
Murine Trophoblast Stem Cells and Their Differentiated Cells Attenuate Zika Virus In Vitro by Reducing Glycosylation of the Viral Envelope Protein

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Ethics Statement and Biosafety
2.2. Viruses, Cells, and Animals
2.3. Cells
2.4. Mice
2.5. Attachment Assay
2.6. Plaque Assay
2.7. Quantitative PCR (qPCR)
2.8. Heparin Sepharose Bead Binding Assay
2.9. Concentrating of ZIKV and Protein Glycosylation Assay
2.10. Immunoblotting Assay for HEXA
2.11. qPCR Array
2.12. Statistical Analyses
3. Results
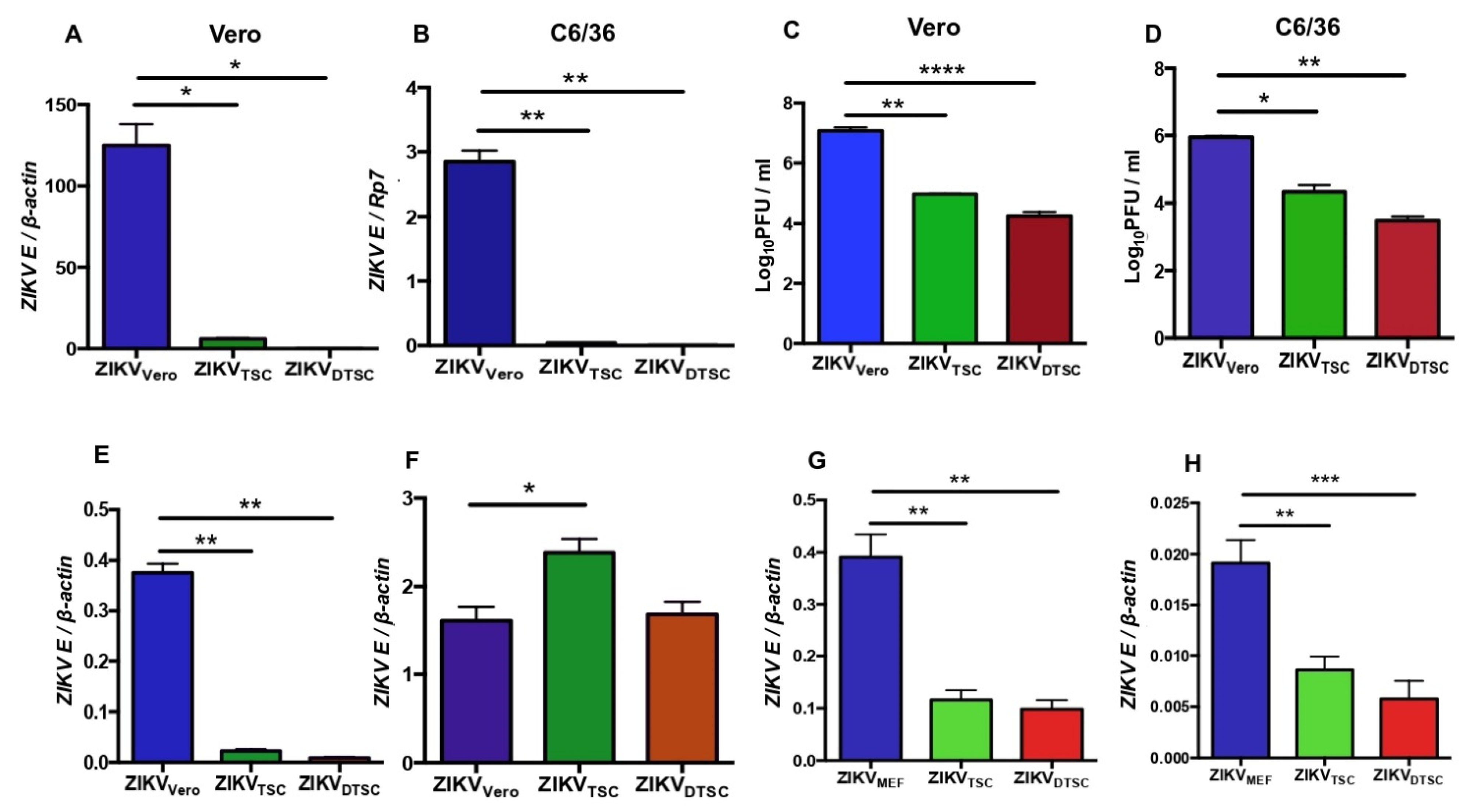
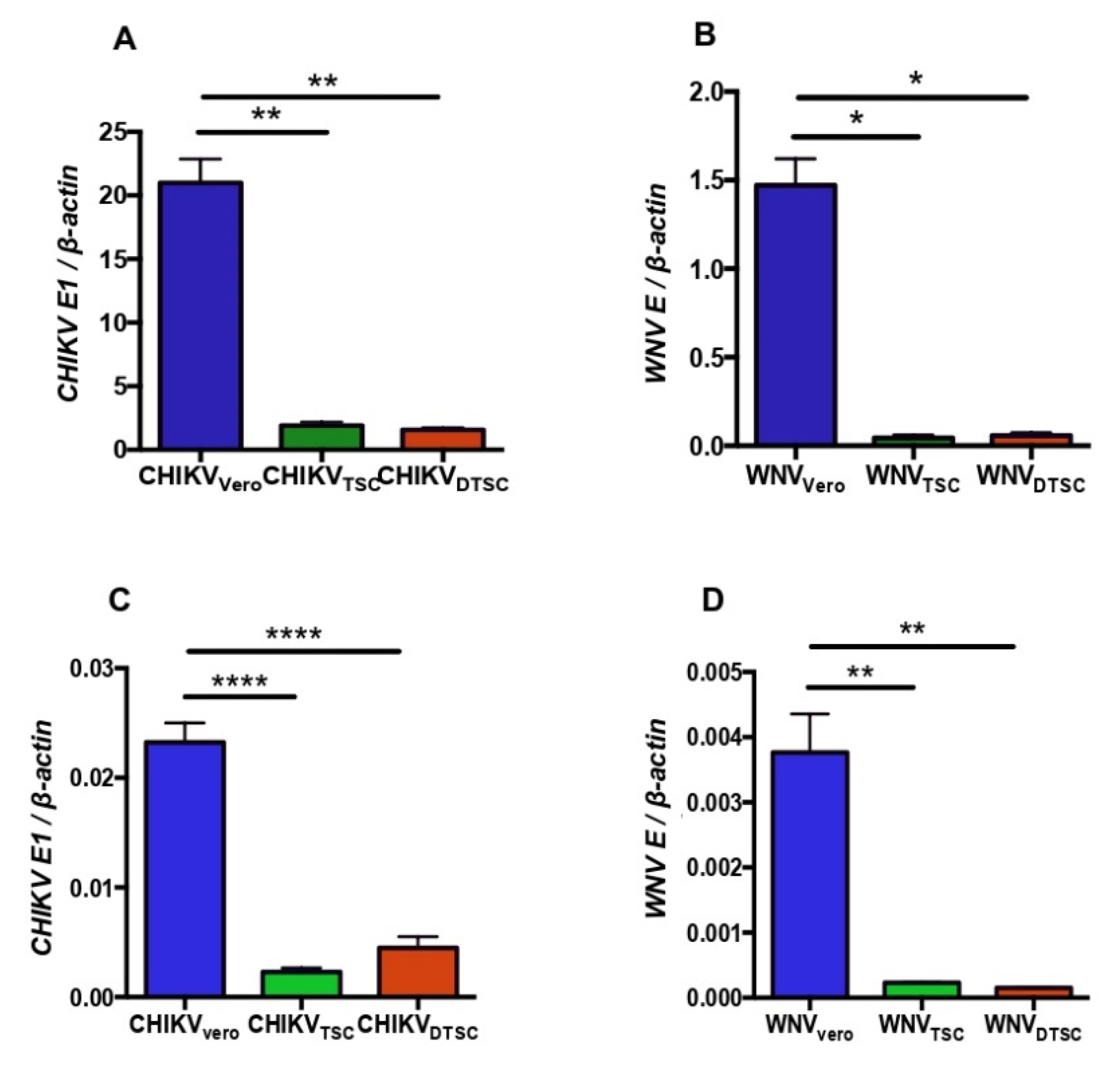
3.1. ZIKV Propagated in TSCs and DTSCs Exhibits Reduced Infectivity In Vitro
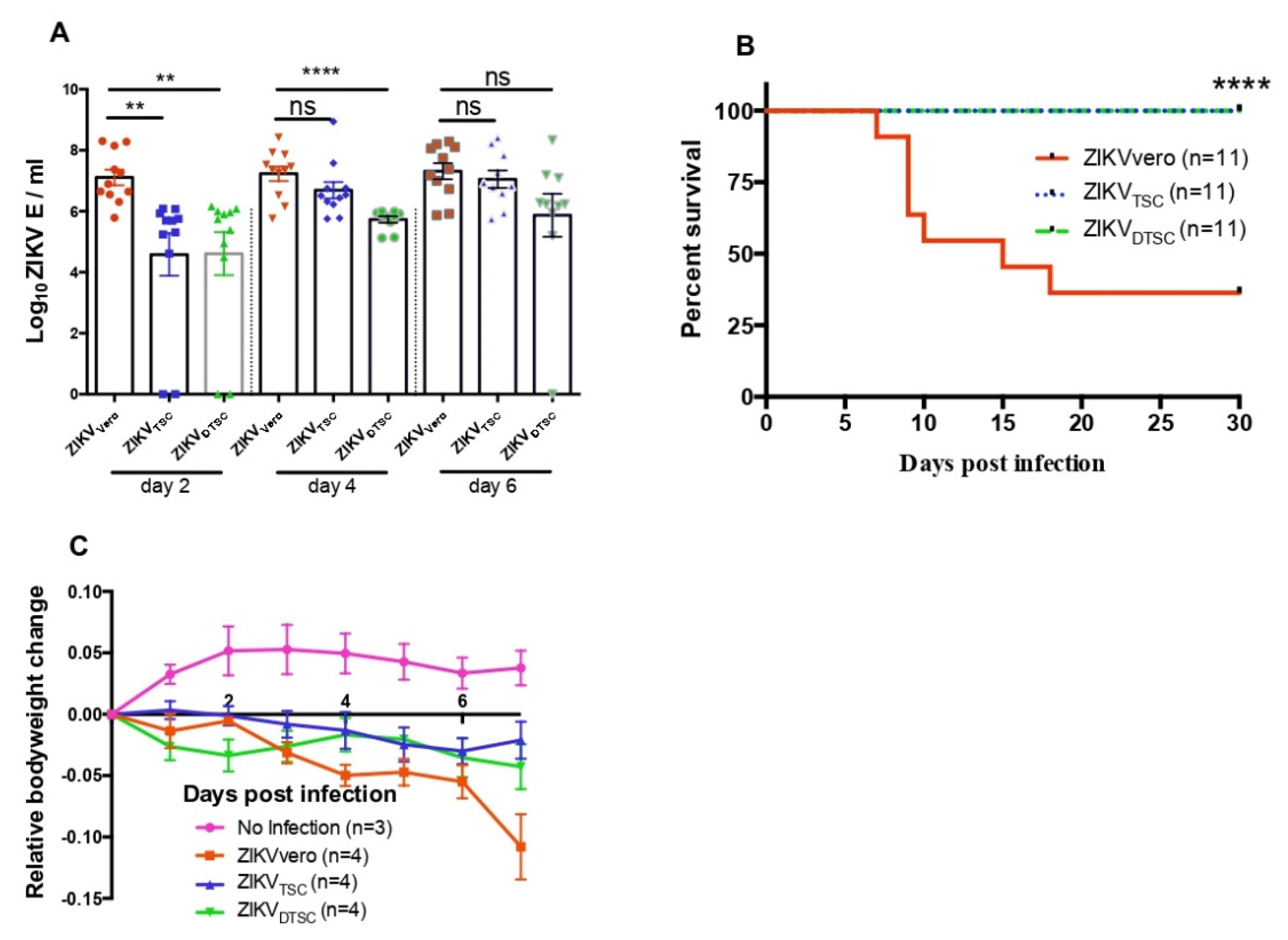
3.2. ZIKVTSC and ZIKVDTSC Exhibit Attenuated Infectivity in Ifnar1−/− Mice
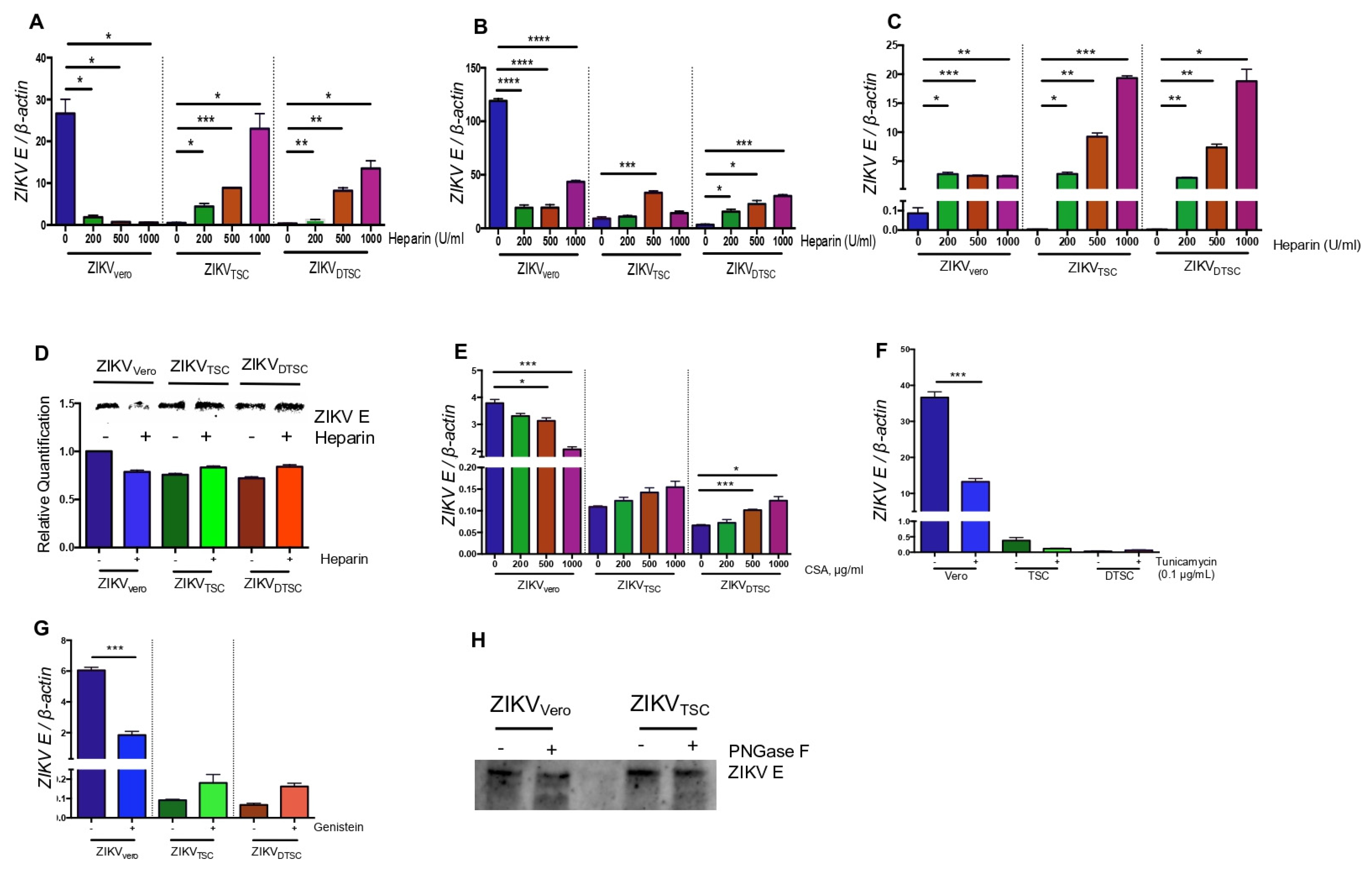
3.3. ZIKVTSC and ZIKVDTSC Have Reduced Glycosylation on E Proteins
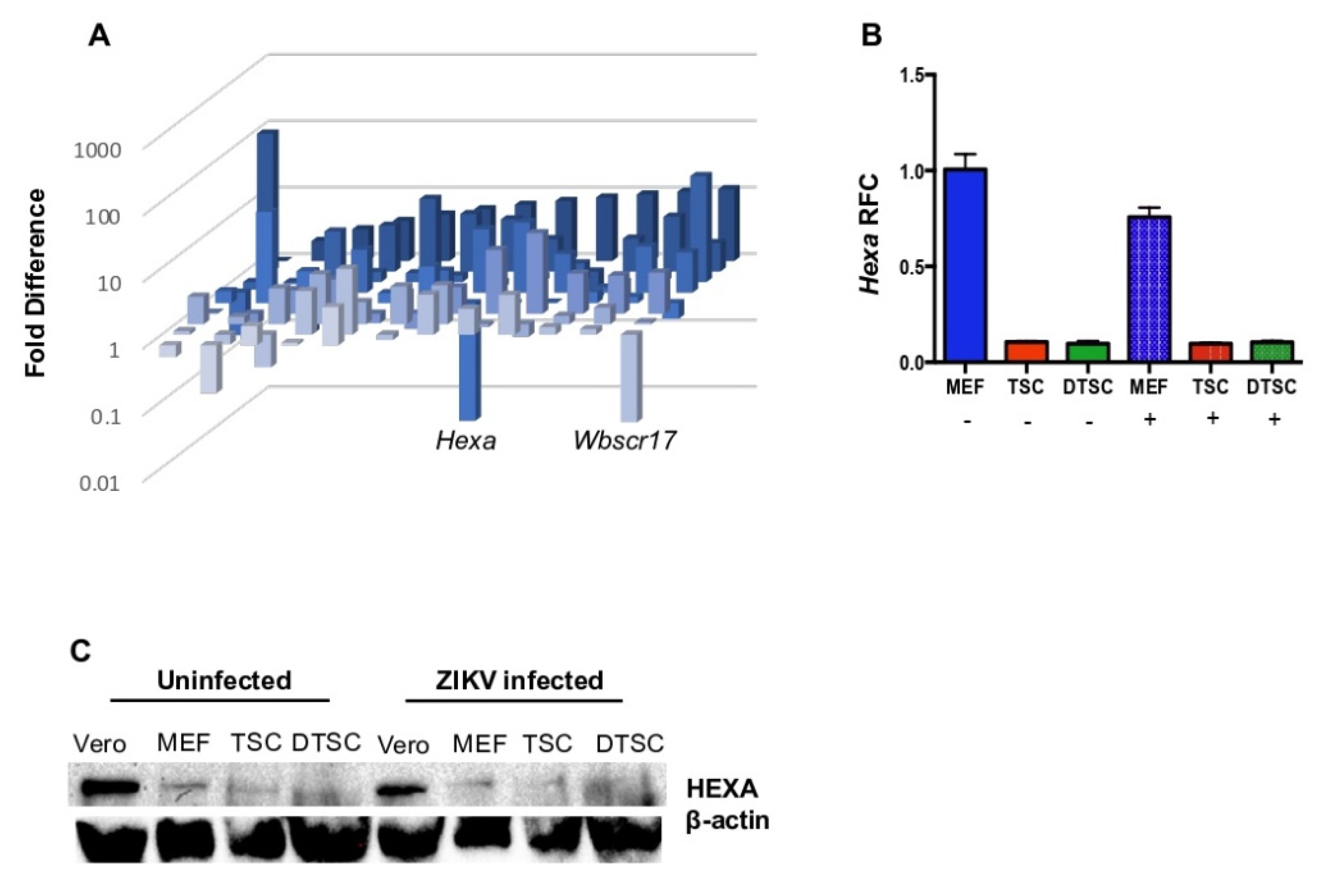
3.4. The Expression of Hexa Was Decreased in TSCs and DTSCs
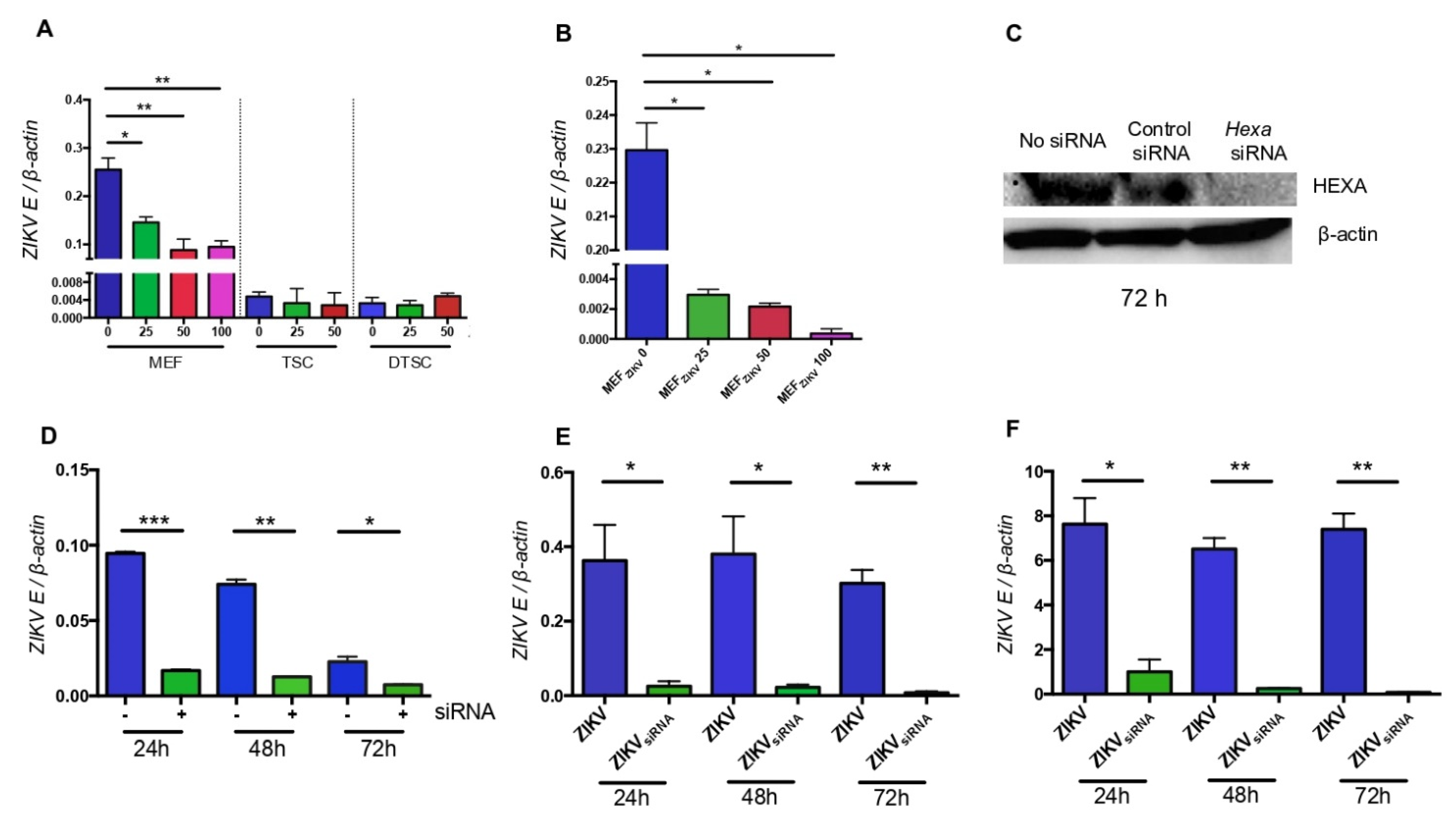
3.5. HEXA Contributes to the Glycosylation of ZIKV in TSCs and DTSCs
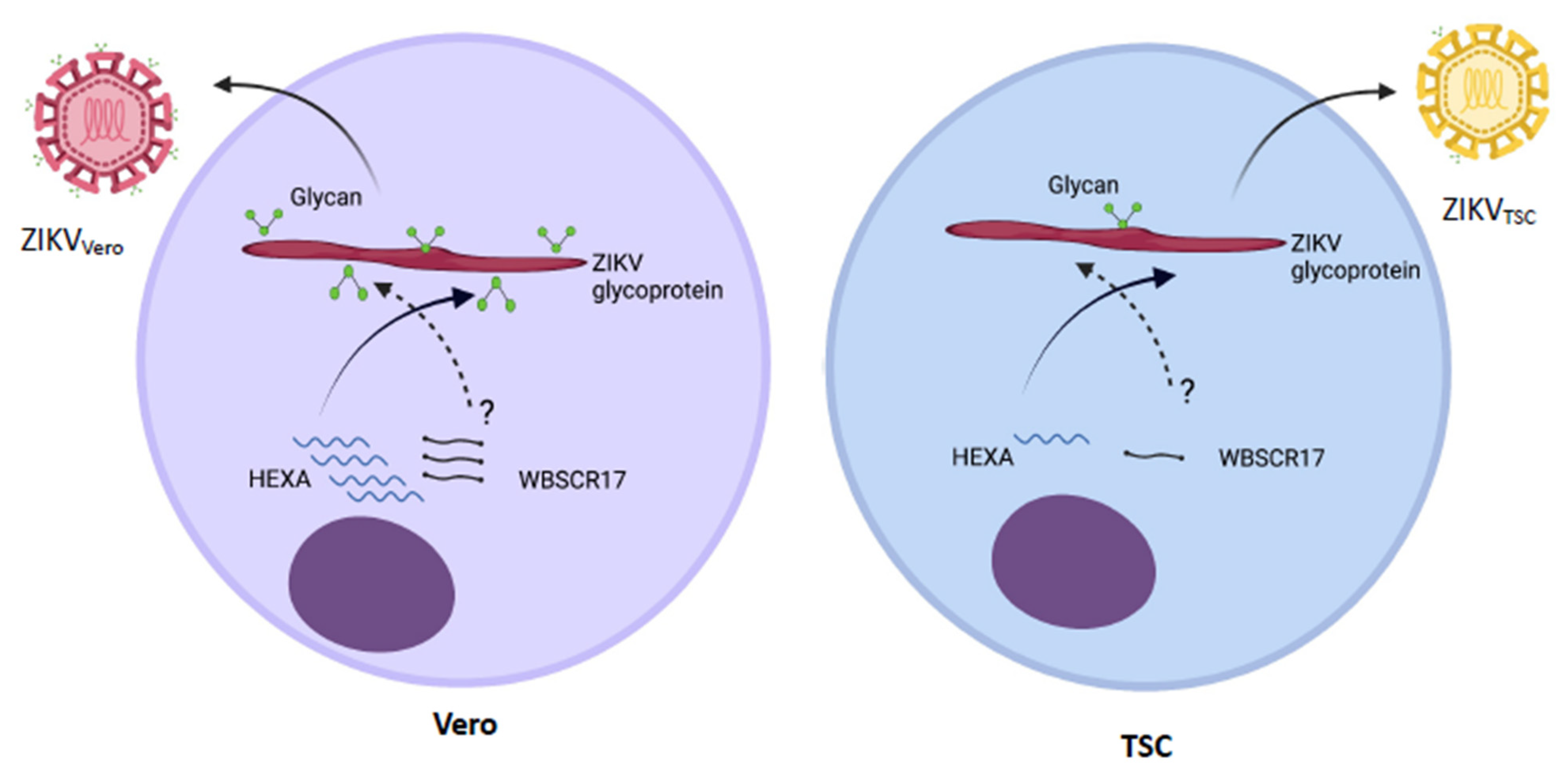
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lazear, H.; Govero, J.; Smith, A.M.; Platt, D.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, F.; Thompson, E.A.; Vig, P.J.S.; Leis, A.A. Current Understanding of West Nile Virus Clinical Manifestations, Immune Responses, Neuroinvasion, and Immunotherapeutic Implications. Pathogens 2019, 8, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [Green Version]
- Brasil, P.; Pereira, J.P.; Moreira, M.E., Jr.; Ribeiro Nogueira, R.M.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334. [Google Scholar] [CrossRef]
