
Inhibitors of the Plasmodium falciparum Hsp90 towards Selective Antimalarial Drug Design: The Past, Present and Future


Abstract
:1. Introduction
1.1. Plasmodium Falciparum Life Cycle
1.2. Antimalarial Drug Resistance
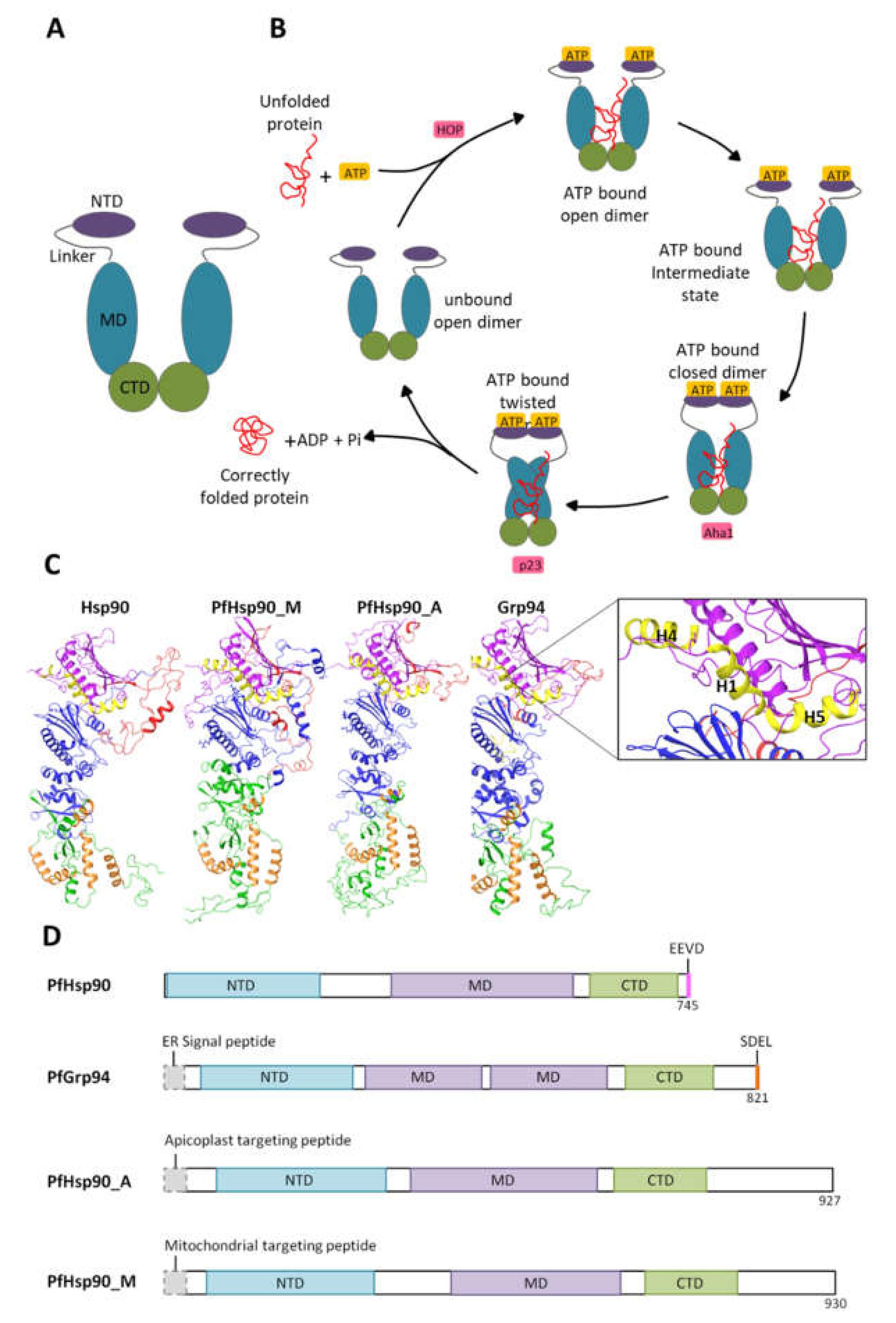
2. Heat Shock Protein 90 and Its Functional Cycle
3. Plasmodial Hsp90s
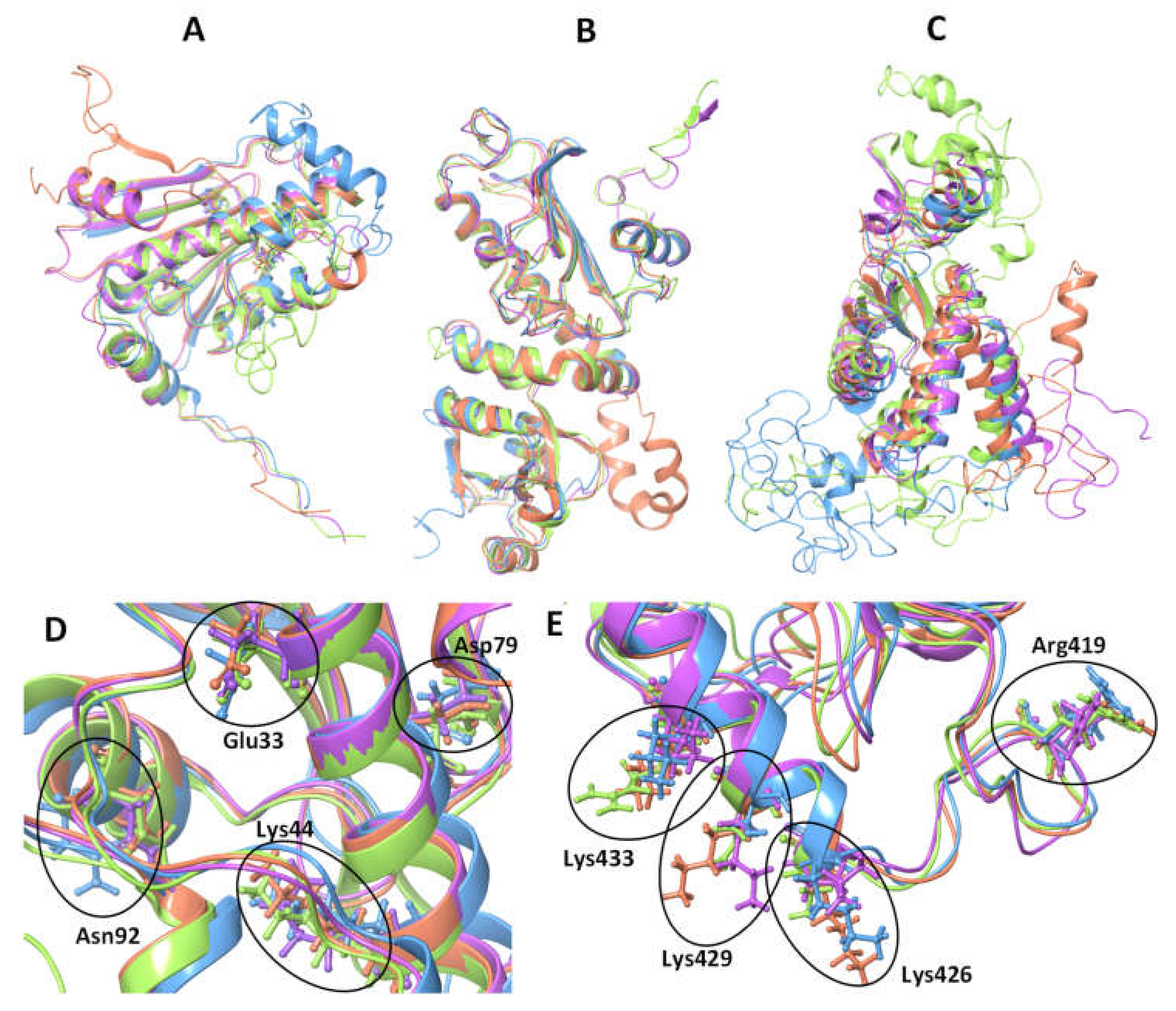
3.1. Cytosolic P. falciparum Hsp90
3.2. Co-Chaperones of Cytosolic PfHsp90
3.3. Plasmodium falciparum Endoplasmic Reticulum Hsp90 (PfGrp94)
3.4. P. falciparum Apicoplast Hsp90
3.5. P. falciparum Mitochondrial Hsp90
4. P. falciparum Hsp90 as Drug Targets
4.1. Inhibitors of the Hsp90 N-Terminal Domain
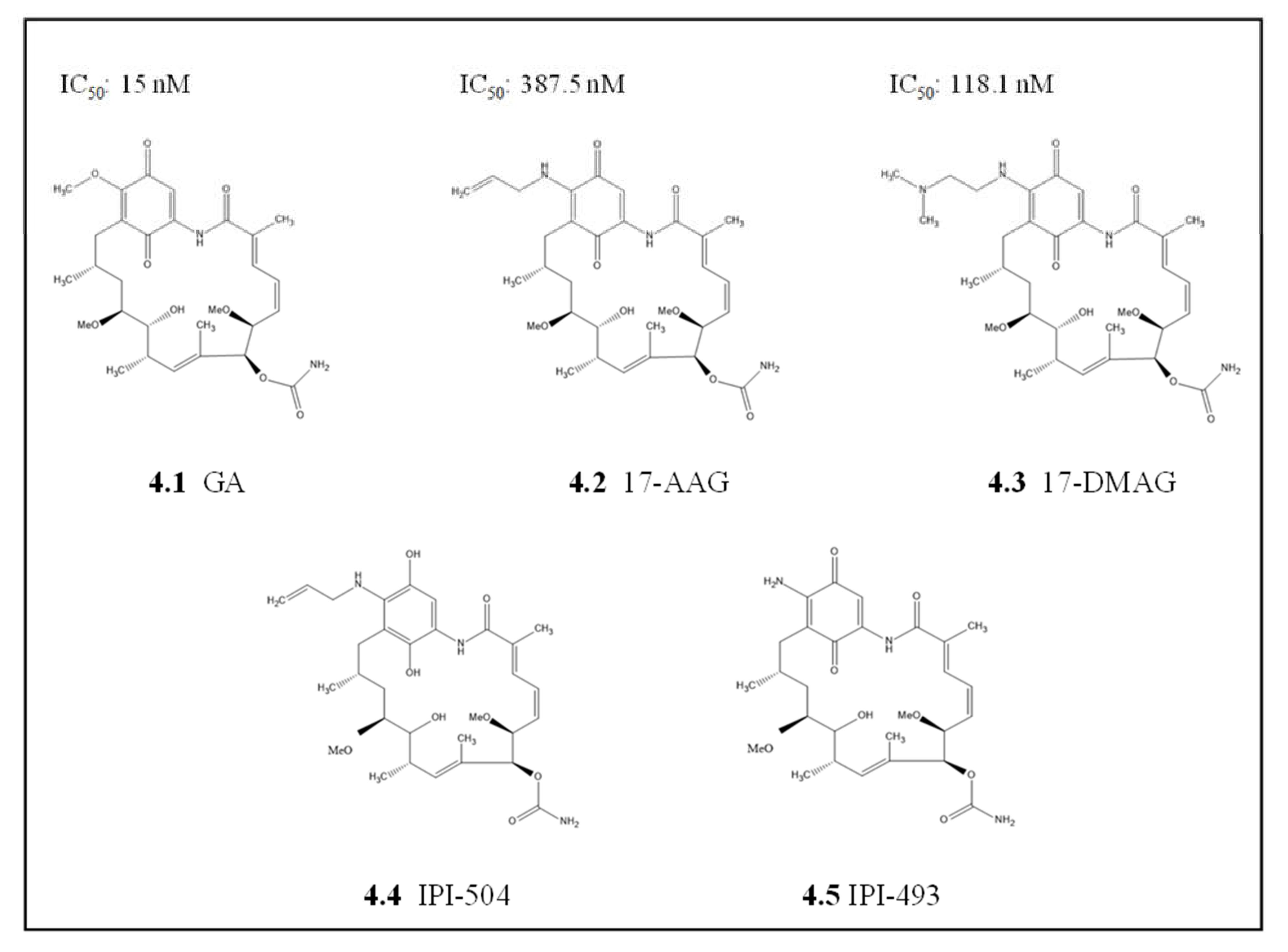
4.1.1. Geldanamycin and Its Derivatives
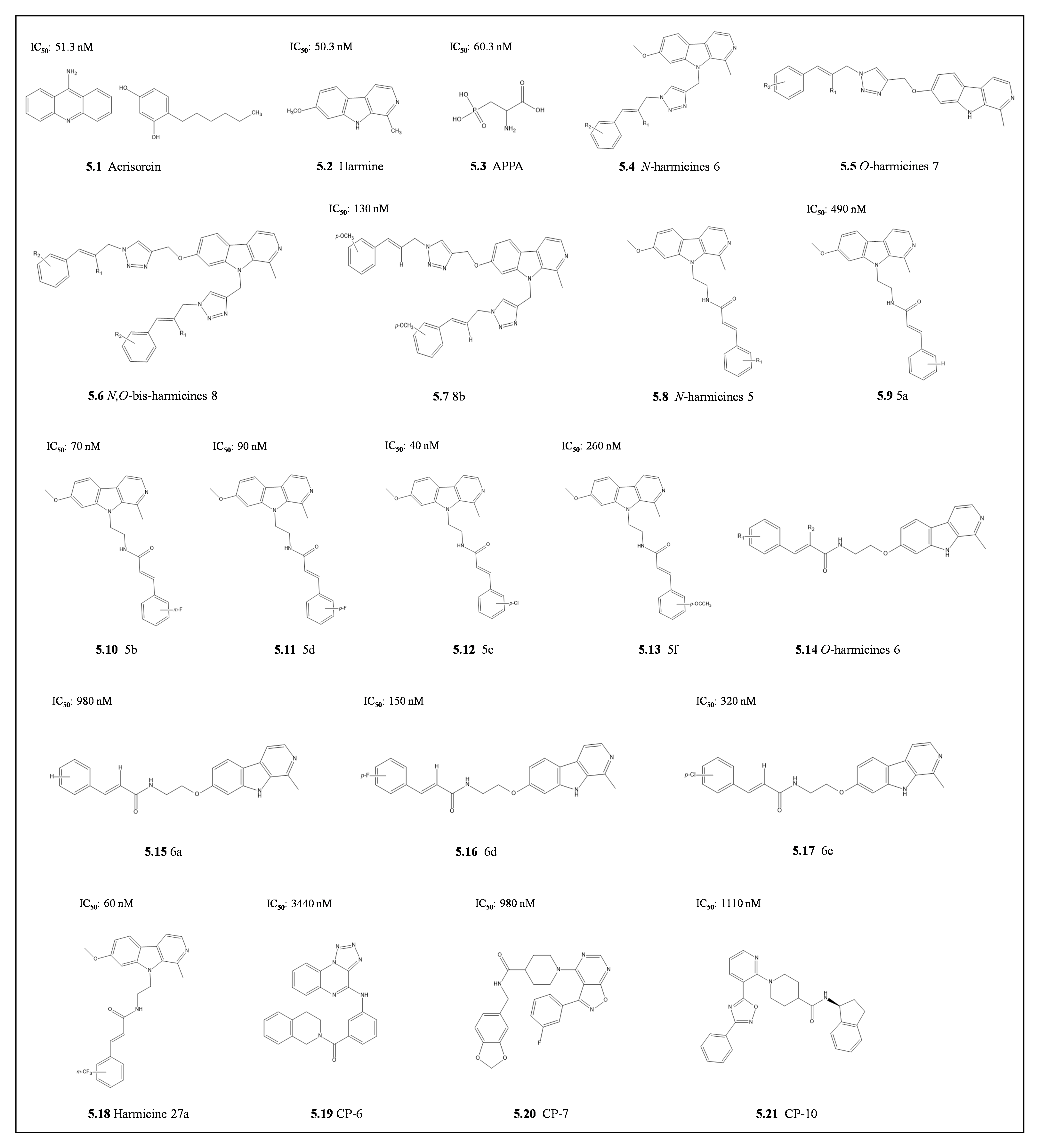
4.1.2. Harmine and Its Derivatives
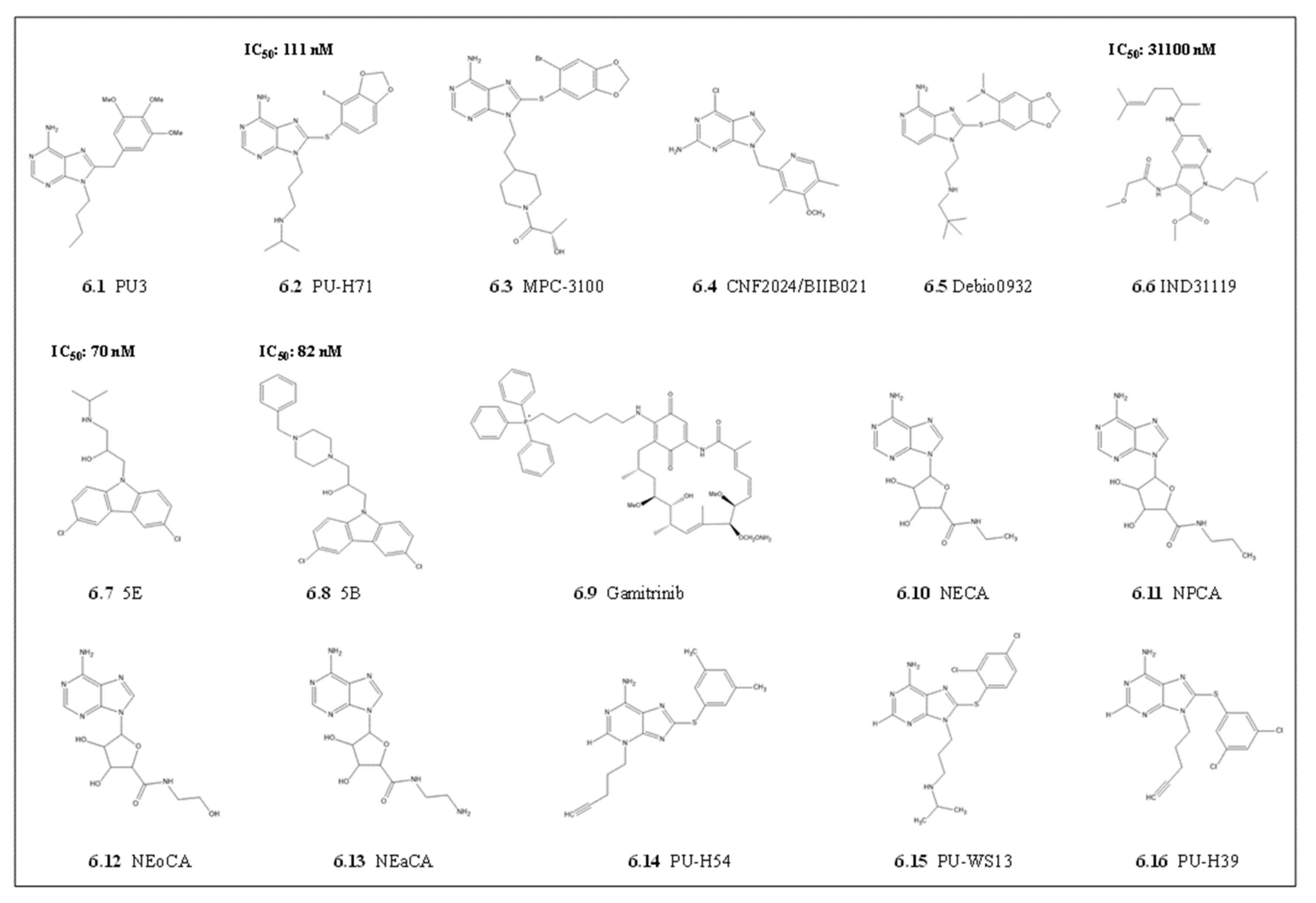
4.1.3. Purine Scaffolds
4.1.4. Other In Silico Derived Scaffolds
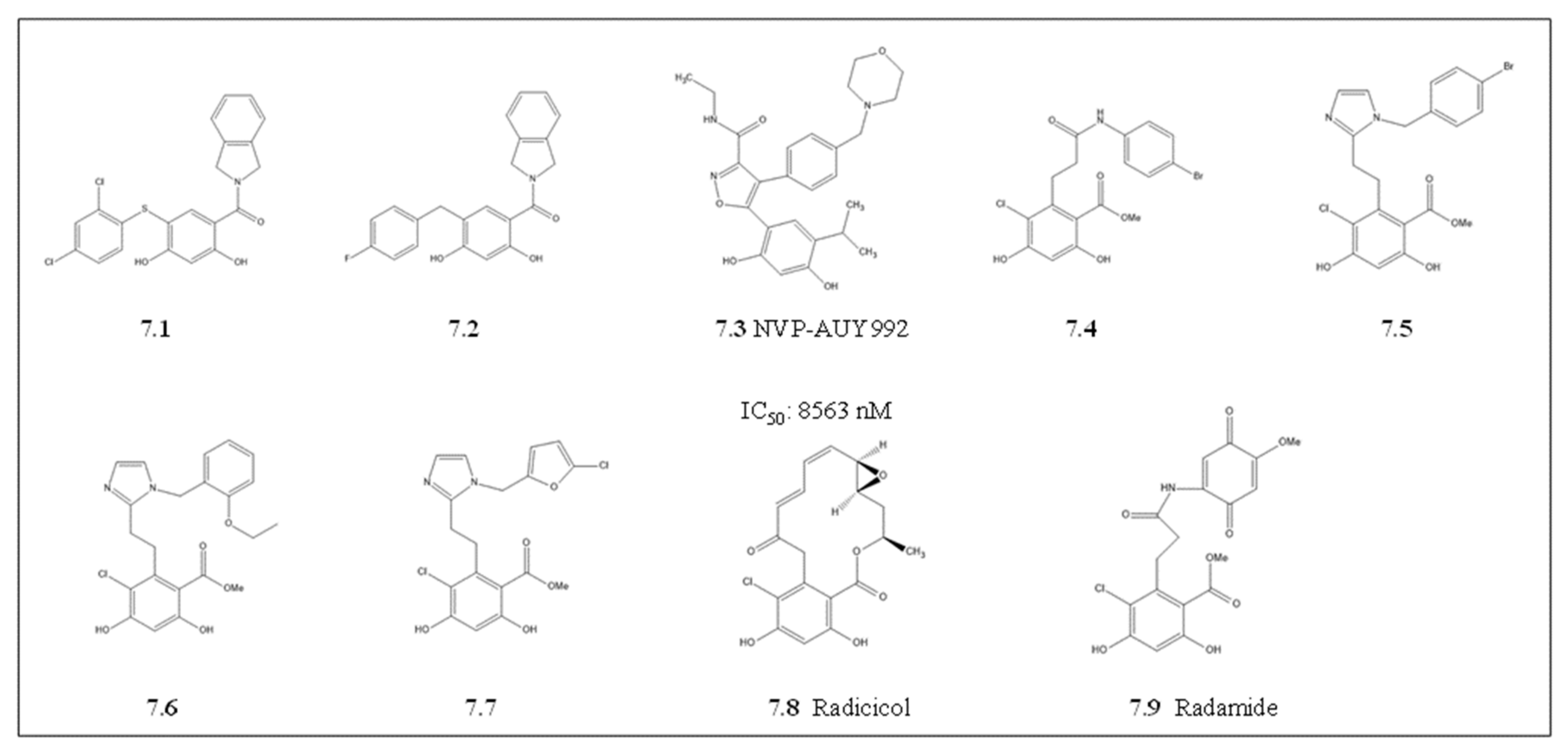
4.1.5. Radicicol and Its Derivatives
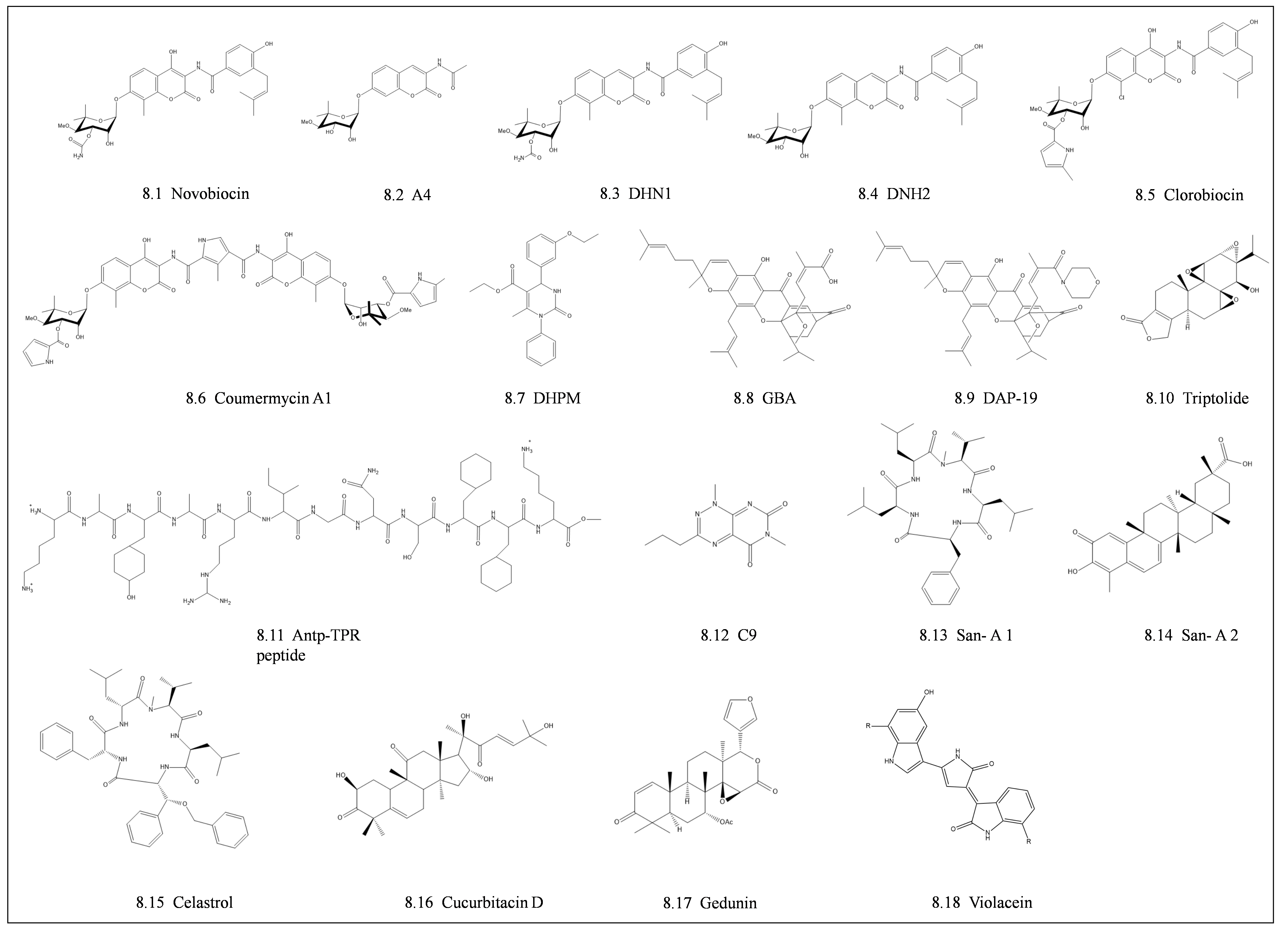
4.2. Middle Domain and C-Terminal Domain Inhibitors
4.3. Inhibitors Targeting Co-Chaperone Interactions
5. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. World Malaria Report, 2020. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2020 (accessed on 23 September 2021).
- Behzad, N.; Behrens, R.H. Malaria: An update for physicians. Infect. Dis. Clin. N. Am. 2012, 26, 243–259. [Google Scholar] [CrossRef]
- Langlois, A.-C.; Manzoni, G.; Vincensini, L.; Coppée, R.; Marinach, C.; Guérin, M.; Huby, T.; Carrière, V.; Cosset, F.-L.; Dreux, M.; et al. Molecular determinants of SR-B1-dependent Plasmodium sporozoite entry into hepatocytes. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Warrell, D.A.; Gilles, H.M. Essential Malariology, 4th ed.; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Malaguarnera, L.; Musumeci, S. The immune response to Plasmodium falciparum malaria. Lancet Infect. Dis. 2002, 2, 472–478. [Google Scholar] [CrossRef]
- Belachew, E.B. Immune Response and Evasion Mechanisms of Plasmodium falciparum Parasites. J. Immunol. Res. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.; Munguti, K.; et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Pavithra, S.R.; Banumathy, G.; Joy, O.; Singh, V.; Tatu, U. Recurrent Fever Promotes Plasmodium falciparum Development in Human Erythrocytes. J. Biol. Chem. 2004, 279, 46692–46699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tintó-Font, E.; Michel-Todó, L.; Russell, T.J.; Casas-Vila, N.; Conway, D.J.; Bozdech, Z.; Llinás, M.; Cortés, A. A heat-shock response regulated by the PfAP2-HS transcription factor protects human malaria parasites from febrile temperatures. Nat. Microbiol. 2021, 6, 1163–1174. [Google Scholar]
- Shahinas, D.; Folefoc, A.; Taldone, T.; Chiosis, G.; Crandall, I.; Pillai, D.R. A Purine Analog Synergizes with Chloroquine (CQ) by Targeting Plasmodium falciparum Hsp90 (PfHsp90). PLoS ONE 2013, 8, e75446. [Google Scholar] [CrossRef] [Green Version]
- Su, X.Z.; Kirkman, L.A.; Fujioka, H.; Wellems, T.E. Complex polymorphisms in an ∼330 kDa protein are linked to chloroquine-resistant P. falciparum in Southeast Asia and Africa. Cell 1997, 91, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Zininga, T. Characterization of heat shock protein 70-z (PfHsp70-z) from Plasmodium falciparum. Doctoral Dissertation, University of Venda, Venda, South Africa, 2015. Available online: https://univendspace.univen.ac.za/handle/11602/619 (accessed on 23 September 2021).
- Spiegelberg, D.; Abramenkovs, A.; Mortensen, A.C.J.; Lundsten, S.; Nestor, M.; Stenerlöw, B. The HSP90 inhibitor Onalespib exerts synergistic anti-cancer effects when combined with radiotherapy: An in vitro and in vivo approach. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, F.H.S.M.M.B.J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Honoré, F.; Méjean, V.; Genest, O. Hsp90 Is Essential under Heat Stress in the Bacterium Shewanella oneidensis. Cell Rep. 2017, 19, 680–687. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar] [CrossRef] [PubMed]
- Dollins, D.E.; Warren, J.J.; Immormino, R.M.; Gewirth, D.T. Structures of GRP94-Nucleotide Complexes Reveal Mechanistic Differences between the hsp90 Chaperones. Mol. Cell 2007, 28, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearl, L.H.; Prodromou, C. Structure and in vivo function of Hsp90. Curr. Opin. Struct. Biol. 2000, 10, 46–51. [Google Scholar] [CrossRef]
