
Application of Induced Pluripotent Stem Cells for Disease Modeling and 3D Model Construction: Focus on Osteoarthritis


Abstract
:1. Introduction
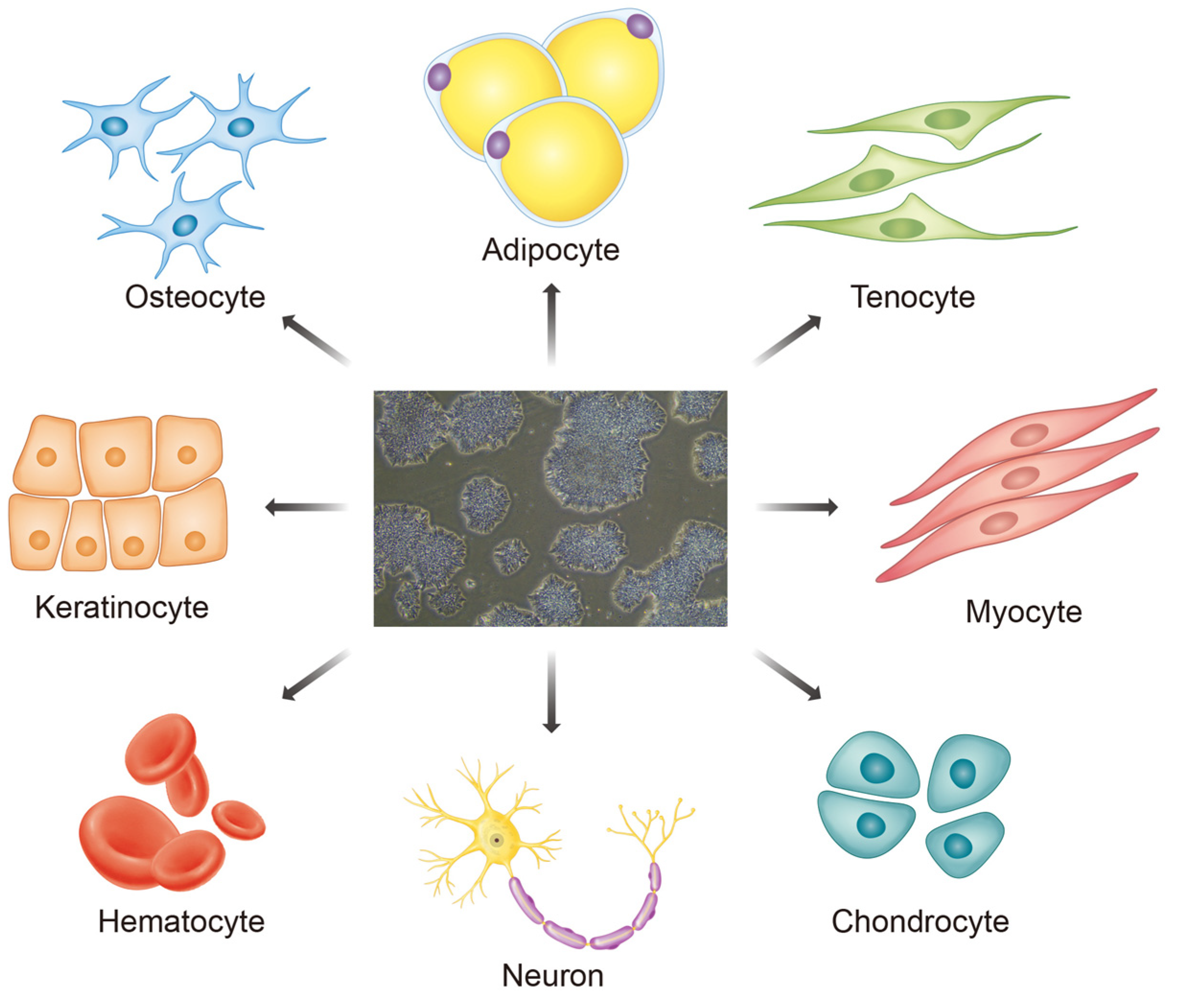
2. Induced Pluripotent Stem Cells (iPSCs) and Their Advantages
Challenges in iPSCs
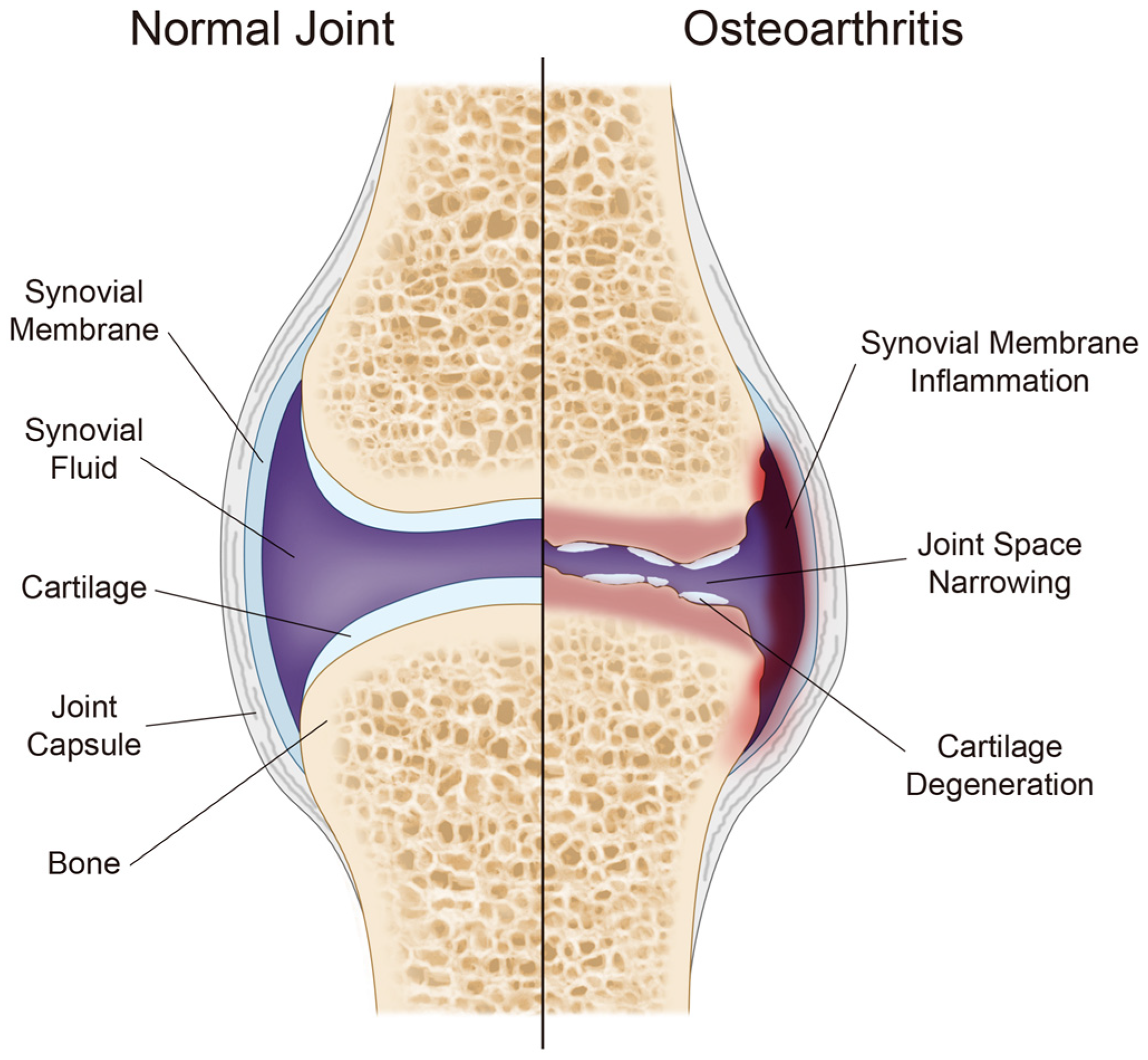
3. Osteoarthritis
4. iPSC Disease Modeling
iPSC Disease Modeling in Various Fields
5. iPSC Disease Modeling in Arthritic Diseases
6. Generating iPSCs from Patients with OA
6.1. iPSC Disease Modeling in OA
6.2. iPSC Disease Modeling in Early-Onset OA
7. iPSC-Derived 3D Model Construction
7.1. iPSC-Derived 3D Model Construction in Various Fields
7.2. iPSC-Derived OA-Related 3D Model Construction
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Medvedev, S.P.; Shevchenko, A.I.; Zakian, S.M. Induced Pluripotent Stem Cells: Problems and Advantages when Applying them in Regenerative Medicine. Acta Nat. 2010, 2, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.G. Embryo-derived stem cells: Of mice and men. Annu. Rev. Cell Dev. Biol. 2001, 17, 435–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Eguizabal, C.; Aran, B.; Chuva de Sousa Lopes, S.M.; Geens, M.; Heindryckx, B.; Panula, S.; Popovic, M.; Vassena, R.; Veiga, A. Two decades of embryonic stem cells: A historical overview. Hum. Reprod. Open 2019, 2019, hoy024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, B.; Parham, L. Ethical issues in stem cell research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Subbaraman, N. NIH reverses Trump-era restrictions on fetal-tissue research. Nature 2021. online ahead of print. [Google Scholar] [CrossRef]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef] [Green Version]
- Byrne, J.A.; Pedersen, D.A.; Clepper, L.L.; Nelson, M.; Sanger, W.G.; Gokhale, S.; Wolf, D.P.; Mitalipov, S.M. Producing primate embryonic stem cells by somatic cell nuclear transfer. Nature 2007, 450, 497–502. [Google Scholar] [CrossRef]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.S.; Sritanaudomchai, H.; et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.J.; Rim, Y.A.; Nam, Y.; Ju, J.H. Recent Developments in Clinical Applications of Mesenchymal Stem Cells in the Treatment of Rheumatoid Arthritis and Osteoarthritis. Front. Immunol. 2021, 12, 631291. [Google Scholar] [CrossRef]
- Liu, Z.; Cai, Y.; Wang, Y.; Nie, Y.; Zhang, C.; Xu, Y.; Zhang, X.; Lu, Y.; Wang, Z.; Poo, M.; et al. Cloning of Macaque Monkeys by Somatic Cell Nuclear Transfer. Cell 2018, 172, 881–887.e7. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later: For Cardiac Applications. Circ. Res. 2017, 120, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Ebert, A.D.; Liang, P.; Wu, J.C. Induced pluripotent stem cells as a disease modeling and drug screening platform. J. Cardiovasc. Pharmacol. 2012, 60, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Deszcz, I.; Lis-Nawara, A.; Grelewski, P.; Dragan, S.; Bar, J. Utility of direct 3D co-culture model for chondrogenic differentiation of mesenchymal stem cells on hyaluronan scaffold (Hyaff-11). Regen. Biomater. 2020, 7, 543–552. [Google Scholar] [CrossRef]
- Scotti, C.; Gobbi, A.; Nakamura, N.; Peretti, G.M. Stem Cells for Cartilage Regeneration: A Roadmap to the Clinic. Stem Cells Int. 2018, 2018, 7348560. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, W.; Fu, X.; Xu, Y. The Immunogenicity and Immune Tolerance of Pluripotent Stem Cell Derivatives. Front. Immunol. 2017, 8, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorecka, J.; Kostiuk, V.; Fereydooni, A.; Gonzalez, L.; Luo, J.; Dash, B.; Isaji, T.; Ono, S.; Liu, S.; Lee, S.R.; et al. The potential and limitations of induced pluripotent stem cells to achieve wound healing. Stem Cell Res. Ther. 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.; Wanczyk, H.; Jensen, T.; Finck, C. Assessment of iPSC teratogenicity throughout directed differentiation toward an alveolar-like phenotype. Differentiation 2019, 105, 45–53. [Google Scholar] [CrossRef]
- Gutierrez-Aranda, I.; Ramos-Mejia, V.; Bueno, C.; Munoz-Lopez, M.; Real, P.J.; Macia, A.; Sanchez, L.; Ligero, G.; Garcia-Parez, J.L.; Menendez, P. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem Cells 2010, 28, 1568–1570. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Chen, M.; Yang, B.; Zhang, F.; Cao, K. Intramyocardial transplantation of undifferentiated rat induced pluripotent stem cells causes tumorigenesis in the heart. PLoS ONE 2011, 6, e19012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2008, 2, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.O.; Moon, S.H.; Jeong, H.C.; Yi, J.Y.; Lee, T.H.; Shim, S.H.; Rhee, Y.H.; Lee, S.H.; Oh, S.J.; Lee, M.Y.; et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, E3281–E3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedel, A.; Beliveau, F.; Lamrissi-Garcia, I.; Rousseau, B.; Moranvillier, I.; Rucheton, B.; Guyonnet-Duperat, V.; Cardinaud, B.; de Verneuil, H.; Moreau-Gaudry, F. Preventing Pluripotent Cell Teratoma in Regenerative Medicine Applied to Hematology Disorders. Stem Cells Transl. Med. 2017, 6, 382–393. [Google Scholar] [CrossRef]