- van der Linden, V.; Pessoa, A.; Dobyns, W.; Barkovich, A.J.; Junior, H.V.; Filho, E.L.; Ribeiro, E.M.; Leal, M.C.; Coimbra, P.P.; Aragao, M.F.; et al. Description of 13 Infants Born During October 2015-January 2016 with Congenital Zika Virus Infection Without Microcephaly at Birth—Brazil. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1343–1348. [Google Scholar] [CrossRef]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guil-Lain-Barre Syndrome Outbreak Associated with Zika Virus Infection in French Polynesia: A Case-Control Study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- Bai, F.; Thompson, E.A. West Nile Virus (Flaviviridae). Encycl. Virol. 2021, 2, 884–890. [Google Scholar] [CrossRef]
- Liu, J.; Thorp, S.C. Cell Surface Heparan Sulfate and its Roles in Assisting Viral Infections. Med. Res. Rev. 2001, 22, 1–25. [Google Scholar] [CrossRef]
- Olofsson, S.; Bergström, T. Glycoconjugate Glycans as Viral Receptors. Ann. Med. 2005, 37, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue Virus Infectivity Depends on Envelope Protein Binding to Target Cell Heparan Sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lobigs, M. Substitutions at the Putative Receptor-Binding Site of an Encephalitic Fla-Vivirus Alter Virulence and Host Cell Tropism and Reveal a Role for Glycosaminoglycans in Entry. J. Virol. 2000, 74, 8867–8875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Hall, R.A.; Lobigs, M. Common E Protein Determinants for Attenuation of Glycosamino-Glycan-Binding Variants F Japanese Encephalitis and West Nile Viruses. J. Virol. 2004, 78, 8271–8280. [Google Scholar] [CrossRef] [Green Version]
- Acharya, D.; Paul, A.; Anderson, J.F.; Huang, F.; Bai, F. Loss of Glycosaminoglycan Receptor Binding after Mosquito Cell Passage Reduces Chikungunya Virus Infectivity. PLoS Negl. Trop. Dis. 2015, 9, e0004139. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Zhao, J.; Liu, X.; Fraser, K.; Lin, L.; Zhang, X.; Zhang, F.; Dordick, J.S.; Linhardt, R.J. Interaction of Zika Virus Envelope Protein with Glycosaminoglycans. Biochemistry 2017, 56, 1151–1162. [Google Scholar] [CrossRef]