- López, A.; Elimelech, A.R.; Klimm, K.; Sattler, M. The Charged Linker Modulates the Conformations and Molecular Interactions of Hsp90. ChemBioChem 2020, 22, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.L.; Lopez, A.; Lawatscheck, J.; Luo, Q.; Rutz, D.A.; Gamiz-Hernandez, A.P.; Sattler, M.; Buchner, J.; Kaila, V.R.I. Conformational dynamics modulate the catalytic activity of the molecular chaperone Hsp90. Nat. Commun. 2020, 11, 1410–1412. [Google Scholar] [CrossRef]
- Perez-Riba, A.; Itzhaki, L.S. The tetratricopeptide-repeat motif is a versatile platform that enables diverse modes of molecular recognition. Curr. Opin. Struct. Biol. 2019, 54, 43–49. [Google Scholar] [CrossRef]
- Newstead, S.; Barr, F. Molecular basis for KDEL-mediated retrieval of escaped ER-resident proteins—SWEET talking the COPs. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Chiosis, G.; Dickey, C.A.; Johnson, J.L. A global view of Hsp90 functions. Nat. Struct. Mol. Biol. 2013, 20, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, L.; You, Q.-D.; Xu, X.-L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2019, 63, 1798–1822. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C. Structure and Functional Relationships of Hsp90. Curr. Cancer Drug Targets 2003, 3, 301–323. [Google Scholar] [CrossRef]
- Silva, N.S.; Torricillas, M.S.; Minari, K.; Barbosa, L.R.; Seraphim, T.V.; Borges, J.C. Solution structure of Plasmodium falciparum Hsp90 indicates a high flexible dimer. Arch. Biochem. Biophys. 2020, 690, 108468. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Lee, B.L.; Wajda, B.; Spyracopoulos, L. Nucleotide Binding and Active Site Gate Dynamics for the Hsp90 Chaperone ATPase Domain from Benchtop and High Field 19F NMR Spectroscopy. J. Phys. Chem. B 2020, 124, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- Radli, M.; Rüdiger, S.G. Dancing with the Diva: Hsp90–Client Interactions. J. Mol. Biol. 2018, 430, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Shahinas, D.; Pillai, D.R. Role of Hsp90 in Plasmodium falciparum malaria. In Heat Shock Proteins of Malaria; Springer: Dordrecht, The Netherlands, 2021; pp. 125–140. [Google Scholar]
- Gewirth, D.T. Paralog Specific Hsp90 Inhibitors—A Brief History and a Bright Future. Curr. Top. Med. Chem. 2016, 16, 2779–2791. [Google Scholar] [CrossRef]
- Zininga, T.; Shonhai, A. Small Molecule Inhibitors Targeting the Heat Shock Protein System of Human Obligate Protozoan Parasites. Int. J. Mol. Sci. 2019, 20, 5930. [Google Scholar] [CrossRef] [Green Version]
- Acharya, P.; Kumar, R.; Tatu, U. Chaperoning a cellular upheaval in malaria: Heat shock proteins in Plasmodium falciparum. Mol. Biochem. Parasitol. 2007, 153, 85–94. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Mollapour, M.; Prodromou, C.; Lee, C.-T.; Panaretou, B.; Yoshida, S.; Mayer, M.; Neckers, L.M. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. USA 2012, 109, 2937–2942. [Google Scholar] [CrossRef] [Green Version]
- Corbett, K.; Berger, J.M. Structure of the ATP-binding domain of Plasmodium falciparum Hsp90. Proteins: Struct. Funct. Bioinform. 2010, 78, 2738–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, K.; Saito, H.; Nagao, H. Decomposition analysis of free energy profile for Hsp90-ADP association. Mol. Simul. 2015, 42, 896–901. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, C.; Chen, W.; Xu, Y.; Shi, Y.; Wen, Y.; Zhang, N. A Dynamic View of ATP-coupled Functioning Cycle of Hsp90 N-terminal Domain. Sci. Rep. 2015, 5, srep09542. [Google Scholar] [CrossRef] [Green Version]
- Seraphim, T.V.; Chakafana, G.; Shonhai, A.; Houry, W.A. Plasmodium falciparum R2TP complex: Driver of parasite Hsp90 function. Biophys. Rev. 2019, 11, 1007–1015. [Google Scholar] [CrossRef]
- Jahn, M.; Rehn, A.; Pelz, B.; Hellenkamp, B.; Richter, K.; Rief, M.; Buchner, J.; Hugel, T. The charged linker of the molecular chaperone Hsp90 modulates domain contacts and biological function. Proc. Natl. Acad. Sci. USA 2014, 111, 17881–17886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, T.O.; Zeng, X.; Pellarin, R.; Bonomi, M.; Sali, A.; Kelly, M.J.; Chu, F.; Agard, D.A. Elucidating the Mechanism of Substrate Recognition by the Bacterial Hsp90 Molecular Chaperone. J. Mol. Biol. 2014, 426, 2393–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Beebe, K.; Chavez, J.D.; Boysen, M.; Lu, Y.; Zuehlke, A.D.; Keramisanou, D.; Trepel, J.B.; Prodromou, C.; Mayer, M.P.; et al. Hsp90 middle domain phosphorylation initiates a complex conformational program to recruit the ATPase-stimulating cochaperone Aha1. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Elnatan, D.; Betegon, M.; Liu, Y.; Ramelot, T.; Kennedy, M.A.; Agard, D.A. Symmetry broken and rebroken during the ATP hydrolysis cycle of the mitochondrial Hsp90 TRAP1. eLife 2017, 6, e25235. [Google Scholar] [CrossRef]