- Wuputra, K.; Ku, C.C.; Wu, D.C.; Lin, Y.C.; Saito, S.; Yokoyama, K.K. Prevention of tumor risk associated with the reprogramming of human pluripotent stem cells. J. Exp. Clin. Cancer Res. 2020, 39, 100. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Nudel, N.; Benvenisty, N. Immunologic and chemical targeting of the tight-junction protein Claudin-6 eliminates tumorigenic human pluripotent stem cells. Nat. Commun. 2013, 4, 1992. [Google Scholar] [CrossRef]
- Tang, C.; Lee, A.S.; Volkmer, J.P.; Sahoo, D.; Nag, D.; Mosley, A.R.; Inlay, M.A.; Ardehali, R.; Chavez, S.L.; Pera, R.R.; et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011, 29, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Moradi, S.; Mahdizadeh, H.; Saric, T.; Kim, J.; Harati, J.; Shahsavarani, H.; Greber, B.; Moore, J.B.T. Research and therapy with induced pluripotent stem cells (iPSCs): Social, legal, and ethical considerations. Stem Cell Res. Ther. 2019, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- Jha, B.S.; Farnoodian, M.; Bharti, K. Regulatory considerations for developing a phase I investigational new drug application for autologous induced pluripotent stem cells-based therapy product. Stem Cells Transl. Med. 2021, 10, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Turkiewicz, A.; Petersson, I.F.; Bjork, J.; Hawker, G.; Dahlberg, L.E.; Lohmander, L.S.; Englund, M. Current and future impact of osteoarthritis on health care: A population-based study with projections to year 2032. Osteoarthr. Cartil. 2014, 22, 1826–1832. [Google Scholar] [CrossRef] [Green Version]
- Rejas-Gutierrez, J.; Sicras-Mainar, A.; Darba, J. Future projections of opioid use and cost in patients with chronic osteoarthritis pain in Spain. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211010599. [Google Scholar] [CrossRef] [PubMed]
- Rim, Y.A.; Ju, J.H. The Role of Fibrosis in Osteoarthritis Progression. Life 2021, 11, 3. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.; Lee, K.; Ju, J.H. Recent Updates of Diagnosis, Pathophysiology, and Treatment on Osteoarthritis of the Knee. Int. J. Mol. Sci. 2021, 22, 2619. [Google Scholar] [CrossRef]
- Grassel, S.; Muschter, D. Recent advances in the treatment of osteoarthritis. F1000Research 2020, 9, 325. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Aiello, F.C.; Szychlinska, M.A.; Di Rosa, M.; Castrogiovanni, P.; Mobasheri, A. Osteoarthritis in the XXIst century: Risk factors and behaviours that influence disease onset and progression. Int. J. Mol. Sci. 2015, 16, 6093–6112. [Google Scholar] [CrossRef]
- Ramos, Y.F.; den Hollander, W.; Bovee, J.V.; Bomer, N.; van der Breggen, R.; Lakenberg, N.; Keurentjes, J.C.; Goeman, J.J.; Slagboom, P.E.; Nelissen, R.G.; et al. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; The RAAK study. PLoS ONE 2014, 9, e103056. [Google Scholar] [CrossRef] [Green Version]
- Reynard, L.N.; Loughlin, J. Genetics and epigenetics of osteoarthritis. Maturitas 2012, 71, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Tachmazidou, I.; Hatzikotoulas, K.; Southam, L.; Esparza-Gordillo, J.; Haberland, V.; Zheng, J.; Johnson, T.; Koprulu, M.; Zengini, E.; Steinberg, J.; et al. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet. 2019, 51, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Boer, C.G.; Hatzikotoulas, K.; Southam, L.; Stefansdottir, L.; Zhang, Y.; Coutinho de Almeida, R.; Wu, T.T.; Zheng, J.; Hartley, A.; Teder-Laving, M.; et al. Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell 2021, 184, 4784–4818.e17. [Google Scholar] [CrossRef]
- Chen, D.; Shen, J.; Zhao, W.; Wang, T.; Han, L.; Hamilton, J.L.; Im, H.J. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017, 5, 16044. [Google Scholar] [CrossRef]
- Reyes, C.; Leyland, K.M.; Peat, G.; Cooper, C.; Arden, N.K.; Prieto-Alhambra, D. Association between Overweight and Obesity and Risk of Clinically Diagnosed Knee, Hip, and Hand Osteoarthritis: A Population-Based Cohort Study. Arthritis Rheumatol. 2016, 68, 1869–1875. [Google Scholar] [CrossRef] [Green Version]
- Eymard, F.; Parsons, C.; Edwards, M.H.; Petit-Dop, F.; Reginster, J.Y.; Bruyere, O.; Richette, P.; Cooper, C.; Chevalier, X. Diabetes is a risk factor for knee osteoarthritis progression. Osteoarthr. Cartil. 2015, 23, 851–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grskovic, M.; Javaherian, A.; Strulovici, B.; Daley, G.Q. Induced pluripotent stem cells—Opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011, 10, 915–929. [Google Scholar] [CrossRef]
- Pasterkamp, G.; van der Laan, S.W.; Haitjema, S.; Foroughi Asl, H.; Siemelink, M.A.; Bezemer, T.; van Setten, J.; Dichgans, M.; Malik, R.; Worrall, B.B.; et al. Human Validation of Genes Associated with a Murine Atherosclerotic Phenotype. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1240–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Sances, S.; Workman, M.J.; Svendsen, C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 2020, 26, 309–329. [Google Scholar] [CrossRef]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis. Model. Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, S.J. Disease modelling using human iPSCs. Hum. Mol. Genet. 2016, 25, R173–R181. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef]