- Carbaugh, D.L.; Baric, R.S.; Lazear, H.M. Envelope Protein Glycosylation Mediates Zika Virus Pathogenesis. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of Glycosylation in Enveloped Virus Pathobiology. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef]
- Cross, J.; Werb, Z.; Fisher, S. Implantation and the Placenta: Key Pieces of the Development Puzzle. Science 1994, 266, 1508–1518. [Google Scholar] [CrossRef]
- Roberts, R.M.; Fisher, S.J. Trophoblast Stem Cells. Biol. Reprod. 2011, 84, 412–421. [Google Scholar] [CrossRef]
- Mor, G.; Aldo, P.; Alvero, A.B. The Unique Immunological and Microbial Aspects of Pregnancy. Nat. Rev. Immunol. 2017, 17, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Ades, E.A.; Soriano-Arandes, A.; Alarcon, A.; Bonfante, F.; Thorne, C.; Peckham, C.S.; Giaquinto, C. Vertical Transmission of Zika Virus and its Outcomes: A Bayesian Synthesis of Prospective Studies. Lancet Infect. Dis. 2020, 21, 537–545. [Google Scholar] [CrossRef]
- Routhu, N.K.; Lehoux, S.D.; Rouse, E.A.; Bidokhti, M.R.M.; Giron, L.B.; Anzurez, A.; Reid, S.P.; Abdel-Mohsen, M.; Cummings, R.D.; Byrareddy, S.N. Glycosylation of Zika Virus is Important in Host-Virus Interaction and Pathogenic Potential. Int. J. Mol. Sci. 2019, 20, 5206. [Google Scholar] [CrossRef] [Green Version]
- Takatsuki, A.; Arima, K.; Tamura GTunicamycin, A. New Antibiotic. I. Isolation and Characterization of Tunicamycin. J. Antibiot. 1971, 24, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, E.; Jakóbkiewicz-Banecka, J.; Baranska, S.; Tylki-Szymanska, A.; Czartoryska, B.; Wegrzyn, A.; Wegrzyn, G. Genistein-Mediated Inhibition of Glycosaminoglycan Synthesis as a Basis for Gene Expression-Targeted Isoflavone Therapy for Mucopolysaccharidoses. Eur. J. Hum. Genet. 2006, 14, 846–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeze, H.H.; Kranz, C. Endoglycosidase and Glycoamidase Release of N-Linked Glycans. Curr. Protoc. Mol. Biol. C. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 A Resolution Cryo-EM Structure of Zika Virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyanathan, K.; Durning, S.; Wells, L. Functional O-GlcNAc Modifications: Implications in Molecular Regulation and Pathophysiology. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 140–163. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-L. The Underdeveloped Innate Immunity in Embryonic Stem Cells: The Molecular Basis and Biological Perspectives from Early Embryogenesis. Am. J. Reprod. Immunol. 2019, 81, e13089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, G.C. Viruses and Interferons. Annu. Rev. Microbiol. 2001, 55, 255–281. [Google Scholar] [CrossRef]
- Zorrilla, C.D.; Garcia Garcia, I.; Garcia Fragoso, L.; De La Vega, A. Zika Virus Infection in Pregnancy: Maternal, Fetal, and Neonatal Considerations. J. Infect. Dis. 2017, 216, S891–S896. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Bos, S.; Li, G.; Wang, S.; Gadea, G.; Desprès, P.; Zhao, R.Y. Probing Molecular Insights into Zika Virus–Host Interactions. Viruses 2018, 10, 233. [Google Scholar] [CrossRef] [Green Version]
- Heil, M.L.; Albee, A.; Strauss, J.H.; Kuhn, R.J. An Amino Acid Substitution in the Coding Region of the E2 Glycoprotein Adapts Ross River Virus to Utilize Heparan Sulfate as an Attachment Moiety. J. Virol. 2001, 75, 6303–6309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimstra, W.B.; Ryman, K.D.; Johnston, R.E. Adaptation of Sindbis Virus to BHK Cells Selects for Use of Heparan Sulfate as an Attachment Receptor. J. Virol. 1998, 72, 7357–7366. [Google Scholar] [CrossRef] [Green Version]
- Smit, J.M.; Waarts, B.L.; Kimata, K.; Klimstra, W.B.; Bittman, R.; Wilschut, J. Adaptation of Alpha-Viruses to Heparan Sulfate: Interaction of Sindbis and Semliki Forest Viruses with Liposomes Containing Lipid-Conjugated Heparin. J. Virol. 2002, 76, 10128–10137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagdonaite, I.; Wandall, H.H. Global Aspects of Viral Glycosylation. Glycobiology 2018, 28, 443–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, P.; Rosner, M.R.; Robbins, P.W. Host-Dependent Variation of Asparagine-Linked Oligosac-Charides at Individual Glycosylation Sites of Sindbis Virus Glycoproteins. J. Biol. Chem. 1983, 258, 2548–2554. [Google Scholar] [CrossRef]
