
Tolerance Induced by Antigen-Loaded PLG Nanoparticles Affects the Phenotype and Trafficking of Transgenic CD4+ and CD8+ T Cells

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Strains
2.2. Peptides
2.3. Nanoparticle Synthesis and Characterization
2.4. Nanoparticle Treatment
2.5. In Vitro Culture for Secreted Cytokine and Proliferation
2.6. Magnetic Cell Separation
2.7. CFSE Dilution
2.8. Flow Cytometry
2.9. BDC-2.5 Adoptive Transfer
2.10. Serum Cytokines/Chemokines
2.11. Statistics
3. Results
3.1. Nanoparticle Synthesis and Characterization
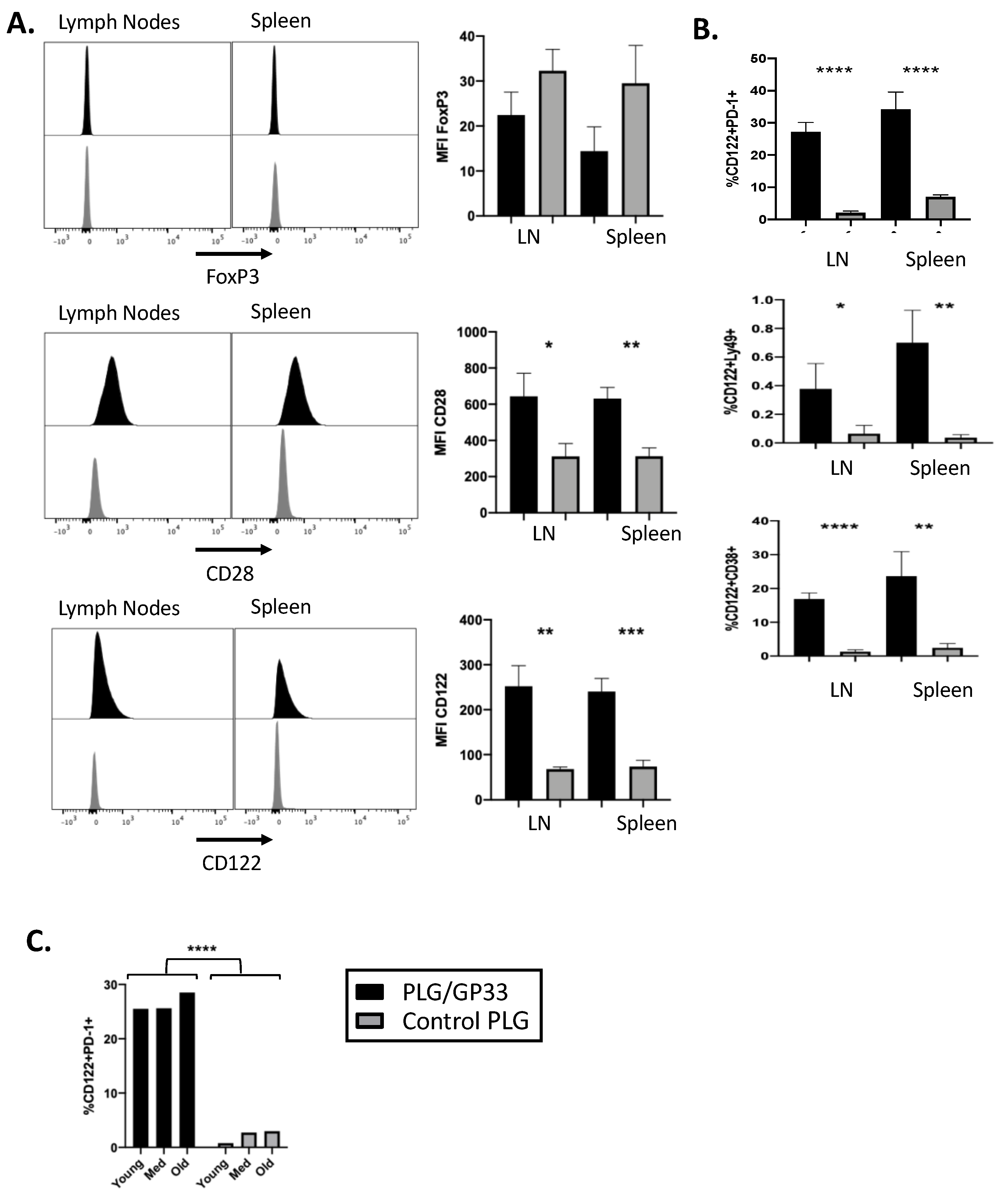
3.2. Antigen-Encapsulating PLG/GP33 Nanoparticles Expand CD8+ Tregs in P14 Transgenic Mice
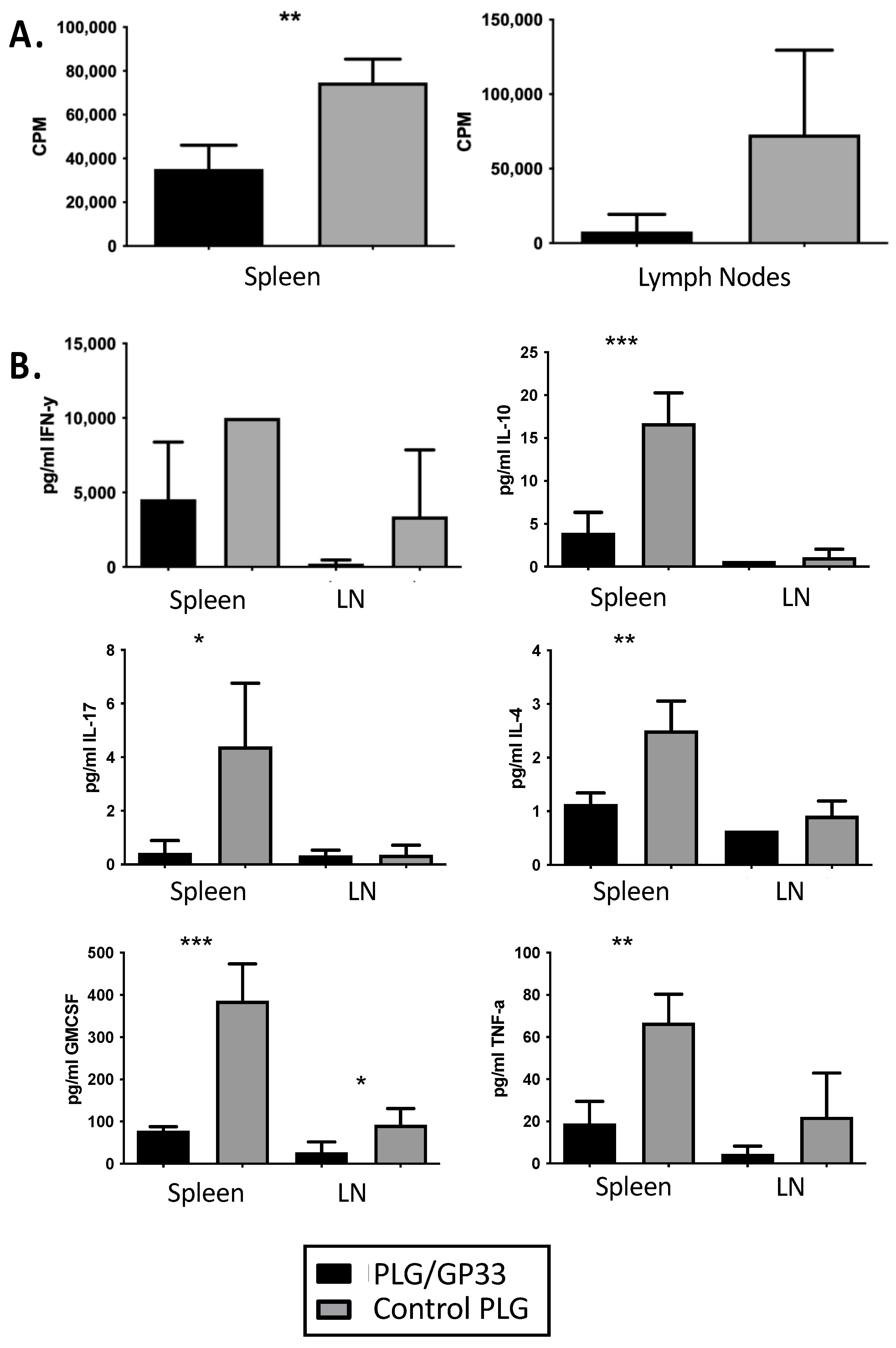
3.3. Recall Culture of PLG/GP33-Treated Splenocytes and Lymphocytes
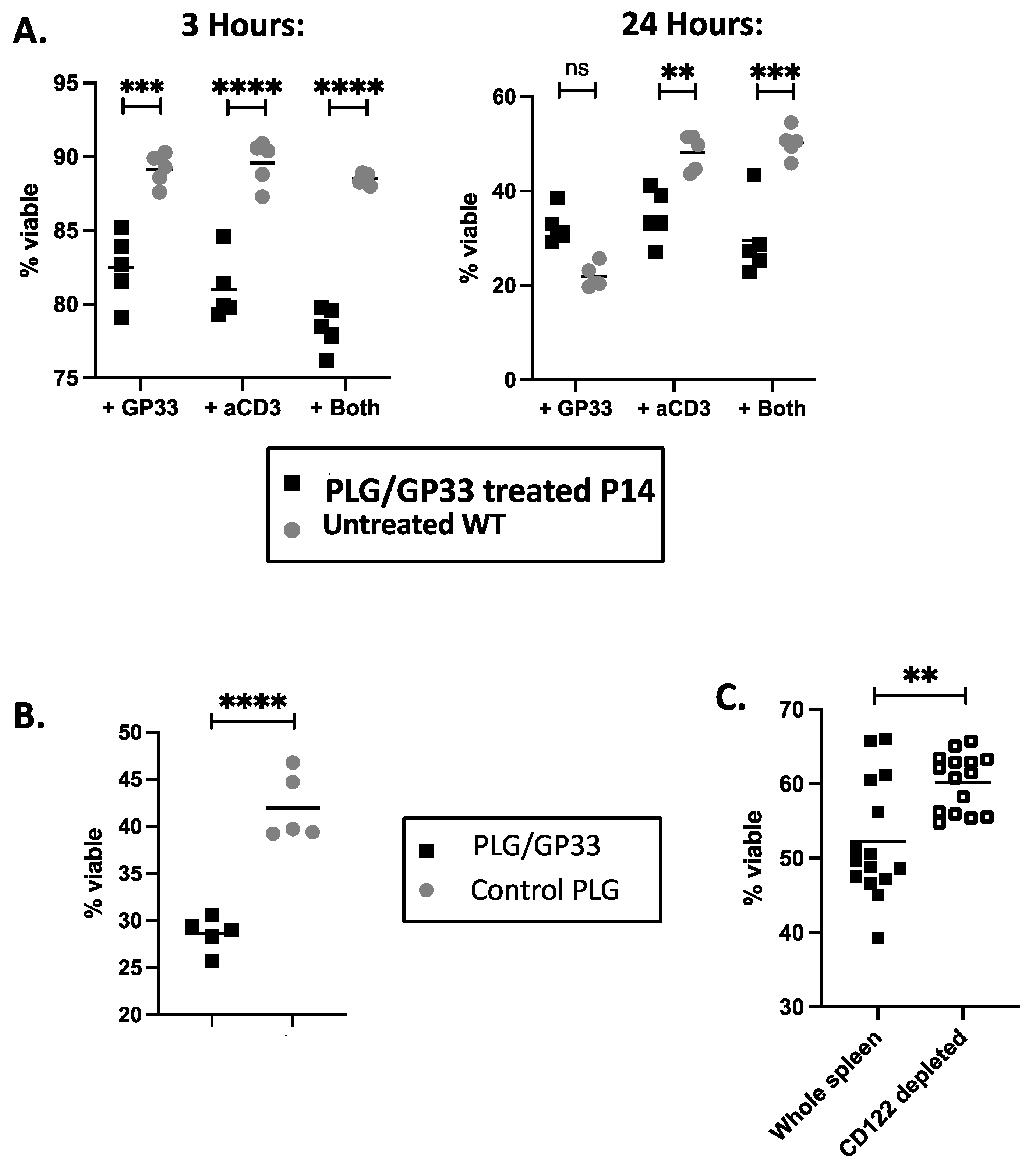
3.4. Expanded CD8+CD122+ Tregs Reduce Viability of CD4+ Cells
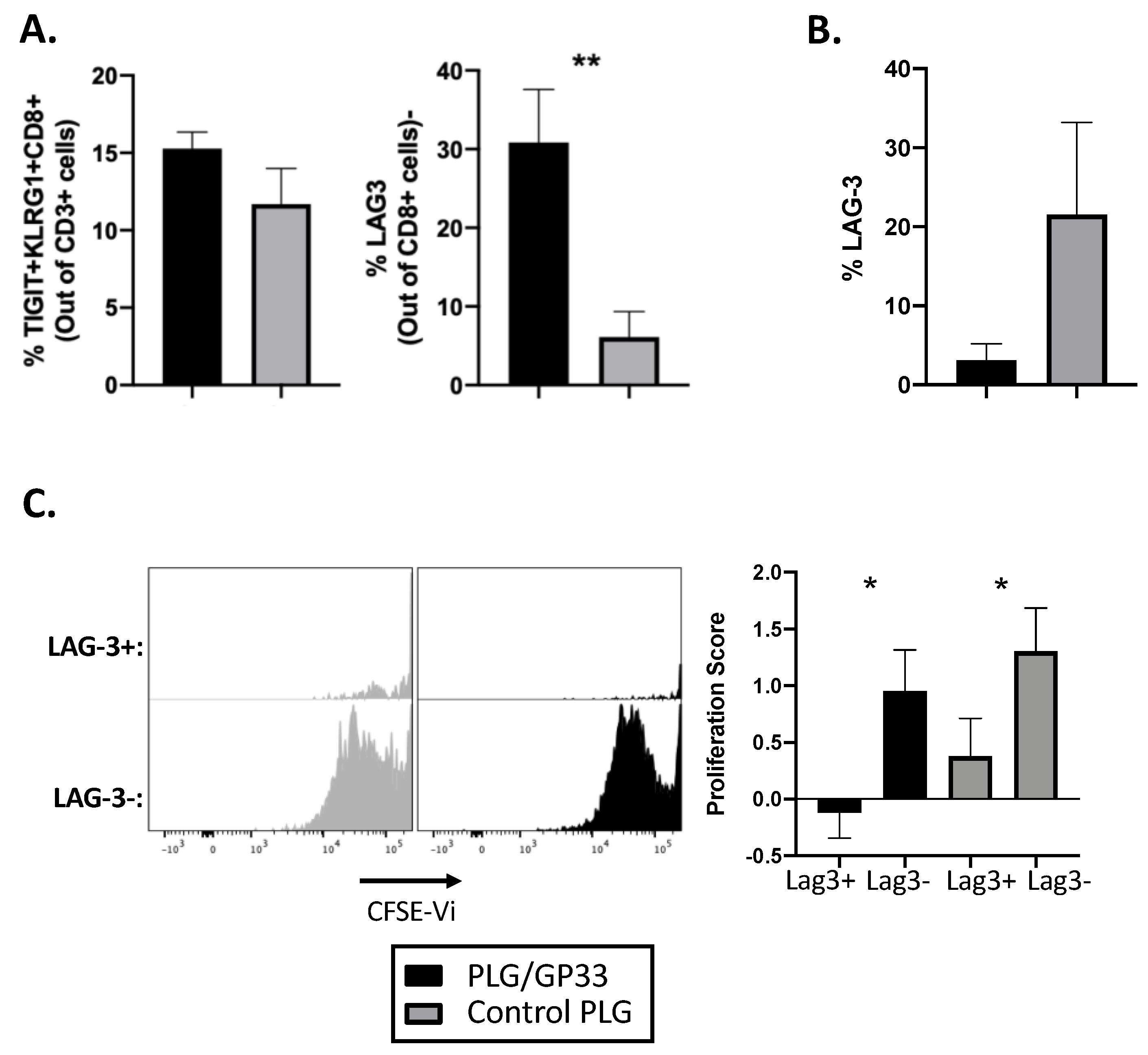
3.5. Antigen-Encapsulating PLG/GP33 Nanoparticles Induce Partial Exhaustion
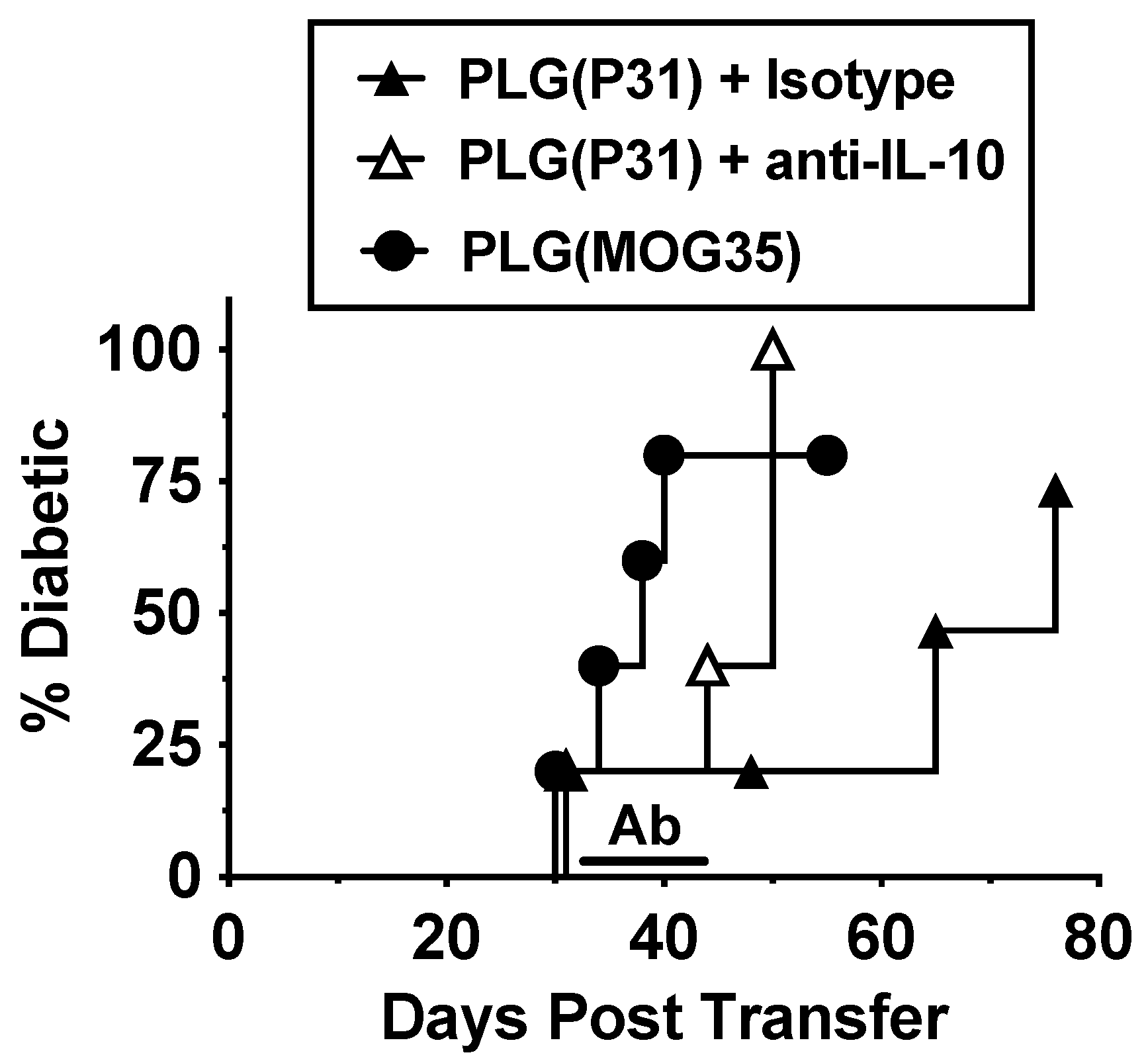
3.6. Tolerance in BDC-2.5 Transfer Model Is Partly Due to IL-10
3.7. Single Injection of Antigen-Loaded Nanoparticles Does Not Significantly Alter Chemokine Receptor Expression
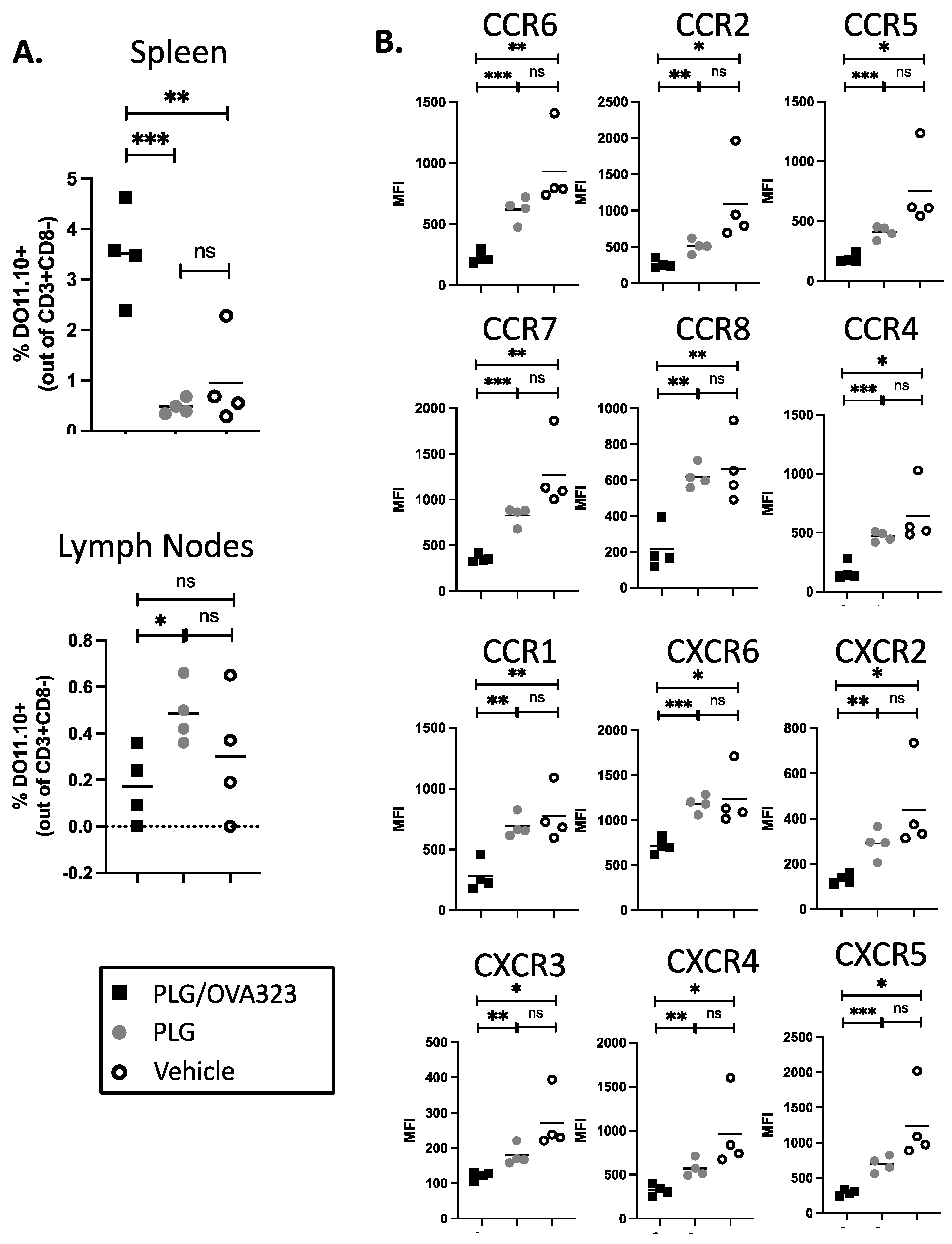
3.8. Multiple Injections of Antigen-Loaded Nanoparticles Sequester Transgenic CD4+ T Cells in Secondary Lymphoid Tissue and Alter Expression of Chemokine Receptors
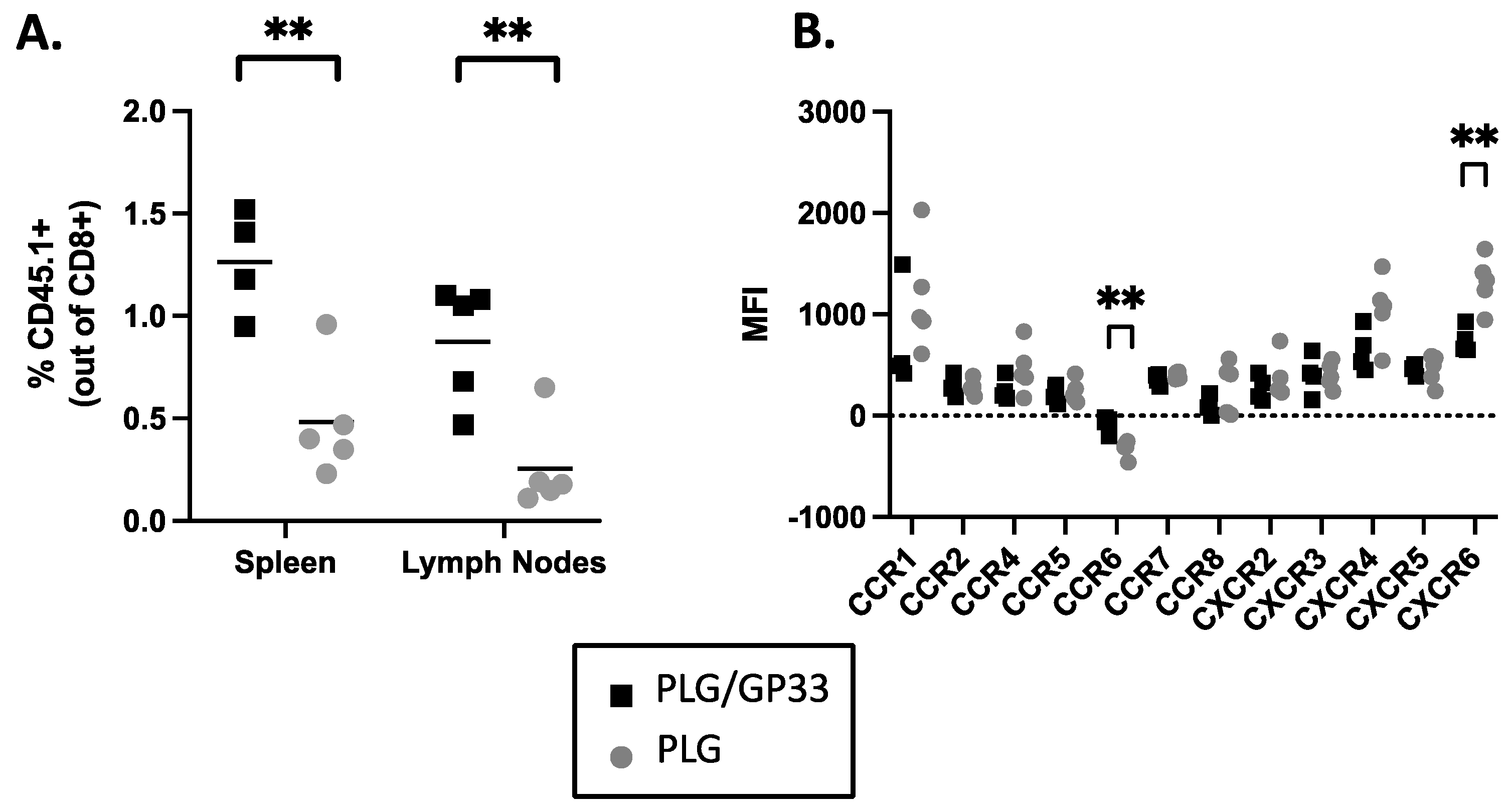
3.9. Multiple Injections of Antigen-Loaded Nanoparticles Sequester Transgenic CD8+ T Cells in Secondary Lymphoid Tissue and Alter Expression of Chemokine Receptors
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neef, T.; Miller, S.D. Tolerogenic Nanoparticles to Treat Islet Autoimmunity. Curr. Diabetes Rep. 2017, 17, 84. [Google Scholar] [CrossRef] [PubMed]