- Mosqueira, D.; Smith, J.G.W.; Bhagwan, J.R.; Denning, C. Modeling Hypertrophic Cardiomyopathy: Mechanistic Insights and Pharmacological Intervention. Trends Mol. Med. 2019, 25, 775–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Bos, J.M.; Ye, D.; Tester, D.J.; Hrstka, S.; Maleszewski, J.J.; Ommen, S.R.; Nishimura, R.A.; Schaff, H.V.; Kim, C.S.; et al. Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Patient with MYL2-R58Q-Mediated Apical Hypertrophic Cardiomyopathy Show Hypertrophy, Myofibrillar Disarray, and Calcium Perturbations. J. Cardiovasc. Transl. Res. 2019, 12, 394–403. [Google Scholar] [CrossRef]
- Grandy, R.; Tomaz, R.A.; Vallier, L. Modeling Disease with Human Inducible Pluripotent Stem Cells. Annu. Rev. Pathol. 2019, 14, 449–468. [Google Scholar] [CrossRef]
- Corbett, J.L.; Duncan, S.A. iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front. Med. 2019, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Cayo, M.A.; Cai, J.; DeLaForest, A.; Noto, F.K.; Nagaoka, M.; Clark, B.S.; Collery, R.F.; Si-Tayeb, K.; Duncan, S.A. JD induced pluripotent stem cell-derived hepatocytes faithfully recapitulate the pathophysiology of familial hypercholesterolemia. Hepatology 2012, 56, 2163–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, J.; Pene, V.; Tolosa, L.; Villaret, M.; Luce, E.; Fourrier, A.; Heslan, J.M.; Saheb, S.; Bruckert, E.; Gomez-Lechon, M.J.; et al. Low-density lipoprotein receptor-deficient hepatocytes differentiated from induced pluripotent stem cells allow familial hypercholesterolemia modeling, CRISPR/Cas-mediated genetic correction, and productive hepatitis C virus infection. Stem Cell Res. Ther. 2019, 10, 221. [Google Scholar] [CrossRef] [Green Version]
- Ge, W.; Song, Y.; Chu, M.; Liu, Y.; Yang, B.; Wang, K.; Yu, B.; Song, C.; Wang, Y.; Yang, J. Generation of a human iPSC line CIBi009-A from a patient with familial hypercholesterolemia carrying variants of LDLR c.T1241G and APOB c.G1618T. Stem Cell Res. 2021, 53, 102347. [Google Scholar] [CrossRef]
- Teeple, E.; Jay, G.D.; Elsaid, K.A.; Fleming, B.C. Animal models of osteoarthritis: Challenges of model selection and analysis. AAPS J. 2013, 15, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinnerling, K.; Rosas, C.; Soto, L.; Thomas, R.; Aguillon, J.C. Humanized Mouse Models of Rheumatoid Arthritis for Studies on Immunopathogenesis and Preclinical Testing of Cell-Based Therapies. Front. Immunol. 2019, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, L.; Yu, F.F.; Wang, S.; Wu, C.; Qu, C.; Lammi, M.J.; Guo, X. The potential of induced pluripotent stem cells as a tool to study skeletal dysplasias and cartilage-related pathologic conditions. Osteoarthr. Cartil. 2017, 25, 616–624. [Google Scholar] [CrossRef]
- McCoy, A.M. Animal Models of Osteoarthritis: Comparisons and Key Considerations. Vet. Pathol. 2015, 52, 803–818. [Google Scholar] [CrossRef]
- Vincent, T.L.; Williams, R.O.; Maciewicz, R.; Silman, A.; Garside, P.; Arthritis Research UK Animal Models Working Group. Mapping pathogenesis of arthritis through small animal models. Rheumatology 2012, 51, 1931–1941. [Google Scholar] [CrossRef] [Green Version]
- Sokka, T.; Toloza, S.; Cutolo, M.; Kautiainen, H.; Makinen, H.; Gogus, F.; Skakic, V.; Badsha, H.; Peets, T.; Baranauskaite, A.; et al. Women, men, and rheumatoid arthritis: Analyses of disease activity, disease characteristics, and treatments in the QUEST-RA study. Arthritis Res. Ther. 2009, 11, R7. [Google Scholar] [PubMed] [Green Version]
- Xu, B.; Lin, J. Characteristics and risk factors of rheumatoid arthritis in the United States: An NHANES analysis. PeerJ 2017, 5, e4035. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S.; Saigo, K.; Morinobu, A.; Koshiba, M.; Kuntz, K.M.; et al. Meta-analysis: Diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann. Intern. Med. 2007, 146, 797–808. [Google Scholar] [CrossRef]
- Boeters, D.M.; Mangnus, L.; Ajeganova, S.; Lindqvist, E.; Svensson, B.; Toes, R.E.M.; Trouw, L.A.; Huizinga, T.W.J.; Berenbaum, F.; Morel, J.; et al. The prevalence of ACPA is lower in rheumatoid arthritis patients with an older age of onset but the composition of the ACPA response appears identical. Arthritis Res. Ther. 2017, 19, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Woude, D.; Alemayehu, W.G.; Verduijn, W.; de Vries, R.R.; Houwing-Duistermaat, J.J.; Huizinga, T.W.; Toes, R.E. Gene-environment interaction influences the reactivity of autoantibodies to citrullinated antigens in rheumatoid arthritis. Nat. Genet. 2010, 42, 814–816. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.; Choi, J.; Jung, H.; Lee, K.; Kang, J.; Park, N.; Rim, Y.A.; Nam, Y.; Ju, J.H. Recapitulation of methotrexate hepatotoxicity with induced pluripotent stem cell-derived hepatocytes from patients with rheumatoid arthritis. Stem Cell Res. Ther. 2018, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Son, M.J.; Son, M.Y.; Seol, B.; Kim, J.; Park, J.; Kim, J.H.; Kim, Y.H.; Park, S.A.; Lee, C.H. Generation of human induced pluripotent stem cells from osteoarthritis patient-derived synovial cells. Arthritis Rheum. 2011, 63, 3010–3021. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Yi, H.; Diecke, S.; Kim, J.; Jung, H.; Rim, Y.A.; Jung, S.M.; Kim, M.; Kim, Y.G. Generation of disease-specific induced pluripotent stem cells from patients with rheumatoid arthritis and osteoarthritis. Arthritis Res. Ther. 2014, 16, R41. [Google Scholar] [CrossRef] [Green Version]
