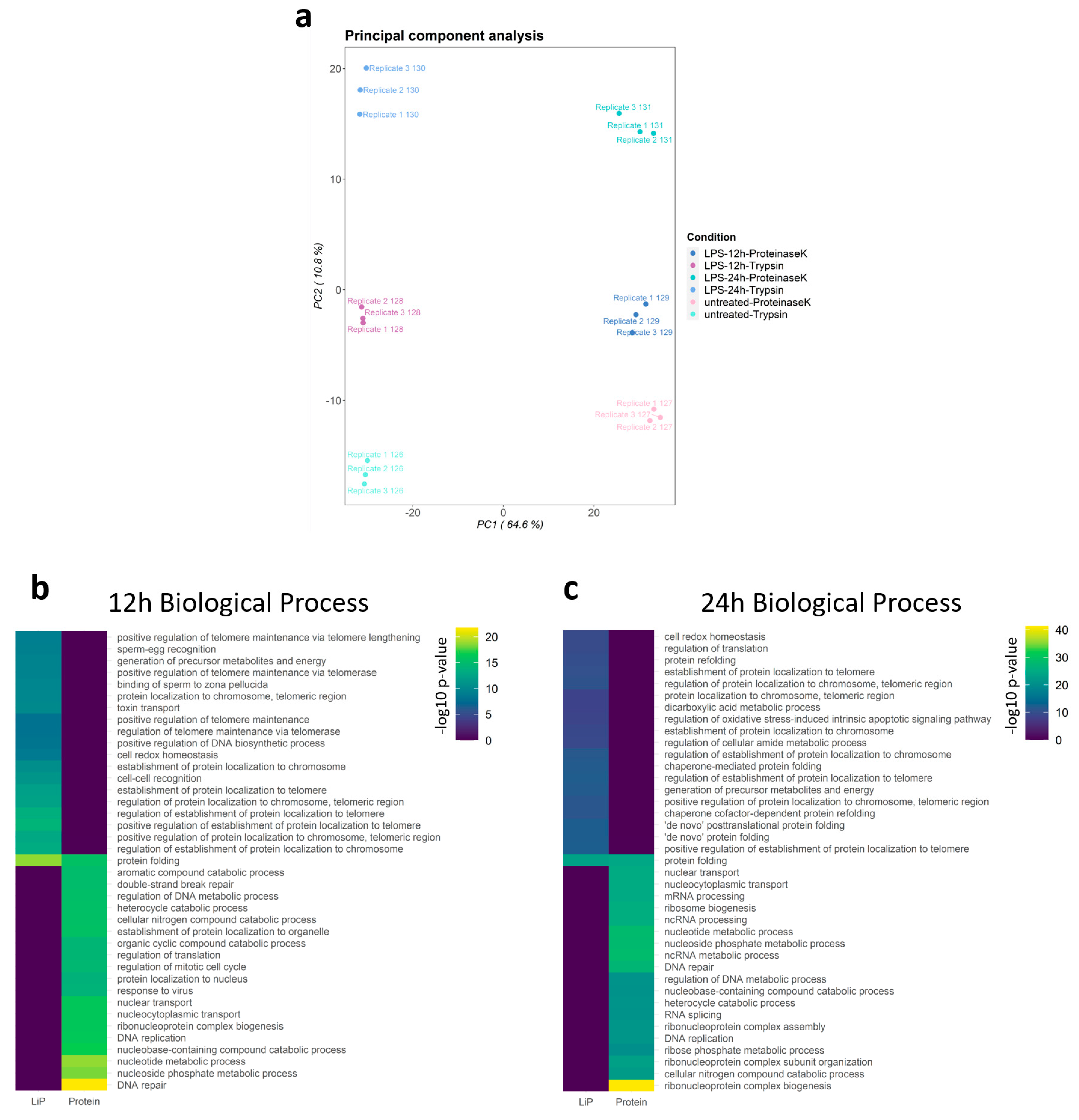
3.2. Landscape of the Structure-Ome during Macrophage Activation
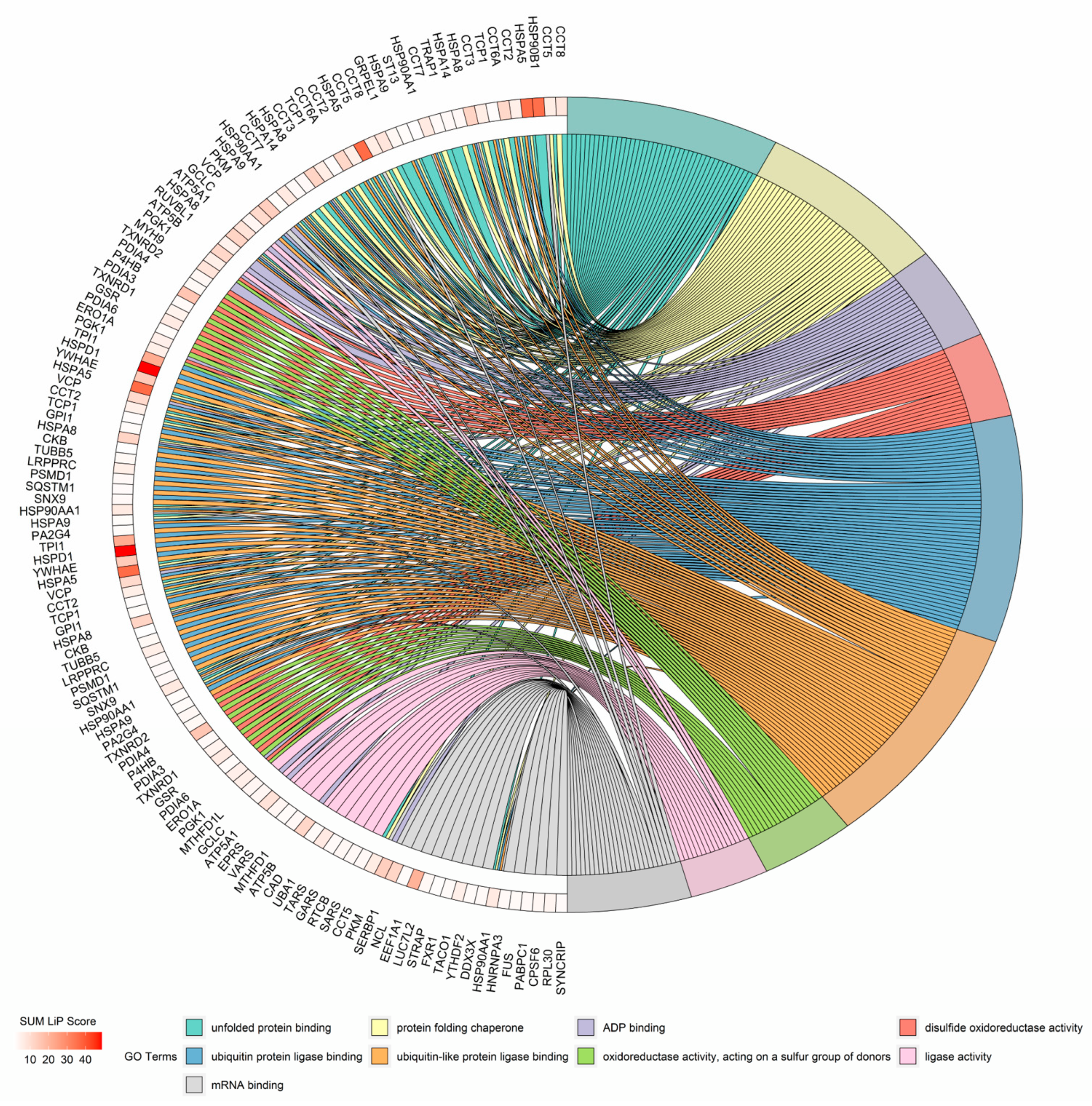
3.3. GO Analysis of Structural Reads from Changed Proteins during Macrophage Activation
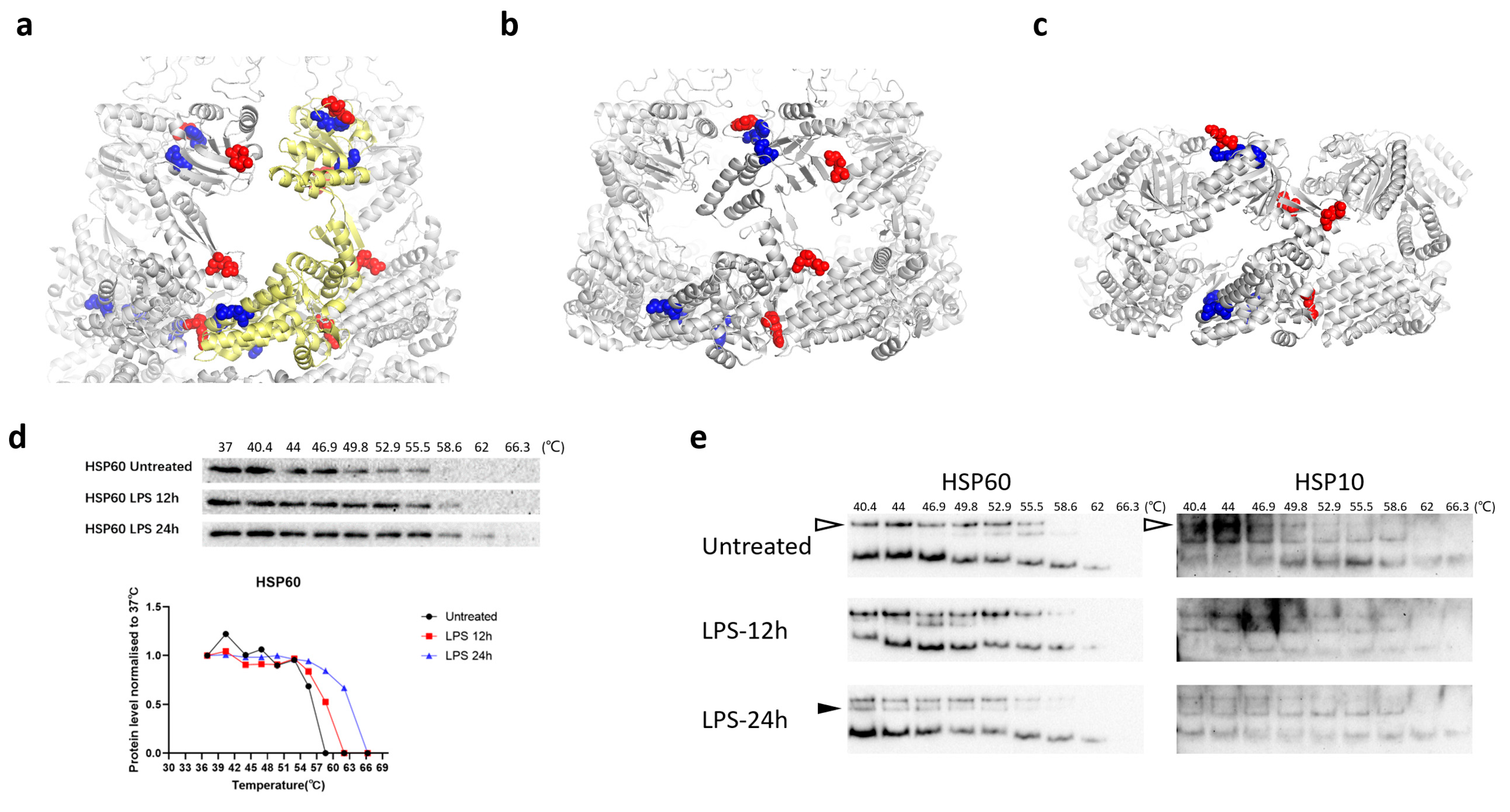
3.4. Structural Changes in HSP60 during Macrophage Activation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.Q.; Chen, Y.Q.; Fisher, J.H.; Wang, M.H. Activation of the RON receptor tyrosine kinase by macrophage-stimulating protein inhibits inducible cyclooxygenase-2 expression in murine macrophages. J. Biol. Chem. 2002, 277, 38104–38110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.V. Toll signaling pathways in the innate immune response. Curr. Opin. Immunol. 2000, 12, 13–19. [Google Scholar] [CrossRef]
- Canton, M.; Sanchez-Rodriguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; De Franceschi, G.; Kahraman, A.; Soste, M.; Melnik, A.; Boersema, P.J.; De Laureto, P.P.; Nikolaev, Y.; Oliveira, A.P.; Picotti, P. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014, 32, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, V.; Hauser, T.; Piazza, I.; Pepelnjak, M.; Malinovska, L.; Fuhrer, T.; Li, Y.; Dörig, C.; Boersema, P.; Gillet, L.; et al. Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ. Cell 2021, 184, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. Chaperone-mediated protein folding. Physiol. Rev. 1999, 79, 425–449. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Llorente, Y.; Jebara, F.; Patra, M.; Malik, R.; Nisemblat, S.; Chomsky-Hecht, O.; Parnas, A.; Azem, A.; Hirsch, J.A.; Ubarretxena-Belandia, I. Structural basis for active single and double ring complexes in human mitochondrial Hsp60-Hsp10 chaperonin. Nat. Commun. 2020, 11, 1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viitanen, P.V.; Lorimer, G.H.; Seetharam, R.; Gupta, R.S.; Oppenheim, J.; Thomas, J.O.; Cowan, N.J. Mammalian mitochondrial chaperonin 60 functions as a single toroidal ring. J. Biol. Chem. 1992, 26, 695–698. [Google Scholar] [CrossRef]
- Nielsen, K.L.; Cowan, N.J. A single ring is sufficient for productive chaperonin-mediated folding in vivo. Mol. Cell 1998, 2, 93–99. [Google Scholar] [CrossRef]
- Viitanen, P.V.; Lorimer, G.; Bergmeier, W.; Weiss, C.; Kessel, M.; Goloubinoff, P. Purification of mammalian mitochondrial chaperonin 60 through in vitro reconstitution of active oligomers. Methods Enzymol. 1998, 290, 203–217. [Google Scholar]
- Nisemblat, S.; Yaniv, O.; Parnas, A.; Frolow, F.; Azem, A. Crystal structure of the human mitochondrial chaperonin symmetrical football complex. Proc. Natl. Acad. Sci. USA 2015, 112, 6044–6049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebl, D.P.; Feasey, M.C.; Hesketh, E.L.; Ranson, N.A.; Wurdak, H.; Sobott, F.; Bon, R.S.; Muench, S.P. Cryo-EM structure of human mitochondrial HSPD1. iScience 2021, 24, 102022. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Isserlin, R.; Emili, A.; Burniston, J.G. Exercise-responsive phosphoproteins in the heart. J. Mol. Cell Cardiol. 