- Moroni, E.; Agard, D.A.; Colombo, G. The Structural Asymmetry of Mitochondrial Hsp90 (Trap1) Determines Fine Tuning of Functional Dynamics. J. Chem. Theory Comput. 2018, 14, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Hawle, P.; Siepmann, M.; Harst, A.; Siderius, M.; Reusch, H.P.; Obermann, W.M.J. The Middle Domain of Hsp90 Acts as a Discriminator between Different Types of Client Proteins. Mol. Cell. Biol. 2006, 26, 8385–8395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, N.S.M.; Bertolino-Reis, D.E.; Dores-Silva, P.R.; Anneta, F.B.; Seraphim, T.V.; Barbosa, L.; Borges, J.C. Structural studies of the Hsp70/Hsp90 organizing protein of Plasmodium falciparum and its modulation of Hsp70 and Hsp90 ATPase activities. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1868, 140282. [Google Scholar] [CrossRef]
- Röhl, A.; Wengler, D.; Madl, T.; Lagleder, S.; Tippel, F.; Herrmann, M.; Hendrix, J.; Richter, K.; Hack, G.; Schmid, A.; et al. Hsp90 regulates the dynamics of its cochaperone Sti1 and the transfer of Hsp70 between modules. Nat. Commun. 2015, 6, 6655. [Google Scholar] [CrossRef] [Green Version]
- Prodromou, C.; Siligardi, G.; O'Brien, R.; Woolfson, D.N.; Regan, L.; Panaretou, B.; Ladbury, J.; Piper, P.W.; Pearl, L. Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J. 1999, 18, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Chua, C.-S.; Low, H.; Sim, T.-S. Co-chaperones of Hsp90 in Plasmodium falciparumand their concerted roles in cellular regulation. Parasitology 2014, 141, 1177–1191. [Google Scholar] [CrossRef]
- Sahasrabudhe, P.; Rohrberg, J.; Biebl, M.M.; Rutz, D.A.; Buchner, J. The Plasticity of the Hsp90 Co-chaperone System. Mol. Cell 2017, 67, 947–961. [Google Scholar] [CrossRef] [Green Version]
- Biebl, M.M.; Riedl, M.; Buchner, J. Hsp90 Co-chaperones Form Plastic Genetic Networks Adapted to Client Maturation. Cell Rep. 2020, 32, 108063. [Google Scholar] [CrossRef]
- Gitau, G.W.; Mandal, P.; Blatch, G.; Przyborski, J.M.; Shonhai, A. Characterisation of the Plasmodium falciparum Hsp70–Hsp90 organising protein (PfHop). Cell Stress Chaperon- 2011, 17, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Makumire, S.; Zininga, T.; Vahokoski, J.; Kursula, I.; Shonhai, A. Biophysical analysis of Plasmodium falciparum Hsp70-Hsp90 organising protein (PfHop) reveals a monomer that is characterised by folded segments connected by flexible linkers. PLoS ONE 2020, 15, e0226657. [Google Scholar] [CrossRef] [PubMed]
- Siligardi, G.; Panaretou, B.; Meyer, P.R.; Singh, S.; Woolfson, D.N.; Piper, P.W.; Pearl, L.; Prodromou, C. Regulation of Hsp90 ATPase Activity by the Co-chaperone Cdc37p/p50. J. Biol. Chem. 2002, 277, 20151–20159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopf, F.H.; Huber, E.M.; Dodt, C.; Lopez, A.; Biebl, M.M.; Rutz, D.A.; Mühlhofer, M.; Richter, G.; Madl, T.; Sattler, M.; et al. The Co-chaperone Cns1 and the Recruiter Protein Hgh1 Link Hsp90 to Translation Elongation via Chaperoning Elongation Factor 2. Mol. Cell 2019, 74, 73–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, M.; Afrin, F.; Tuteja, R. Identification of R2TP complex of Leishmania donovani and Plasmodium falciparum using genome wide in-silico analysis. Commun. Integr. Biol. 2013, 6, e26005. [Google Scholar] [CrossRef] [PubMed]
- Henri, J.; Chagot, M.E.; Bourguet, M.; Abel, Y.; Terral, G.; Maurizy, C.; Aigueperse, C.; Georgescauld, F.; Vandermoere, F.; Saint-Fort, R.; et al. Deep structural analysis of RPAP3 and PIH1D1, two components of the HSP90 co-chaperone R2TP complex. Structure 2018, 26, 1196–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, C.-S.; Low, H.; Goo, K.-S.; Sim, T.S. Characterization of Plasmodium falciparum co-chaperone p23: Its intrinsic chaperone activity and interaction with Hsp90. Cell. Mol. Life Sci. 2010, 67, 1675–1686. [Google Scholar] [CrossRef]
- da Silva, N.S.M.; Seraphim, T.V.; Minari, K.; Barbosa, L.; Borges, J.C. Comparative studies of the low-resolution structure of two p23 co-chaperones for Hsp90 identified in Plasmodium falciparum genome. Int. J. Biol. Macromol. 2018, 108, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.S.; Low, H.; Lehming, N.; Sim, T. Molecular analysis of Plasmodium falciparum co-chaperone Aha1 supports its interaction with and regulation of Hsp90 in the malaria parasite. Int. J. Biochem. Cell Biol. 2012, 44, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Lindenthal, C.; Klinkert, M.-Q. Identification and biochemical characterisation of a Protein Phosphatase 5 homologue from Plasmodium falciparum. Mol. Biochem. Parasitol. 2002, 120, 257–268. [Google Scholar] [CrossRef]