- Castro-Vinuelas, R.; Sanjurjo-Rodriguez, C.; Pineiro-Ramil, M.; Hermida-Gomez, T.; Rodriguez-Fernandez, S.; Oreiro, N.; de Toro, J.; Fuentes, I.; Blanco, F.J.; Diaz-Prado, S. Generation and characterization of human induced pluripotent stem cells (iPSCs) from hand osteoarthritis patient-derived fibroblasts. Sci. Rep. 2020, 10, 4272. [Google Scholar] [CrossRef] [Green Version]
- Saitta, B.; Passarini, J.; Sareen, D.; Ornelas, L.; Sahabian, A.; Argade, S.; Krakow, D.; Cohn, D.H.; Svendsen, C.N.; Rimoin, D.L. Patient-derived skeletal dysplasia induced pluripotent stem cells display abnormal chondrogenic marker expression and regulation by BMP2 and TGFbeta1. Stem Cells Dev. 2014, 23, 1464–1478. [Google Scholar] [CrossRef] [Green Version]
- Willard, V.P.; Diekman, B.O.; Sanchez-Adams, J.; Christoforou, N.; Leong, K.W.; Guilak, F. Use of cartilage derived from murine induced pluripotent stem cells for osteoarthritis drug screening. Arthritis Rheumatol. 2014, 66, 3062–3072. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Morioka, M.; Kishi, H.; Kimura, T.; Yahara, Y.; Okada, M.; Fujita, K.; Sawai, H.; Ikegawa, S.; Tsumaki, N. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature 2014, 513, 507–511. [Google Scholar] [CrossRef]
- Xu, M.; Stattin, E.L.; Shaw, G.; Heinegard, D.; Sullivan, G.; Wilmut, I.; Colman, A.; Onnerfjord, P.; Khabut, A.; Aspberg, A.; et al. Chondrocytes Derived from Mesenchymal Stromal Cells and Induced Pluripotent Cells of Patients with Familial Osteochondritis Dissecans Exhibit an Endoplasmic Reticulum Stress Response and Defective Matrix Assembly. Stem Cells Transl. Med. 2016, 5, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, Z.; Li, E.N.; Li, X.; Del Duke, C.J.; Shen, H.; Hao, T.; O’Donnell, B.; Bunnell, B.A.; Goodman, S.B.; et al. Osteochondral Tissue Chip Derived From iPSCs: Modeling OA Pathologies and Testing Drugs. Front. Bioeng. Biotechnol. 2019, 7, 411. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Park, N.; Lee, K.; Jung, H.; Jung, S.M.; Lee, J.; Ju, J.H. Characterization of Early-Onset Finger Osteoarthritis-Like Condition Using Patient-Derived Induced Pluripotent Stem Cells. Cells 2021, 10, 317. [Google Scholar] [CrossRef]
- Diederichs, S.; Tuan, R.S. Functional comparison of human-induced pluripotent stem cell-derived mesenchymal cells and bone marrow-derived mesenchymal stromal cells from the same donor. Stem Cells Dev. 2014, 23, 1594–1610. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, W.S.; Yang, Z.; Liu, H.; Heng, B.C.; Lee, E.H.; Cao, T. Effects of culture conditions and bone morphogenetic protein 2 on extent of chondrogenesis from human embryonic stem cells. Stem Cells 2007, 25, 950–960. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, Y.A.; Nam, Y.; Park, N.; Jung, H.; Jang, Y.; Lee, J.; Ju, J.H. Different Chondrogenic Potential among Human Induced Pluripotent Stem Cells from Diverse Origin Primary Cells. Stem Cells Int. 2018, 2018, 9432616. [Google Scholar] [CrossRef] [Green Version]
- Rim, Y.A.; Nam, Y.; Park, N.; Jung, H.; Lee, K.; Lee, J.; Ju, J.H. Chondrogenic Differentiation from Induced Pluripotent Stem Cells Using Non-Viral Minicircle Vectors. Cells 2020, 9, 582. [Google Scholar] [CrossRef]
- Nam, Y.; Rim, Y.A.; Ju, J.H. Chondrogenic Pellet Formation from Cord Blood-derived Induced Pluripotent Stem Cells. J. Vis. Exp. 2017, 124, 55988. [Google Scholar] [CrossRef]
- Nam, Y.; Rim, Y.A.; Jung, S.M.; Ju, J.H. Cord blood cell-derived iPSCs as a new candidate for chondrogenic differentiation and cartilage regeneration. Stem Cell Res. Ther. 2017, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, Y.A.; Nam, Y.; Park, N.; Lee, J.; Park, S.H.; Ju, J.H. Repair potential of nonsurgically delivered induced pluripotent stem cell-derived chondrocytes in a rat osteochondral defect model. J. Tissue Eng. Regen. Med. 2018, 12, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Oikonomopoulos, A.; Sayed, N.; Wu, J.C. Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development 2018, 145, dev156166. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Schaffer, D.V. A fully defined and scalable 3D culture system for human pluripotent stem cell expansion and differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, E5039–E5048. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.M.; Hochedlinger, K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat. Cell Biol. 2011, 13, 497–505. [Google Scholar] [CrossRef]
- Soman, S.S.; Vijayavenkataraman, S. Applications of 3D Bioprinted-Induced Pluripotent Stem Cells in Healthcare. Int. J. Bioprint. 2020, 6, 280. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Takebe, T.; Zhang, B.; Radisic, M. Synergistic Engineering: Organoids Meet Organs-on-a-Chip. Cell Stem Cell 2017, 21, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-chip: Recent breakthroughs and future prospects. Biomed. Eng. Online 2020, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- McMinn, P.; Guckenberger, D.J.; Beebe, D.J. Induced Pluripotent Stem Cells on a Chip: A Self-Contained, Accessible, Pipette-less iPSC Culturing and Differentiation Kit. SLAS Technol. 2021, 26, 80–91. [Google Scholar] [PubMed]
- Zhu, J.; Marchant, R.E. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devices 2011, 8, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Andersson, H.; Martinsson, S.; Sabirsh, A.; Jonebring, A.; Wang, Q.D.; Plowright, A.T.; Drowley, L. Aligned nanofiber scaffolds improve functionality of cardiomyocytes differentiated from human induced pluripotent stem cell-derived cardiac progenitor cells. Sci. Rep. 2020, 10, 13575. [Google Scholar] [CrossRef]