- Neef, T.; Miller, S.D. Novel delivery mechanisms for antigen-specific immunotherapy. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Francis, D.M.; Manspeaker, M.P.; Archer, P.A.; Sestito, L.F.; Heiler, A.J.; Schudel, A.; Thomas, S.N. Drug-eluting immune checkpoint blockade antibody-nanoparticle conjugate enhances locoregional and systemic combination cancer immunotherapy through T lymphocyte targeting. Biomaterials. 2021, 279, 121184. [Google Scholar] [CrossRef]
- Lv, S.; Song, K.; Yen, A.; Peeler, D.J.; Nguyen, D.C.; Olshefsky, A.; Sylvestre, M.; Srinivasan, S.; Stayton, P.S.; Pun, S.H. Well-Defined Mannosylated Polymer for Peptide Vaccine Delivery with Enhanced Antitumor Immunity. Adv. Healthc. Mater. 2021, 2101651. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.; Luo, X.; King, N.J.; et al. Microparticles bearing en-cephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalo-myelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Neef, T.; Xu, D.; Podojil, J.R.; Getts, D.R.; Shea, L.D.; Miller, S.D. Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. J. Autoimmun. 2018, 89, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Jamison, B.L.; Neef, T.; Goodspeed, A.; Bradley, B.; Baker, R.L.; Miller, S.D.; Haskins, K. Nanoparticles containing an insulin-ChgA hybrid peptide protect from transfer of autoimmune diabetes by shifting the balance between effector T cells and regulatory T cells. J. Immunol. 2019, 203, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Freitag, T.L.; Podojil, J.R.; Pearson, R.M.; Fokta, F.J.; Sahl, C.; Messing, M.; Andersson, L.C.; Leskinen, K.; Saavalainen, P.; Hoover, L.I.; et al. Gliadin nanoparticles induce immune tolerance to gliadin in mouse models of celiac disease. Gastroenterology 2020, 158, 1667–1681.e12. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, S.; Nomura, T.; Sakaguchi, S. Pillars Article: Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Penaloza-MacMaster, P.; Kamphorst, A.O.; Wieland, A.; Araki, K.; Iyer, S.S.; West, E.E.; O’Mara, L.; Yang, S.; Konieczny, B.T.; Sharpe, A.H.; et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J. Exp. Med. 2014, 211, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, D.; Nie, G.D.; Dai, Z. CD8+CD122+ T-Cells: A Newly Emerging Regulator with Central Memory Cell Phenotypes. Front. Immunol. 2015, 6, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieyra-Lobato, M.R.; Vela-Ojeda, J.; Montiel-Cervantes, L.; Lopez-Santiago, R.; Moreno-Lafont, M.C. Description of CD8(+) regulatory T lymphocytes and their specific intervention in graft-versus-host and infectious diseases, autoimmunity, and cancer. J. Immunol. Res. 2018, 2018, 3758713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafian, N.; Chitnis, T.; Salama, A.D.; Zhu, B.; Benou, C.; Yuan, X.; Clarkson, M.R.; Sayegh, M.H.; Khoury, S.J. Regulatory functions of CD8+CD28− T cells in an autoimmune disease model. J. Clin. Investig. 2003, 112, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Ishida, Y.; Rifa’I, M.; Shi, Z.; Isobe, K.-I.; Suzuki, H. Essential role of CD8+CD122+ regulatory T cells in the recovery from experimental autoimmune encephalomyelitis. J. Immunol. 2008, 180, 825–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endharti, A.T.; Rifa’I, M.; Shi, Z.; Fukuoka, Y.; Nakahara, Y.; Kawamoto, Y.; Takeda, K.; Isobe, K.; Suzuki, H. Cutting edge: CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J. Immunol. 2005, 175, 7093–7097. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Han, G.; Song, L.; Wang, J.; Chen, G.; Xu, R.; Yu, M.; Qian, J.; Shen, B.; Li, Y. CD8+ regulatory T cells are responsible for GAD-IgG gene-transferred tolerance induction in NOD mice. Immunology 2009, 126, 123–131. [Google Scholar] [CrossRef]