2017, 111, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Reinhard, F.B.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Walter, W.; Sanchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Gao, W.; Mu, H.; Qin, T.; Long, F.; Ren, L.; Tang, H.; Liu, J.; Zeng, M. HSP60 Regulates Monosodium Urate Crystal-Induced Inflammation by Activating the TLR4-NF-kappaB-MyD88 Signaling Pathway and Disrupting Mitochondrial Function. Oxid. Med. Cell Longev. 2020, 2020, 8706898. [Google Scholar] [CrossRef] [PubMed]
- Malik, J.A.; Lone, R. Heat shock proteins with an emphasis on HSP 60. Mol. Biol. Rep. 2021, 48, 6959–6969. [Google Scholar] [CrossRef]
- Xu, Z.; Horwich, A.L.; Sigler, P.B. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature 1997, 388, 741–750. [Google Scholar] [CrossRef]
- Braig, K.; Otwinowski, Z.; Hegde, R.; Boisvert, D.C.; Joachimiak, A.; Horwich, A.L.; Sigler, P.B. The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature 1994, 371, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Vilasi, S.; Bulone, D.; Caruso Bavisotto, C.; Campanella, C.; Marino Gammazza, A.; San Biagio, P.L.; Cappello, F.; Conway de Macario, E.; Macario, A.J. Chaperonin of Group I: Oligomeric Spectrum and Biochemical and Biological Implications. Front. Mol. Biosci. 2017, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Enriquez, A.S.; Li, J.; Rodriguez, A.; Holguin, B.; Von Salzen, D.; Bhatt, J.M.; Bernal, R.A. MitCHAP-60 and Hereditary Spastic Paraplegia SPG-13 Arise from an Inactive hsp60 Chaperonin that Fails to Fold the ATP Synthase beta-Subunit. Sci. Rep. 2019, 9, 12300. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Names | Protein Description | 24 h Sum LiP Score |