- Zhu, X.; Sun, L.; He, Y.; Wei, H.; Hong, M.; Liu, F.; Liu, Q.; Cao, Y.; Cui, L. Plasmodium berghei serine/threonine protein phosphatase PP5 plays a critical role in male gamete fertility. Int. J. Parasitol. 2019, 49, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Zuehlke, A.; Tenge, V.; Langworthy, J.C. Mutation of essential Hsp90 co-chaperones SGT1 or CNS1 renders yeast hypersensitive to overexpression of other co-chaperones. Curr. Genet. 2014, 60, 265–276. [Google Scholar] [CrossRef]
- Alag, R.; Bharatham, N.; Dong, A.; Hills, T.; Harikishore, A.; Widjaja, A.; Shochat, S.G.; Hui, R.; Yoon, H.S. Crystallographic structure of the tetratricopeptide repeat domain of Plasmodium falciparum FKBP35 and its molecular interaction with Hsp90 C-terminal pentapeptide. Protein Sci. 2009, 18, 2115–2124. [Google Scholar] [CrossRef] [Green Version]
- Bianchin, A.; Allemand, F.; Bell, A.; Chubb, A.J.; Guichou, J.-F. Two crystal structures of the FK506-binding domain of Plasmodium falciparum FKBP35 in complex with rapamycin at high resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1319–1327. [Google Scholar] [CrossRef]
- Gupta, S.; Jadaun, A.; Kumar, V.P.; Raj, U.; Varadwaj, P.; Rao, A.R. Exploration of new drug-like inhibitors for serine/threonine protein phosphatase 5 of Plasmodium falciparum: A docking and simulation study. J. Biomol. Struct. Dyn. 2015, 33, 2421–2441. [Google Scholar] [CrossRef]
- Banumathy, G.; Singh, V.; Pavithra, S.R.; Tatu, U. Heat Shock Protein 90 Function Is Essential for Plasmodium falciparum Growth in Human Erythrocytes. J. Biol. Chem. 2003, 278, 18336–18345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, S.; Kar, B.; Kumar, R.; Adams, B.; Barik, S. A novel tetratricopeptide repeat (TPR) containing PP5 serine/threonine protein phosphatase in the malaria parasite, Plasmodium falciparum. BMC Microbiol. 2001, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, S.K.; Suhre, M.; Wegele, H.; Buchner, J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 2006, 25, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Adams, B.; Musiyenko, A.; Shulyayeva, O.; Barik, S. The FK506-binding protein of the malaria parasite, Plasmodium falciparum, is a FK506-sensitive chaperone with FK506-independent calcineurin-inhibitory activity. Mol. Biochem. Parasitol. 2005, 141, 163–173. [Google Scholar] [CrossRef]
- Shaw, P.E. Peptidyl-prolyl isomerases: A new twist to transcription. EMBO Rep. 2002, 3, 521–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huck, J.D.; Que, N.; Hong, F.; Li, Z.; Gewirth, D.T. Structural and Functional Analysis of GRP94 in the Closed State Reveals an Essential Role for the Pre-N Domain and a Potential Client-Binding Site. Cell Rep. 2017, 20, 2800–2809. [Google Scholar] [CrossRef] [Green Version]
- Biswas, C.; Ostrovsky, O.; Makarewich, C.A.; Wanderling, S.; Gidalevitz, T.; Argon, Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem. J. 2007, 405, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Houry, W.A. Chapter 9—Chaperones and proteases of Plasmodium falciparum. In Heat Shock Proteins of Malaria; Shonhai, A., Blatch, G.L., Eds.; Springer: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- Chu, F.; Maynard, J.C.; Chiosis, G.; Nicchitta, C.V.; Burlingame, A.L. Identification of novel quaternary domain interactions in the Hsp90 chaperone, GRP94. Protein Sci. 2006, 15, 1260–1269. [Google Scholar] [CrossRef] [Green Version]
- Edkins, A.L.; Boshoff, A. Chapter 2—General structural and functional features of molecular chaperones. In Heat Shock Proteins of Malaria; Shonhai, A., Blatch, G.L., Eds.; Springer: New York, NY, USA, 2014; pp. 5–46. [Google Scholar] [CrossRef]
- Florentin, A.; Cobb, D.W.; Kudyba, H.M.; Muralidharan, V. Directing traffic: Chaperone-mediated protein transport in malaria parasites. Cell. Microbiol. 2020, 22, e13215. [Google Scholar] [CrossRef]
- Spillman, N.J.; Beck, J.R.; Goldberg, D.E. Protein Export into Malaria Parasite–Infected Erythrocytes: Mechanisms and Functional Consequences. Annu. Rev. Biochem. 2015, 84, 813–841. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hebert, D.N. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przyborski, J.M.; Nyboer, B.; Lanzer, M. Ticket to ride: Export of proteins to the Plasmodium falciparum-infected erythrocyte. Mol. Microbiol. 2016, 101, 1–11. [Google Scholar]