- Del Carmen Ortuno-Costela, M.; Garcia-Lopez, M.; Cerrada, V.; Gallardo, M.E. iPSCs: A powerful tool for skeletal muscle tissue engineering. J. Cell. Mol. Med. 2019, 23, 3784–3794. [Google Scholar] [CrossRef]
- Gu, Q.; Zhu, H.; Chen, L.; Shuai, L.; Fang, J.; Wu, J.; Liu, L.; Li, W.; Dai, J.; Hao, J.; et al. Three dimensional collagen scaffolds promote iPSC induction with higher pluripotency. Protein Cell 2016, 7, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Sasai, Y. Cytosystems dynamics in self-organization of tissue architecture. Nature 2013, 493, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Sapiens, M.A.; Reza-Zaldivar, E.E.; Cevallos, R.R.; Marquez-Aguirre, A.L.; Gazarian, K.; Canales-Aguirre, A.A. A Three-Dimensional Alzheimer’s Disease Cell Culture Model Using iPSC-Derived Neurons Carrying A246E Mutation in PSEN1. Front. Cell. Neurosci. 2020, 14, 151. [Google Scholar] [CrossRef]
- Kobolak, J.; Teglasi, A.; Bellak, T.; Janstova, Z.; Molnar, K.; Zana, M.; Bock, I.; Laszlo, L.; Dinnyes, A. Human Induced Pluripotent Stem Cell-Derived 3D-Neurospheres are Suitable for Neurotoxicity Screening. Cells 2020, 9, 1122. [Google Scholar] [CrossRef]
- Woodruff, G.; Phillips, N.; Carromeu, C.; Guicherit, O.; White, A.; Johnson, M.; Zanella, F.; Anson, B.; Lovenberg, T.; Bonaventure, P.; et al. Screening for modulators of neural network activity in 3D human iPSC-derived cortical spheroids. PLoS ONE 2020, 15, e0240991. [Google Scholar] [CrossRef]
- Costamagna, G.; Andreoli, L.; Corti, S.; Faravelli, I. iPSCs-Based Neural 3D Systems: A Multidimensional Approach for Disease Modeling and Drug Discovery. Cells 2019, 8, 1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaspyropoulos, A.; Tsolaki, M.; Foroglou, N.; Pantazaki, A.A. Modeling and Targeting Alzheimer’s Disease with Organoids. Front. Pharmacol. 2020, 11, 396. [Google Scholar] [CrossRef]
- Zuppinger, C. 3D Cardiac Cell Culture: A Critical Review of Current Technologies and Applications. Front. Cardiovasc. Med. 2019, 6, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, P.; Jackson, C.B.; Ozhathil, L.C.; Agarkova, I.; Galindo, C.L.; Sawyer, D.B.; Suter, T.M.; Zuppinger, C. 3D Co-culture of hiPSC-Derived Cardiomyocytes with Cardiac Fibroblasts Improves Tissue-Like Features of Cardiac Spheroids. Front. Mol. Biosci. 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Lei, W.; Hu, S. Cardiac organoid—A promising perspective of preclinical model. Stem Cell Res. Ther. 2021, 12, 272. [Google Scholar] [CrossRef] [PubMed]
- Shinnawi, R.; Shaheen, N.; Huber, I.; Shiti, A.; Arbel, G.; Gepstein, A.; Ballan, N.; Setter, N.; Tijsen, A.J.; Borggrefe, M.; et al. Modeling Reentry in the Short QT Syndrome with Human-Induced Pluripotent Stem Cell-Derived Cardiac Cell Sheets. J. Am. Coll. Cardiol. 2019, 73, 2310–2324. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.J.; Li, Y.; Kerr, C.M.; Yao, J.; Beeson, G.C.; Coyle, R.C.; Chen, X.; Jia, J.; Damon, B.; Wilson, R.; et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 2020, 4, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Miyagawa, S.; Fukushima, S.; Saito, A.; Ito, E.; Harada, A.; Matsuura, R.; Iseoka, H.; Sougawa, N.; Mochizuki-Oda, N.; et al. Development of In Vitro Drug-Induced Cardiotoxicity Assay by Using Three-Dimensional Cardiac Tissues Derived from Human Induced Pluripotent Stem Cells. Tissue Eng. Part C Methods 2018, 24, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchetto, C.; Vitiello, L.; de Windt, L.J.; Rampazzo, A.; Calore, M. Modeling Cardiovascular Diseases with hiPSC-Derived Cardiomyocytes in 2D and 3D Cultures. Int. J. Mol. Sci. 2020, 21, 3404. [Google Scholar] [CrossRef]
- Archer, C.R.; Sargeant, R.; Basak, J.; Pilling, J.; Barnes, J.R.; Pointon, A. Characterization and Validation of a Human 3D Cardiac Microtissue for the Assessment of Changes in Cardiac Pathology. Sci. Rep. 2018, 8, 10160. [Google Scholar] [CrossRef]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling Cardiac Disease Mechanisms Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Progress, Promises and Challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Nagata, S.; Ozawa, F.; Nie, M.; Takeuchi, S. 3D culture of functional human iPSC-derived hepatocytes using a core-shell microfiber. PLoS ONE 2020, 15, e0234441. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Shafagh, R.Z.; Hendriks, D.F.G.; Ingelman-Sundberg, M. 3D Primary Hepatocyte Culture Systems for Analyses of Liver Diseases, Drug Metabolism, and Toxicity: Emerging Culture Paradigms and Applications. Biotechnol. J. 2019, 14, e1800347. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Yi, X.; Liao, W.; Chen, Q.; Yang, W.; Li, Y.; Li, S.; Gao, Y.; Peng, Q.; Zhou, S. Advancements in stem cell-derived hepatocyte-like cell models for hepatotoxicity testing. Stem Cell Res. Ther. 2021, 12, 84. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Deng, P.; Chen, W.; Guo, Y.; Tao, T.; Qin, J. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, G.; Sjogren, A.K.; Barragan, I.; Sabirsh, A.; Sartipy, P.; Synnergren, J.; Bjorquist, P.; Ingelman-Sundberg, M.; Andersson, T.B.; Edsbagge, J. Long-term chronic toxicity testing using human pluripotent stem cell-derived hepatocytes. Drug Metab. Dispos. 2014, 42, 1401–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayo, M.A.; Mallanna, S.K.; Di Furio, F.; Jing, R.; Tolliver, L.B.; Bures, M.; Urick, A.; Noto, F.K.; Pashos, E.E.; Greseth, M.D.; et al. A Drug Screen using Human iPSC-Derived Hepatocyte-like Cells Reveals Cardiac Glycosides as a Potential Treatment for Hypercholesterolemia. Cell Stem Cell 2017, 20, 478–489.e5. [Google Scholar] [CrossRef] [Green Version]