- Dai, Z.; Wan, N.; Zhang, S.; Moore, Y.; Wan, F. Cutting Edge: Programmed Death-1 Defines CD8+CD122+ T Cells as Regulatory versus Memory T Cells. J. Immunol. 2010, 185, 803–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elizondo, D.M.; Andargie, T.E.; Haddock, N.L.; da Silva, R.L.L.; de Moura, T.R.; Lipscomb, M.W. IL-10 producing CD8(+) CD122(+) PD-1(+) regulatory T cells are expanded by dendritic cells silenced for allograft inflammatory Factor-1. J. Leukoc. Biol. 2019, 105, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izawa, A.; Yamaura, K.; Albin, M.J.; Jurewicz, M.; Tanaka, K.; Clarkson, M.R.; Ueno, T.; Habicht, A.; Freeman, G.J.; Yagita, H.; et al. A Novel Alloantigen-Specific CD8+PD1+ Regulatory T Cell Induced by ICOS-B7h Blockade In Vivo. J. Immunol. 2007, 179, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Wang, X.; Radfar, S.; Sproule, T.J.; Roopenian, D.C.; Cantor, H. CD8+T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6-Yaa mice. Proc. Natl. Acad. Sci. USA 2011, 108, 2010–2015. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Bamford, R.N.; Waldmann, T.A. IL-15-dependent CD8+CD122+T cells ameliorate experimental autoimmune encephalomyelitis by modulating IL-17 production by CD4+T cells. Eur. J. Immunol. 2014, 44, 3330–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, B.; Witkowski, L.; Ellwart, J.; Seissler, J. CD8+ CD122+ PD-1- effector cells promote the development of diabetes in NOD mice. J. Leukoc. Biol. 2015, 97, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocks, B.T.; Wilson, C.S.; Marshall, A.F.; Hoopes, E.M.; Moore, D.J. Regulation of Diabetogenic Immunity by IL-15–Activated Regulatory CD8 T Cells in Type 1 Diabetes. J. Immunol. 2019, 203, 158–166. [Google Scholar] [CrossRef]