- Strauss, J.H.; Burge, B.W.; Darnell, J.E. Carbohydrate Content of the Membrane Protein of Sindbis Virus. J. Mol. Biol. 1970, 47, 437–448. [Google Scholar] [CrossRef]
- Mitsuhashi, J.; Nakasone, S.; Horie, Y. Sterol-Free Eukaryotic Cells from Continuous Cell Lines of Insects. Cell Biol. Int. Rep. 1983, 7, 1057–1062. [Google Scholar] [CrossRef]
- Klimstra, W.B.; Nangle, E.M.; Smith, M.S.; Yurochko, A.D.; Ryman, K.D. DC-SIGN and L-SIGN can Act as Attachment Receptors for Alphaviruses and Distinguish Between Mosquito Cell- and Mammali-An Cell-Derived Viruses. J. Virol. 2003, 77, 12022–12032. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, P.; Robbins, P.W. Regulation of Asparagine-Linked Oligosaccharide Processing. Oligosac-Charide Processing in Aedes Albopictus Mosquito Cells. J. Biol. Chem. 1984, 259, 2375–2382. [Google Scholar] [CrossRef]
- Leavitt, R.; Schlesinger, S.; Kornfeld, S. Tunicamycin Inhibits Glycosylation and Multiplication of Sindbis and Vesicular Stomatitis Viruses. J. Virol. 1977, 21, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Gwon, Y.-D.; Zusinaite, E.; Merits, A.; Överby, A.K.; Evander, M. N-Glycosylation in the Pre-Membrane Protein is Essential for the Zika Virus Life Cycle. Viruses 2020, 12, 925. [Google Scholar] [CrossRef]
- Torres, C.R.; Hart, G.W. Topography and Polypeptide Distribution of Terminal N-Acetylglucosamine Residues on the Surfaces of Intact Lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Blomberg, M.A.; Hart, G.W. Glycosylation of Nuclear and Cytoplasmic Proteins. Purification and Characterization of a Uridine Diphospho-N-Acetylglucosamine: Polypeptide Beta-N-acetylglucosaminyltransferase. J. Biol. Chem. 1992, 267, 9005–9013. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Holt, G.D.; Hart, G.W. Enzymatic Addition of O-GlcNAc to Nuclear and Cytoplasmic Proteins. Identification of a Uridine Diphospho-N-Acetylglucosamine:Peptide Beta-N-Acetylglucosaminyltransferase. J. Biol. Chem. 1990, 265, 2563–2568. [Google Scholar] [CrossRef]