- Nuciforo, S.; Heim, M.H. Organoids to model liver disease. JHEP Rep. 2021, 3, 100198. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef] [PubMed]
- Akbari, S.; Sevinc, G.G.; Ersoy, N.; Basak, O.; Kaplan, K.; Sevinc, K.; Ozel, E.; Sengun, B.; Enustun, E.; Ozcimen, B.; et al. Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling. Stem Cell Rep. 2019, 13, 627–641. [Google Scholar] [CrossRef] [Green Version]
- Fiorotto, R.; Amenduni, M.; Mariotti, V.; Fabris, L.; Spirli, C.; Strazzabosco, M. Liver diseases in the dish: iPSC and organoids as a new approach to modeling liver diseases. Biochim Biophys Acta BBA—Mol. Basis Dis. 2019, 1865, 920–928. [Google Scholar] [CrossRef]
- Diekman, B.O.; Christoforou, N.; Willard, V.P.; Sun, H.; Sanchez-Adams, J.; Leong, K.W.; Guilak, F. Cartilage tissue engineering using differentiated and purified induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 19172–19177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teramura, T.; Onodera, Y.; Mihara, T.; Hosoi, Y.; Hamanishi, C.; Fukuda, K. Induction of mesenchymal progenitor cells with chondrogenic property from mouse-induced pluripotent stem cells. Cell Reprogram. 2010, 12, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, S.P.; Grigor’eva, E.V.; Shevchenko, A.I.; Malakhova, A.A.; Dementyeva, E.V.; Shilov, A.A.; Pokushalov, E.A.; Zaidman, A.M.; Aleksandrova, M.A.; Plotnikov, E.Y.; et al. Human induced pluripotent stem cells derived from fetal neural stem cells successfully undergo directed differentiation into cartilage. Stem Cells Dev. 2011, 20, 1099–1112. [Google Scholar] [CrossRef]
- Yamashita, A.; Morioka, M.; Yahara, Y.; Okada, M.; Kobayashi, T.; Kuriyama, S.; Matsuda, S.; Tsumaki, N. Generation of scaffoldless hyaline cartilaginous tissue from human iPSCs. Stem Cell Rep. 2015, 4, 404–418. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.N.; Tam, W.L.; Andrikopoulos, K.S.; Casas-Fraile, L.; Voyiatzis, G.A.; Geris, L.; Luyten, F.P.; Papantoniou, I. Patterned, organoid-based cartilaginous implants exhibit zone specific functionality forming osteochondral-like tissues in vivo. Biomaterials 2021, 273, 120820. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Rim, Y.A.; Yi, H.; Park, N.; Park, S.H.; Ju, J.H. The Generation of Human Induced Pluripotent Stem Cells from Blood Cells: An Efficient Protocol Using Serial Plating of Reprogrammed Cells by Centrifugation. Stem Cells Int. 2016, 2016, 1329459. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Hagg, D.A.; Forsman, A.; Ekholm, J.; Nimkingratana, P.; Brantsing, C.; Kalogeropoulos, T.; Zaunz, S.; Concaro, S.; Brittberg, M.; et al. Cartilage Tissue Engineering by the 3D Bioprinting of iPS Cells in a Nanocellulose/Alginate Bioink. Sci. Rep. 2017, 7, 658. [Google Scholar] [CrossRef] [PubMed]
- Limraksasin, P.; Kondo, T.; Zhang, M.; Okawa, H.; Osathanon, T.; Pavasant, P.; Egusa, H. In Vitro Fabrication of Hybrid Bone/Cartilage Complex Using Mouse Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 581. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, S.K.; Katz, D.B.; Oswald, S.J.; Groneck, L.; Guilak, F. Formation of Osteochondral Organoids from Murine Induced Pluripotent Stem Cells. Tissue Eng. Part A 2021, 27, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Dicks, A.; Steward, N.; Tang, R.; Katz, D.B.; Choi, Y.R.; Guilak, F. Single cell transcriptomic analysis of human pluripotent stem cell chondrogenesis. Nat. Commun. 2021, 12, 362. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Pulai, J.; Loeser, R.F. Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002, 46, 2368–2376. [Google Scholar] [CrossRef]
- Durigova, M.; Troeberg, L.; Nagase, H.; Roughley, P.J.; Mort, J.S. Involvement of ADAMTS5 and hyaluronidase in aggrecan degradation and release from OSM-stimulated cartilage. Eur. Cell Mater. 2011, 21, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.S.M.; Ulici, V.; Kim, C.; Chubinskaya, S.; Loeser, R.F.; Phanstiel, D.H. Transcriptional response of human articular chondrocytes treated with fibronectin fragments: An in vitro model of the osteoarthritis phenotype. Osteoarthr. Cartil. 2021, 29, 235–247. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Year | Reference | OA Type | iPSC Source and Reprogramming Procedure | OA Disease Model Generation Procedure | Study Objective and Results |
|---|---|---|---|---|---|
| 2014s | Saitta et al. [79] | Early-onset OA (skeletal dysplasia) | Human neonatal skin fibroblasts from a patient with lethal metatropic dysplasia were nucleofected using nucleofector II and non-integrating episomal plasmid expression vectors with OSKM factors. | Heterozygous mutation of TRPV4 confirmed in iPSC clones. | Objective: To assess the characteristics of iPSC model with a mutation in TRPV4 (causing skeletal dysplasia). Results: The micromasses of TRPV4-iPSCs grown in chondrogenic differentiation conditions had lower expression of cartilage growth plate markers (COL2A1 (IIA and IIB), Sox9, Aggrecan, COL10A1, and RUNX2), lower GAG expression, and higher expression of osteogenic differentiation marker COL1A1. This study successfully recapitulated skeletal dysplasia. |