- Bahri, R.; Bollinger, A.; Bollinger, T.; Orinska, Z.; Bulfone-Paus, S. Ectonucleotidase CD38 demarcates regulatory, memory-like CD8+ T cells with IFN-gamma-mediated suppressor activities. PLoS ONE. 2012, 7, e45234. [Google Scholar] [CrossRef]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smarr, C.B.; Yap, W.T.; Neef, T.P.; Pearson, R.M.; Hunter, Z.N.; Ifergan, I.; Getts, D.R.; Bryce, P.J.; Shea, L.D.; Miller, S.D. Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization. Proc. Natl. Acad. Sci. USA 2016, 113, 5059–5064. [Google Scholar] [CrossRef] [Green Version]
- Hunter, Z.; McCarthy, D.P.; Yap, W.T.; Harp, C.T.; Getts, D.R.; Shea, L.D.; Miller, S.D. A Biodegradable Nanoparticle Platform for the Induction of Antigen-Specific Immune Tolerance for Treatment of Autoimmune Disease. ACS Nano 2014, 8, 2148–2160. [Google Scholar] [CrossRef]
- Yap, W.T.; Song, W.K.; Chauhan, N.; Scalise, P.N.; Agarwal, R.; Miller, S.D.; Shea, L.D. Quantification of particle-conjugated or particle-encapsulated peptides on interfering reagent backgrounds. Biotechniques 2014, 57, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Ahlen, M.T.; Husebekk, A.; Killie, M.K.; Skogen, B.; Stuge, T.B. T-cell responses associated with neonatal alloimmune thrombocytopenia: Isolation of HPA-1a–specific, HLA-DRB3*0101–restricted CD4+ T cells. Blood 2009, 113, 3838–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.P.; Yap, J.W.; Harp, C.T.; Song, W.K.; Chen, J.; Pearson, R.M.; Miller, S.D.; Shea, L.D. An antigen-encapsulating na-noparticle platform for T(H)1/17 immune tolerance therapy. Nanomedicine 2017, 13, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.M.; Casey, L.M.; Hughes, K.R.; Wang, L.Z.; North, M.G.; Getts, D.R.; Miller, S.D.; Shea, L.D. Controlled delivery of single or multiple antigens in tolerogenic nanoparticles using peptide-polymer bioconjugates. Mol. Ther. 2017, 25, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifa’I, M.; Kawamoto, Y.; Nakashima, I.; Suzuki, H. Essential Roles of CD8+CD122+ Regulatory T