- Dong, D.L.; Hart, G.W. Purification and Characterization of an O-GlcNAc Selective N-acetyl-beta-D-glucosaminidase from Rat Spleen Cytosol. J. Biol. Chem. 1994, 269, 19321–19330. [Google Scholar] [CrossRef]
- Madey, B.; Niemiro, A.; Danowska, A.; Forycki, Z.; Pawelec, D. Farmer’s lung. Wiad Lek 1975, 28, 203–207. [Google Scholar]
- Kearse, K.P.; Hart, G.W. Topology of O-linked N-acetylglucosamine in Murine Lymphocytes. Arch. Biochem. Biophys. 1991, 290, 543–548. [Google Scholar] [CrossRef]
- Zachara, N.E.; O’Donnell, N.; Cheung, W.D.; Mercer, J.J.; Marth, J.D.; Hart, G.W. Dynamic O-GlcNAc Modification of Nucleocytoplasmic Proteins in Response to Stress. A Survival Response of Mammali-An Cells. J. Biol. Chem. 2004, 279, 30133–30142. [Google Scholar] [CrossRef] [Green Version]
- Abramowitz, L.K.; Harly, C.; Das, A.; Bhandoola, A.; Hanover, J.A. Blocked O-GlcNAc Cycling Dis-Rupts Mouse Hematopoeitic Stem Cell Maintenance and Early T Cell Development. Sci. Rep. 2019, 9, 12569. [Google Scholar] [CrossRef]
- Fuentes-Garcia, G.; Castaneda-Patlan, M.C.; Vercoutter-Edouart, A.S.; Lefebvre, T.; Robles-Flores, M. O-GlcNAcylation is Involved in the Regulation of Stem Cell Markers Expression in Colon Cancer Cells. Front. Endocrinol. 2019, 10, 289. [Google Scholar] [CrossRef]
- Andres, L.M.; Blong, I.W.; Evans, A.C.; Rumachik, N.G.; Yamaguchi, T.; Pham, N.D.; Thompson, P.; Kohler, J.J.; Bertozzi, C.R. Chemical Modulation of Protein O-GlcNAcylation via OGT Inhibition Pro-motes Human Neural Cell Differentiation. ACS Chem. Biol. 2017, 12, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Boehmelt, G.; Wakeham, A.; Elia, A.; Sasaki, T.; Plyte, S.; Potter, J.; Yang, Y.; Tsang, E.; Ruland, J.; Iscove, N.N.; et al. Decreased UDP-GlcNAc Levels Abrogate Proliferation Control in EMeg32-Deficient Cells. EMBO J. 2000, 19, 5092–5104. [Google Scholar] [CrossRef] [Green Version]