| 2014 | Willard et al. [80] | Primary OA | Tail fibroblasts from adult C57BL/6 mice were transduced using single doxycycline-inducible lentiviral vector expressing mouse cDNA for OSKM factors. | The iPSC-derived cartilage model was treated with IL-1α in a serum-free chondrogenic medium for 3 days. | Objective: To construct iPSC-derived cartilage for an in vitro OA model. Results: IL-1α-treated cartilage models showed OA characteristics (increase in inflammatory and catabolic genes, decrease in tissue elastic modulus, and loss of GAG). The five therapeutic agents (IL-4, Metalloproteinase 3, NS398, SC514, and GM6001) improved OA conditions. |
| 2014 | Yamashita et al. [81] | Early-onset OA (skeletal dysplasia) | Human dermal fibroblasts from patients with thanatophoric dysplasia type I (TD1) recapitulated the disease phenotypes. | Inherited heterozygous mutation (R248C) in the FGFR3 gene was confirmed in all samples. | Objective: To test the clinical efficacy of statin treatment in skeletal dysplasia patients. Results: The TD1 iPSCs formed abnormal chondrocyte particles that replicated TD1 phenotypes (lower GAG, FGFR3, cartilage matrix gene expressions). While the FGFR3-neutralizing antibody was induced partial recovery of cartilage formation, statin was able to successfully induce cartilage formation in TD1-iPSC-derived cartilage. This result was obtained by controlling phosphorylated MAPK production. Hence, iPSC-derived models could be used for drug screening and closely examine pathology. |
| 2016 | Xu et al. [82] | Early-onset OA (osteochondritis dissecans) | Human dermal fibroblasts from patients with familial osteochondritis dissecans were transfected using retrovirus with OSKM factors. | Inherited | Objective: To determine if cartilage models derived from BM-MSCs and iPSCs could recapitulate the phenotypes of familial osteochondritis dissecans (FOCD). Results: The FOCD-iPSC-derived cartilage displayed identical disease phenotypes in the chondrogenic cultures of primary MSCs. Both showed GAG abundance, aggrecan shortage in ECM, and aggrecan intracellular localization in early/late chondrocytes. The similarities in the disease phenotypes, such as abnormal aggrecan processing, were evident. |
| 2019 | Lin et al. [83] | Primary OA | Human bone marrow-derived MSCs from femoral heads were transduced using lentiviral vector with OSKM factors. | IL-1β was added to the chondrogenic medium that was perfused into the top of the iPSC construct during the fabrication of osteochondral tissue chips for 28 days. | Objective: To construct iPSC-derived microphysiological osteochondral tissue chips that can recapitulate OA conditions. Results: The IL-1β treatment created an OA model with a lower expression of COL2 and ACAN, a decrease in the GAG, and an increase in both cartilage-degenerating enzymes and proinflammatory cytokines. The therapeutic effect of celecoxib in the OA chip model demonstrated decreased expression of catabolic and inflammatory factors in addition to its osteoprotective effect. |
| 2021 | Rim et al. [84] | Early-Onset Finger OA | Human dermal fibroblasts from a patient with radiographic early-onset finger OA-like condition (efOA-like condition) were transduced using Sendai virus with OSKM factors. | Inherited reprogrammed iPSCs contained a mutation in exon 17 of the aggrecan gene. | Objective: To construct an iPSC model of early-onset finger OA and characterize it. Results: The chondrogenic pellets from the patient with efOA-like condition displayed increase in size and vacuole-like morphologies. The abnormal size could be due to the overexpression of hypertrophic and chondrogenic markers. Hence, iPSC-derived disease models could serve as an effective tool to understand OA pathology. |
| Year | Reference | iPSC Source and Reprogramming Procedure | Cartilage Model Construction Procedure | Study Results |
|---|---|---|---|---|
| 2014 | Willard et al. [80] | Tail fibroblasts from adult C57BL/6 mice were transduced with single doxycycline-inducible lentiviral vector containing OSKM factors. | The iPSCs were placed in a high-density micromass culture with a serum-free chondrogenic medium (including BMP-4 and dexamethasone). Upon micromass digestion, the GFP+ cells were separated and expanded in a chondrogenic medium (with fetal bovine serum and basic fibroblast growth factor). These cells were then centrifuged for pellet formation before being cultured in a serum-free chondrogenic medium with TGFβ3 and dexamethasone for cartilage model generation. | The iPSC-derived cartilage model was successfully generated and was then treated with IL-1α to recapitulate the OA environment. The OA model was used to test the clinical efficacy of current OA drugs. |