- Ninagawa, S.; George, G.; Mori, K. Mechanisms of productive folding and endoplasmic reticulum-associated degradation of glycoproteins and non-glycoproteins. Biochim. Biophys. Acta (BBA) Gen. Subj. 2020, 1865, 129812. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, S.; Grover, M.; Tatu, U. Endoplasmic Reticulum Stress Triggers Gametocytogenesis in the Malaria Parasite. J. Biol. Chem. 2014, 289, 16662–16674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo-Solano, C.; Dong, C.; Sanchez, C.G.; Pizarro, J.C. Identification and characterization of the antiplasmodial activity of Hsp90 inhibitors. Malar. J. 2017, 16, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1823, 774–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, J.C.; Pham, T.; Zheng, T.; Jockheck-Clark, A.; Rankin, H.B.; Newgard, C.B.; Spana, E.P.; Nicchitta, C.V. Gp93, the Drosophila GRP94 ortholog, is required for gut epithelial homeostasis and nutrient assimilation-coupled growth control. Dev. Biol. 2010, 339, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Sheiner, L.; Vaidya, A.B.; McFadden, G.I. The metabolic roles of the endosymbiotic organelles of Toxoplasma and Plasmodium spp. Curr. Opin. Microbiol. 2013, 16, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallo, N.; Fellows, J.; Johnson, C.; Sheiner, L. Protein Import into the Endosymbiotic Organelles of Apicomplexan Parasites. Genes 2018, 9, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, N.; Froehlich, J.E.; Zhang, H.; Cheng, C.-L. The chlorate-resistant and photomorphogenesis-defective mutant cr88 encodes a chloroplast-targeted HSP90. Plant J. 2003, 33, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Fan, P.; Jiang, P.; Lv, S.; Chen, X.; Li, Y. Chloroplast-targeted Hsp90 plays essential roles in plastid development and embryogenesis in Arabidopsis possibly linking with VIPP1. Physiol. Plant. 2013, 150, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Kloehn, J.; Lacour, C.E.; Soldati-Favre, D. The metabolic pathways and transporters of the plastid organelle in Apicomplexa. Curr. Opin. Microbiol. 2021, 63, 250–258. [Google Scholar] [CrossRef]
- Sato, S.; Wilson, R. Organelle-specific cochaperonins in apicomplexan parasites. Mol. Biochem. Parasitol. 2005, 141, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Heiny, S.R.; Pautz, S.; Recker, M.; Przyborski, J.M. Protein Traffic to the Plasmodium falciparum Apicoplast: Evidence for a Sorting Branch Point at the Golgi. Traffic 2014, 15, 1290–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro, J.C.; Wernimont, A.K.; Lew, J.; Hutchinson, A.; Artz, J.D.; Amaya, M.F.; Plotnikova, O.; Vedadi, M.; Kozieradzki, I.; Weigelt, J.; et al. Structural Genomics Consortium (SGC)Crystal structure of N-terminal domain of Plasmodium falciparum Hsp90 (PF14_0417) in complex with AMPPN. Available online: https://www.rcsb.org/structure/3IED (accessed on 23 September 2021). [CrossRef]
- Vedadi, M.; Lew, J.; Artz, J.; Amani, M.; Zhao, Y.; Dong, A.; Wasney, G.A.; Gao, M.; Hills, T.; Brokx, S.; et al. Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol. Biochem. Parasitol. 2007, 151, 100–110. [Google Scholar] [CrossRef]
- Pesce, E.-R.; Cockburn, I.; Goble, J.; Stephens, L.; Blatch, G. Malaria heat shock proteins: Drug targets that chaperone other drug targets. Infect. Disord. Drug Targets 2010, 10, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Im, C.-N.; Lee, J.-S.; Zheng, Y.; Seo, J.-S. Iron chelation study in a normal human hepatocyte cell line suggests that tumor necrosis factor receptor-associated protein 1 (TRAP1) regulates production of reactive oxygen species. J. Cell. Biochem. 2006, 100, 474–486. [Google Scholar] [CrossRef]
- Hua, G.; Zhang, Q.; Fan, Z. Heat Shock Protein 75 (TRAP1) Antagonizes Reactive Oxygen Species Generation and Protects Cells from Granzyme M-mediated Apoptosis. J. Biol. Chem. 2007, 282, 20553–20560. [Google Scholar] [CrossRef] [Green Version]
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L.R. TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1823, 767–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, B.-H. TRAP1 regulation of mitochondrial life or death decision in cancer cells and mitochondria-targeted TRAP1 inhibitors. BMB Rep. 2012, 45, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masgras, I.; Sanchez-Martin, C.; Colombo, G.; Rasola, A. The Chaperone TRAP1 As a Modulator of the Mitochondrial Adaptations in Cancer Cells. Front. Oncol. 2017, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.; Dai, L.; Liu, Y.; Lee, J.; Ghahhari, N.M.; Segala, G.; Beebe, K.; Jenkins, L.M.; Lyons, G.C.; Bernasconi, L.; et al. The mitochondrial HSP90 paralog TRAP1 forms an OXPHOS-regulated tetramer and is involved in mitochondrial metabolic homeostasis. BMC Biol. 2020, 18, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elnatan, D.; Agard, D.A. Calcium binding to a remote site can replace magnesium as cofactor for mitochondrial Hsp90 (TRAP1) ATPase activity. J. Biol. Chem. 2018, 293, 13717–13724. [Google Scholar] [CrossRef] [Green Version]