- Slawson, C.; Zachara, N.E.; Vosseller, K.; Cheung, W.D.; Lane, M.D.; Hart, G.W. Perturbations in O-Linked Beta-N-Acetylglucosamine Protein Modification Cause Severe Defects in Mitotic Progression and Cytokinesis. J. Biol. Chem. 2005, 280, 32944–32956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gallup, M.; Zlock, L.; Chen, Y.T.F.; Finkbeiner, W.E.; McNamara, N.A. Pivotal Role of MUC1 Glycosylation by Cigarette Smoke in Modulating Disruption of Airway Adherens Junctions In Vitro. J. Pathol. 2014, 234, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Reddy, G.R.; Wallukat, G.; Xiang, Y.K.; Steinberg, S.F. beta1-adrenergic receptor O-glycosylation regulates N-terminal cleavage and signaling responses in cardiomyocytes. Sci. Rep. 2017, 7, 7890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decourcelle, A.; Very, N.; Djouina, M.; Loison, I.; Thevenet, J.; Body-Malapel, M.; Lelievre, E.; Coqueret, O.; Leprince, D.; El Yazidi-Belkoura, I.; et al. O-GlcNAcylation Links Nutrition to the Ep-igenetic Downregulation of UNC5A during Colon Carcinogenesis. Cancers 2020, 12, 3168. [Google Scholar] [CrossRef] [PubMed]
- Dersh, D.; Iwamoto, Y.; Argon, Y. Tay–Sachs Disease Mutations in HEXA Target the α Chain of Hexosaminidase A to Endoplasmic Reticulum–Associated Degradation. Mol. Biol. Cell 2016, 27, 3813–3827. [Google Scholar] [CrossRef]
- Bai, F.; Wang, T.; Pal, U.; Bao, F.; Gould, L.H.; Fikrig, E. Use of RNA Interference to Prevent Lethal Murine West Nile Virus Infection. J. Infect. Dis. 2005, 191, 1148–1154. [Google Scholar] [CrossRef]