|---|---|---|
| Hspd1 Hsp60 | 60-kDa heat shock protein, mitochondrial | 48.8701551 |
| Hsp90b1 | Heat shock protein 90-kDa beta member 1 | 35.57114236 |
| P4hb | Endoplasmic reticulum resident protein 59 | 15.61354634 |
| Hspa8 Hsc70 Hsc73 | Heat shock cognate 71-kDa protein | 11.97500326 |
| Cct6a Cct6 Cctz Cctz1 | T-complex protein 1 subunit zeta | 11.27109261 |
| Cct8 Cctq | T-complex protein 1 subunit theta | 7.959656493 |
| Hsp90aa1 | Heat shock protein HSP 90-alpha | 6.882919007 |
| Cct5 Ccte | T-complex protein 1 subunit epsilon | 6.202519596 |
| Cct3 Cctg | T-complex protein 1 subunit gamma | 5.911248248 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Wei, Y.; Zhang, H.; Liu, J.; Zong, Z.; Liu, Z.; Zhu, S.; Hou, W.; Chen, Y.; Deng, H. Structural Alternation in Heat Shock Proteins of Activated Macrophages. Cells 2021, 10, 3507. https://doi.org/10.3390/cells10123507
Zhang W, Wei Y, Zhang H, Liu J, Zong Z, Liu Z, Zhu S, Hou W, Chen Y, Deng H. Structural Alternation in Heat Shock Proteins of Activated Macrophages. Cells. 2021; 10(12):3507. https://doi.org/10.3390/cells10123507
Chicago/Turabian StyleZhang, Wenhao, Ying Wei, Huaijin Zhang, Jing Liu, Zhaoyun Zong, Zongyuan Liu, Songbiao Zhu, Wenxuan Hou, Yuling Chen, and Haiteng Deng. 2021. "Structural Alternation in Heat Shock Proteins of Activated Macrophages" Cells 10, no. 12: 3507. https://doi.org/10.3390/cells10123507