- Whitesell, L.; Shifrin, S.D.; Schwab, G.; Neckers, L.M. Benzoquinonoid ansamycins possess selective tumoricidal activity unrelated to src kinase inhibition. Cancer Res. 1992, 52, 1721–1728. [Google Scholar] [PubMed]
- Chène, P. ATPases as drug targets: Learning from their structure. Nat. Rev. Drug Discov. 2002, 1, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Koren, J.; Blagg, B.S.J. The right tool for the job: An overview of Hsp90 inhibitors. In HSF1 and Molecular Chaperones in Biology and Cancer; Mendillo, M.L., Pincus, D., Scherz-Shouval, R., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 135–146. [Google Scholar] [CrossRef]
- Han, J.; Goldstein, L.A.; Hou, W.; Chatterjee, S.; Burns, T.F.; Rabinowich, H. HSP90 inhibition targets autophagy and induces a CASP9-dependent resistance mechanism in NSCLC. Autophagy 2018, 14, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallavi, R.; Roy, N.; Nageshan, R.K.; Talukdar, P.; Pavithra, S.R.; Reddy, R.; Venketesh, S.; Kumar, R.; Gupta, A.K.; Singh, R.K.; et al. Heat Shock Protein 90 as a Drug Target against Protozoan Infections. J. Biol. Chem. 2010, 285, 37964–37975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zininga, T.; Shonhai, A. Are Heat Shock Proteins Druggable Candidates? Am. J. Biochem. Biotechnol. 2014, 10, 208–210. [Google Scholar] [CrossRef] [Green Version]
- Whitesell, L.; Robbins, N.; Huang, D.S.; McLellan, C.A.; Shekhar-Guturja, T.; Leblanc, E.V.; Nation, C.; Hui, R.; Hutchinson, A.; Collins, C.; et al. Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Vali, S.; Pallavi, R.; Kapoor, S.; Tatu, U. Virtual prototyping study shows increased ATPase activity of Hsp90 to be the key determinant of cancer phenotype. Syst. Synth. Biol. 2009, 4, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, O.W.; Sharma, N.; Reynisson, J.; Leung, I.K. Discovery of novel Hsp90 C-terminal domain inhibitors that disrupt co-chaperone binding. Bioorganic Med. Chem. Lett. 2021, 38, 127857. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-K.; Yoon, N.G.; Lee, J.-E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.-H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huck, J.D.; Que, N.L.S.; Immormino, R.M.; Shrestha, L.; Taldone, T.; Chiosis, G.; Gewirth, D.T. NECA derivatives exploit the paralog-specific properties of the site 3 side pocket of Grp94, the endoplasmic reticulum Hsp90. J. Biol. Chem. 2019, 294, 16010–16019. [Google Scholar] [CrossRef] [PubMed]
- Immormino, R.M.; Dollins, D.E.; Shaffer, P.L.; Soldano, K.L.; Walker, M.A.; Gewirth, D.T. Ligand-induced Conformational Shift in the N-terminal Domain of GRP94, an Hsp90 Chaperone. J. Biol. Chem. 2004, 279, 46162–46171. [Google Scholar] [CrossRef] [Green Version]
- Que, N.L.S.; Crowley, V.M.; Duerfeldt, A.S.; Zhao, J.; Kent, C.N.; Blagg, B.S.J.; Gewirth, D.T. Structure Based Design of a Grp94-Selective Inhibitor: Exploiting a Key Residue in Grp94 To Optimize Paralog-Selective Binding. J. Med. Chem. 2018, 61, 2793–2805. [Google Scholar] [CrossRef]
- Cheung, K.-M.J.; Matthews, T.P.; James, K.; Rowlands, M.G.; Boxall, K.J.; Sharp, S.Y.; Maloney, A.; Roe, M.; Prodromou, C.; Pearl, L.; et al. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorganic Med. Chem. Lett. 2005, 15, 3338–3343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.Z.; Ho, D.H.-H.; Wong, R.H.-F. Triptolide, a HSP90 middle domain inhibitor, induces apoptosis in triple manner. Oncotarget 2018, 9, 22301–22315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidera, K.; Patsavoudi, E. Hsp90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anticancer Drug Discov. 2014, 9, 1–20. [Google Scholar] [PubMed]
- Mak, O.W.; Chand, R.; Reynisson, J.; Leung, I.K.H. Identification of Isoform-Selective Ligands for the Middle Domain of Heat Shock Protein 90 (Hsp90). Int. J. Mol. Sci. 2019, 20, 5333. [Google Scholar] [CrossRef] [Green Version]