Cells in the Maintenance of T Cell Homeostasis. J. Exp. Med. 2004, 200, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Ablamunits, V.; Bisikirska, B.; Herold, K.C. Acquisition of regulatory function by human CD8+ T cells treated with anti-CD3 antibody requires TNF. Eur. J. Immunol. 2010, 40, 2891–2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlavaty, K.A.; McCarthy, D.P.; Saito, E.; Yap, W.T.; Miller, S.D.; Shea, L.D. Tolerance induction using nanoparticles bearing HY peptides in bone marrow transplantation. Biomaterials 2016, 76, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.P.; Murray, J.A.; Leffler, D.A.; Getts, D.R.; Bledsoe, A.C.; Smithson, G.; First, M.R.; Morris, A.; Boyne, M.; Elhofy, A.; et al. TAK-101 nanoparticles induce gluten-specific tolerance in celiac disease: A randomized, double-blind, placebo-controlled study. Gastroenterology 2021, 161, 66–80. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Sullivan, N.L.; Ebel, M.; Teague, R.M.; DiPaolo, R.J. Antigen-specific TGF-beta-induced regula-tory T cells secrete chemokines, regulate T cell trafficking, and suppress ongoing autoimmunity. J. Immunol. 2011, 187, 1745–1753. [Google Scholar] [CrossRef]
- Tanwar, S.; Oguz, C.; Metidji, A.; Dahlstrom, E.; Barbian, K.; Kanakabandi, K.; Sykora, L.; Shevach, E.M. Type I IFN signaling in T regulatory cells modulates chemokine production and myeloid derived suppressor cells trafficking during EAE. J. Autoimmun. 2020, 115, 102525. [Google Scholar] [CrossRef]
- Kim, H.J.; Verbinnen, B.; Tang, X.; Lu, L.; Cantor, H. Inhibition of follicular T-helper cells by CD8(+) regula-tory T cells is essential for self tolerance. Nature 2010, 467, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Boyden, A.W.; Brate, A.A.; Stephens, L.M.; Karandikar, N.J. Immune Autoregulatory CD8 T Cells Require IFN-γ Responsiveness to Optimally Suppress Central Nervous System Autoimmunity. J. Immunol. 2020, 205, 359–368. [Google Scholar] [CrossRef]