| 2015 | Yamashita et al. [141] | Human dermal fibroblasts and dental pulp were transduced using episomal factors with OSKM factors. | High-density cell colonies were first formed by culturing iPSCs in a feeder-free medium. These colonies were then cultured in a mesendodermal differentiation medium. Subsequently, the cells were put in a basal medium with various chondrogenic supplementations (combinations of ascorbic acid, BMP2, TGFβ1, GDF5) that generated cartilaginous nodules. Later, these models were placed in suspension culture and chondrogenic medium (for proliferation) to further be examined. | It was concluded that BMP2, TGFβ1, and GDF5 were needed for GFP+ cells. The suspension culture could potentially be used to separate any non-chondrocytic cells for purification purposes. This approach could be used for iPSC differentiation into scaffold-less hyaline cartilage. |
| 2017 | Nam et al. [92] | Human cord blood mononuclear cells were transduced using Sendai virus with OSKM factors. | The iPSCs underwent expansion, resuspension, and incubation to form embryoid bodies (EB). The outgrown cells from EBs were subsequently suspended in a conical tube containing a chondrogenic differentiation medium for pellet generation. | The chondrogenic pellets expressed ECM component proteins and chondrogenic markers. Moreover, the ECM region showed characteristics of hyaline cartilage. Hence, CMBC-derived iPSCs can be used to form cartilage models, which could potentially translate to therapeutic applications. |
| 2017 | Nguyen et al. [144] | Human chondrocytes underwent mRNA-based reprogramming. | The two types of bioink: NFC with alginate and NFC with hyaluronic acid were mixed with iPSCs and/or irradiated chondrocytes. Various combinations were then used for cartilage printing. Once completed, the constructs were cross-linked with either water or CaCl2 before rinsing and incubation. Subsequently, the constructs were placed in a pluripotent medium before undergoing differentiation in a chondrogenic medium. | The NFC/HA bioink did not show the proliferation of cells. Both ratios (80/20 and 60/40) of NFC/A bioink showed cell growth and cluster formations. NFC/A (60/40) models displayed the greatest cell growth and viability in addition to a decrease in tumorigenic expression. Moreover, the model showed the formation of hyaline-like cartilaginous tissue. |
| 2019 | Lin et al. [83] | Human bone marrow-derived MSCs from femoral heads were transduced using lentiviral vectors with OSKM factors. | The iPSCs were first differentiated into iMPCs in a mesenchymal induction medium. The iMPCs were then suspended and placed inside the microbioreactor where the constructs were perfused with a chondrogenic medium on its top side and osteogenic medium on its bottom to form osteochondral tissue chips. The chondral tissue chips were perfused with cell-free mGL solution instead of osteogenic medium. | The osteochondral and chondral tissue chips were successfully generated. The tissue chips were treated with IL-1β to recapitulate the OA environment, model OA pathology, and screen current OA drugs. |
| 2020 | Limraksasin et al. [145] | Mouse gingival fibroblasts were transduced using retrovirus with OSK factors (without c-Myc). | After attaining confluence, the iPSCs formed into 3D-iPSCs spheres in U-bottom-shaped microwell spots per well. The spheres were placed in an ES medium to form predominantly mesenchymal precursor cells and were later placed either in an osteogenic induction medium (OI-iPSCs) or both an osteogenic and chondrogenic medium (OIC-iPSCs) with physical shaking. | OI-iPSCs showed higher expressions of osteogenic markers: Osx and Col1a1 with robust mineralization and some presence of cartilage-like tissues. OCI-iPSCs showed higher expressions of osteogenic marker Ocn and chondrogenic markers: Sox9, COl2a1, Aggrecan, and partial mineralization and strong presence of cartilage tissue. Mechanical stimuli and medium type affect the osteochondral model formation. |
| 2020 | O’Connor et al. [146] | Mouse tail fibroblasts were transduced using single doxycycline-induced lentiviral vector with OSKM factors. | iPSCs were nucleofected with linearized pCOL2-EGFP-SV40-NEO reporter plasmid before being expanded with G418. The G418-resistant clones were then selected to be differentiated in a micromass culture with chondrogenic media. Upon steps of centrifugation, incubation, and resuspension, GFP+ cells were separated to be expanded in chondrogenic differentiation media with TGF-β3 for pellet generation. The pellets were then cultured in chondrogenic and osteogenic media to form osteochondral organoids. | Chondrogenic pellet culture expressed chondrogenic markers and a robust cartilaginous matrix. Osteochondral organoids displayed endochondral ossification. Therefore, osteochondral organoids were able to be generated through a scaffold/bioreactor-free procedure. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, J.J.; Choi, J.; Rim, Y.A.; Nam, Y.; Ju, J.H. Application of Induced Pluripotent Stem Cells for Disease Modeling and 3D Model Construction: Focus on Osteoarthritis. Cells 2021, 10, 3032. https://doi.org/10.3390/cells10113032
Hwang JJ, Choi J, Rim YA, Nam Y, Ju JH. Application of Induced Pluripotent Stem Cells for Disease Modeling and 3D Model Construction: Focus on Osteoarthritis. Cells. 2021; 10(11):3032. https://doi.org/10.3390/cells10113032
Chicago/Turabian StyleHwang, Joel Jihwan, Jinhyeok Choi, Yeri Alice Rim, Yoojun Nam, and Ji Hyeon Ju. 2021. "Application of Induced Pluripotent Stem Cells for Disease Modeling and 3D Model Construction: Focus on Osteoarthritis" Cells 10, no. 11: 3032. https://doi.org/10.3390/cells10113032