- Morra, G.; Neves, M.A.C.; Plescia, C.J.; Tsustsumi, S.; Neckers, L.; Verkhivker, G.; Altieri, D.C.; Colombo, G. Dynamics-Based Discovery of Allosteric Inhibitors: Selection of New Ligands for the C-terminal Domain of Hsp90. J. Chem. Theory Comput. 2010, 6, 2978–2989. [Google Scholar] [CrossRef]
- Bopp, B.; Ciglia, E.; Ouald-Chaib, A.; Groth, G.; Gohlke, H.; Jose, J. Design and biological testing of peptidic dimerization inhibitors of human Hsp90 that target the C-terminal domain. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Posfai, D.; Eubanks, A.L.; Keim, A.I.; Lu, K.-Y.; Wang, G.Z.; Hughes, P.F.; Kato, N.; Haystead, T.A.; Derbyshire, E.R. Identification of Hsp90 Inhibitors with Anti-Plasmodium Activity. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Bisson, W.H.; Mäser, P.; Scapozza, L.; Picard, D. Differences in Conformational Dynamics between Plasmodium falciparum and Human Hsp90 Orthologues Enable the Structure-Based Discovery of Pathogen-Selective Inhibitors. J. Med. Chem. 2014, 57, 2524–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Mäser, P.; Picard, D. Inhibition of Plasmodium falciparum Hsp90 Contributes to the Antimalarial Activities of Aminoalcohol-carbazoles. J. Med. Chem. 2016, 59, 6344–6352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Moser, S.; Hagn, F.; Friedrich, R.; Hainzl, O.; Heller, M.; Schlee, S.; Kessler, H.; Reinstein, J.; Buchner, J. Intrinsic Inhibition of the Hsp90 ATPase Activity. J. Biol. Chem. 2006, 281, 11301–11311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, S.M.; Prodromou, C.; O'Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Zuehlke, A.D.; Moses, M.A.; Neckers, L. Heat shock protein 90: Its inhibition and function. Philos. Trans. R. Soc. B: Biol. Sci. 2017, 373, 20160527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margineantu, D.H.; Emerson, C.B.; Diaz, D.; Hockenbery, D.M. Hsp90 Inhibition Decreases Mitochondrial Protein Turnover. PLoS ONE 2007, 2, e1066. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Ramdhave, A.S.; Patel, D.; Ramya, I.; Nandave, M.; Kharkar, P.S. Targeting heat shock protein 90 for malaria. Mini-Reviews Med. Chem. 2013, 13, 1903–1920. [Google Scholar] [CrossRef] [PubMed]
- Shahinas, D.; Liang, M.; Datti, A.; Pillai, D.R. A Repurposing Strategy Identifies Novel Synergistic Inhibitors of Plasmodium falciparum Heat Shock Protein 90. J. Med. Chem. 2010, 53, 3552–3557. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Musiyenko, A.; Barik, S. Plasmodium falciparum calcineurin and its association with heat shock protein 90: Mechanisms for the antimalarial activity of cyclosporin A and synergism with geldanamycin. Mol. Biochem. Parasitol. 2005, 141, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Musiyenko, A.; Barik, S. The heat shock protein 90 of Plasmodium falciparum and antimalarial activity of its inhibitor, geldanamycin. Malar. J. 2003, 2, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.J.; Caton, E.; Shapiro, T.A. Model System Identifies Kinetic Driver of Hsp90 Inhibitor Activity against African Trypanosomes and Plasmodium falciparum. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuno, A.; Lee, M.-J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. Chaperones 2017, 1709, 423–441. [Google Scholar] [CrossRef]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D'Andrea-Carlino, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 Inhibition Is Effective in Breast Cancer: A Phase II Trial of Tanespimycin (17-AAG) Plus Trastuzumab in Patients with HER2-Positive Metastatic Breast Cancer Progressing on Trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modi, S.; Saura, C.; Henderson, C.; Lin, N.U.; Mahtani, R.; Goddard, J.; Rodenas, E.; Hudis, C.; O’Shaughnessy, J.; Baselga, J. A multicenter trial evaluating retaspimycin HCL (IPI-504) plus trastuzumab in patients with advanced or metastatic HER2-positive breast cancer. Breast Cancer Res. Treat. 2013, 139, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebbins, E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.; Pavletich, N.P. Crystal Structure of an Hsp90–Geldanamycin Complex: Targeting of a Protein Chaperone by an Antitumor Agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Prodromou, C.; Roe, M.; O'Brien, R.; Ladbury, J.; Piper, P.W.; Pearl, L.H. Identification and Structural Characterization of the ATP/ADP-Binding Site in the Hsp90 Molecular Chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Shahinas, D.; MacMullin, G.; Benedict, C.; Crandall, I.; Pillai, D.R. Harmine Is a Potent Antimalarial Targeting Hsp90 and Synergizes with Chloroquine and Artemisinin. Antimicrob. Agents Chemother. 2012, 56, 4207–4213. [Google Scholar] [CrossRef] [Green Version]