- Maulloo, C.D.; Cao, S.; Watkins, E.A.; Raczy, M.M.; Solanki, A.S.; Nguyen, M.; Reda, J.W.; Shim, H.N.; Wilson, D.S.; Swartz, M.A.; et al. lymph node-targeted synthetically glycosylated antigen leads to antigen-specific immunological tolerance. Front. Immunol. 2021, 12, 714842. [Google Scholar] [CrossRef] [PubMed]
- Moser, B. T-Cell Memory: The Importance of Chemokine-Mediated Cell Attraction. Curr. Biol. 2006, 16, R504–R507. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.V.; Nombela-Arrieta, C. Chemokine control of lymphocyte trafficking: A general overview. Immunology 2005, 116, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lord, G.M.; Rao, R.M.; Choe, H.; Sullivan, B.M.; Lichtman, A.H.; Luscinskas, F.W.; Glimcher, L.H. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood 2005, 106, 3432–3439. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Guloglu, F.B.; VanMorlan, A.M.; Rowland, L.M.; Jain, R.; Haymaker, C.L.; Cascio, J.A.; Dhakal, M.; Hoeman, C.M.; Tartar, D.M.; et al. Mechanisms underlying antigen-specific tolerance of stable and convertible Th17 cells during suppression of autoimmune diabetes. Diabetes 2012, 61, 2054–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, A.J.; Guillen, C.; Symon, F.A.; Huynh, T.T.; Berry, M.A.; Entwisle, J.J.; Briskin, M.; Pavord, I.D.; Wardlaw, A. Expression of CXCR6 and its ligand CXCL16 in the lung in health and disease. Clin. Exp. Allergy 2005, 35, 1572–1580. [Google Scholar] [CrossRef]
- Lee, L.N.; Ronan, E.O.; de Lara, C.; Franken, K.L.; Ottenhoff, T.H.; Tchilian, E.Z.; Beverley, P.C. CXCR6 is a marker for protective antigen-specific cells in the lungs after intranasal immunization against Mycobacterium tuberculosis. Infect. Immun. 2011, 79, 3328–3337. [Google Scholar] [CrossRef] [Green Version]