- Qiu, L.-Q.; Lai, W.S.; Stumpo, D.J.; Blackshear, P.J. Mouse Embryonic Fibroblast Cell Culture and Stimulation. Bio-Protocol 2016, 6, e1859. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.M.; Acharya, D.; Neupane, B.; Thompson, E.A.; Gonzalez-Fernandez, G.; Copeland, K.M.; Garrett, M.; Liu, H.; Lopez, M.E.; de Cruz, M.; et al. Congenital Zika Virus Infection in Immunocompetent Mice Causes Postnatal Growth Imped-iment and Neurobehavioral Deficits. Front. Microbiol. 2018, 9, 2028. [Google Scholar] [CrossRef] [PubMed]
- Acharya, D.; Bastola, P.; Le, L.; Paul, A.M.; Fernandez, E.; Diamond, M.S.; Miao, W.; Bai, F. An Ultra-Sensitive Electrogenerated Chemiluminescence-Based Immunoassay for Specific Detection of Zika Virus. Sci. Rep. 2016, 6, 32227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, A.M.; Shi, Y.; Acharya, D.; Douglas, J.R.; Cooley, A.; Anderson, J.F.; Huang, F.; Bai, F. Delivery of Antiviral Small Interfering RNA with Gold Nanoparticles Inhibits Dengue Virus Infection In Vitro. J. Gen. Virol. 2014, 95, 1712–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neupane, B.; Acharya, D.; Nazneen, F.; Gonzalez-Fernandez, G.; Flynt, A.S.; Bai, F. Interleukin-17A Facilitates Chikungunya Virus Infection by Inhibiting IFN-α2 Expression. Front. Immunol. 2020, 11, 2955. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neupane, B.; Fendereski, M.; Nazneen, F.; Guo, Y.-L.; Bai, F. Murine Trophoblast Stem Cells and Their Differentiated Cells Attenuate Zika Virus In Vitro by Reducing Glycosylation of the Viral Envelope Protein. Cells 2021, 10, 3085. https://doi.org/10.3390/cells10113085
Neupane B, Fendereski M, Nazneen F, Guo Y-L, Bai F. Murine Trophoblast Stem Cells and Their Differentiated Cells Attenuate Zika Virus In Vitro by Reducing Glycosylation of the Viral Envelope Protein. Cells. 2021; 10(11):3085. https://doi.org/10.3390/cells10113085
Chicago/Turabian StyleNeupane, Biswas, Mona Fendereski, Farzana Nazneen, Yan-Lin Guo, and Fengwei Bai. 2021. "Murine Trophoblast Stem Cells and Their Differentiated Cells Attenuate Zika Virus In Vitro by Reducing Glycosylation of the Viral Envelope Protein" Cells 10, no. 11: 3085. https://doi.org/10.3390/cells10113085