- Heesch, K.; Raczkowski, F.; Schumacher, V.; Hünemörder, S.; Panzer, U.; Mittrücker, H.-W. The Function of the Chemokine Receptor CXCR6 in the T Cell Response of Mice against Listeria monocytogenes. PLoS ONE 2014, 9, e97701. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticle | Antigen | Transgenic Mouse Strain | Z-Average Diameter (nm) a | Polydispersity Index a | Zeta Potential (mV) a | Antigen Load (μg/mg) b |
|---|---|---|---|---|---|---|
| PLG | None | N/A | 415.1 474.7 559.4 | 0.258 0.309 0.378 | −93.6 +/− 11.7 −84.8 +/− 11.8 −59.7 +/− 7.83 | N/A |
| PLG/GP33 | LCMV GP 33–41 | P14 | 496.4 | 0.276 | −74.7 +/− 7.27 | 3.4 |
| PLG/Ova323 | Ovalbumin 323–339 | DO11.10 | 522.5 | 0.376 | −89.0 +/− 9.69 | 64.5 |
| PLG(P31) | BDC-2.5 mimetope 1040-31 | BDC-2.5 | 464.2 | 0.351 | −84.1 +/− 10.1 | 2.1 |
| PLG(MO35) | MOG 35-55 | N/A | 483.7 | 0.288 | −84.7 +/− 14.3 | 5.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neef, T.; Ifergan, I.; Beddow, S.; Penaloza-MacMaster, P.; Haskins, K.; Shea, L.D.; Podojil, J.R.; Miller, S.D. Tolerance Induced by Antigen-Loaded PLG Nanoparticles Affects the Phenotype and Trafficking of Transgenic CD4+ and CD8+ T Cells. Cells 2021, 10, 3445. https://doi.org/10.3390/cells10123445
Neef T, Ifergan I, Beddow S, Penaloza-MacMaster P, Haskins K, Shea LD, Podojil JR, Miller SD. Tolerance Induced by Antigen-Loaded PLG Nanoparticles Affects the Phenotype and Trafficking of Transgenic CD4+ and CD8+ T Cells. Cells. 2021; 10(12):3445. https://doi.org/10.3390/cells10123445
Chicago/Turabian StyleNeef, Tobias, Igal Ifergan, Sara Beddow, Pablo Penaloza-MacMaster, Kathryn Haskins, Lonnie D. Shea, Joseph R. Podojil, and Stephen D. Miller. 2021. "Tolerance Induced by Antigen-Loaded PLG Nanoparticles Affects the Phenotype and Trafficking of Transgenic CD4+ and CD8+ T Cells" Cells 10, no. 12: 3445. https://doi.org/10.3390/cells10123445