- Perković, I.; Raić-Malić, S.; Fontinha, D.; Prudêncio, M.; de Carvalho, L.P.; Held, J.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Harmicines—harmine and cinnamic acid hybrids as novel antiplasmodial hits. Eur. J. Med. Chem. 2019, 187, 111927. [Google Scholar] [CrossRef]
- Marinović, M.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; De Carvalho, L.P.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Novel Harmicines with Improved Potency against Plasmodium. Molecules 2020, 25, 4376. [Google Scholar] [CrossRef] [PubMed]
- Marinović, M.; Poje, G.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; de Carvalho, L.P.; Tandarić, T.; Vianello, R.; Rajić, Z. Further investigation of harmicines as novel antiplasmodial agents: Synthesis, structure-activity relationship and insight into the mechanism of action. Eur. J. Med. Chem. 2021, 224, 113687. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Everson, N.; Bach, J.; Hammill, J.T.; Falade, M.O.; Rice, A.L.; Guy, R.K.; Eagon, S. Identification of Plasmodium falciparum heat shock 90 inhibitors via molecular docking. Bioorganic Med. Chem. Lett. 2021, 35, 127818. [Google Scholar] [CrossRef]
- Wright, L.; Barril, X.; Dymock, B.; Sheridan, L.; Surgenor, A.; Beswick, M.; Drysdale, M.; Collier, A.; Massey, A.; Davies, N.; et al. Structure-Activity Relationships in Purine-Based Inhibitor Binding to HSP90 Isoforms. Chem. Biol. 2004, 11, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Ochiana, S.O.; Taldone, T.; Chiosis, G. Designing drugs against Hsp90 for cancer therapy. In The Molecular Chaperones Interaction Networks in Protein Folding and Degradation; Springer: New York, NY, USA, 2014; pp. 151–183. [Google Scholar]
- Patel, P.D.; Yan, P.; Seidler, P.M.; Patel, H.J.; Sun, W.; Yang, C.; Que, N.; Taldone, T.; Finotti, P.; Stephani, R.A.; et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol. 2013, 9, 677–684. [Google Scholar] [CrossRef]
- Kang, B.H.; Altieri, D.C. Compartmentalized cancer drug discovery targeting mitochondrial Hsp90 chaperones. Oncogene 2009, 28, 3681–3688. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Park, H.-K.; Jeong, H.; Lim, J.; Lee, A.-J.; Cheon, K.Y.; Kim, C.-S.; Thomas, A.P.; Bae, B.; Kim, N.D.; et al. Development of a Mitochondria-Targeted Hsp90 Inhibitor Based on the Crystal Structures of Human TRAP1. J. Am. Chem. Soc. 2015, 137, 4358–4367. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Siegelin, M.D.; Plescia, J.; Raskett, C.M.; Garlick, D.S.; Dohi, T.; Lian, J.B.; Stein, G.S.; Languino, L.; Altieri, D.C. Preclinical Characterization of Mitochondria-Targeted Small Molecule Hsp90 Inhibitors, Gamitrinibs, in Advanced Prostate Cancer. Clin. Cancer Res. 2010, 16, 4779–4788. [Google Scholar] [CrossRef] [Green Version]
- Schulte, T.W.; Akinaga, S.; Soga, S.; Sullivan, W.; Stensgard, B.; Toft, D.; Neckers, L.M. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998, 3, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Dehner, A.; Furrer, J.; Richter, K.; Schuster, I.; Buchner, J.; Kessler, H. NMR Chemical Shift Perturbation Study of the N-Terminal Domain of Hsp90 upon Binding of ADP, AMP-PNP, Geldanamycin, and Radicicol. ChemBioChem 2003, 4, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.; Pettit, S.N.; Magnolo, S.K.; Sanvoisin, J.; Chen, W.; Wood, S.P.; Freeman, L.D.; Pengelly, R.J.; Hughes, D.E. Fragment Screening Using Capillary Electrophoresis (CEfrag) for Hit Identification of Heat Shock Protein 90 ATPase Inhibitors. J. Biomol. Screen. 2012, 17, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Dutton, B.L.; Kitson, R.R.A.; Parry-Morris, S.; Roe, S.M.; Prodromou, C.; Moody, C.J. Synthesis of macrolactam analogues of radicicol and their binding to heat shock protein Hsp90. Org. Biomol. Chem. 2014, 12, 1328–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proisy, N.; Sharp, S.Y.; Boxall, K.; Connelly, S.; Roe, M.; Prodromou, C.; Slawin, A.; Pearl, L.; Workman, P.; Moody, C. Inhibition of Hsp90 with Synthetic Macrolactones: Synthesis and Structural and Biological Evaluation of Ring and Conformational Analogs of Radicicol. Chem. Biol. 2006, 13, 1203–1215. [Google Scholar] [CrossRef] [Green Version]
- Chalapareddy, S.; Bhattacharyya, M.K.; Mishra, S.; Bhattacharyya, S. Radicicol Confers Mid-Schizont Arrest by Inhibiting Mitochondrial Replication in Plasmodium falciparum. Antimicrob. Agents Chemother. 2014, 58, 4341–4352. [Google Scholar] [CrossRef] [Green Version]
- Chalapareddy, S.; Chakrabarty, S.; Bhattacharyya, M.K.; Bhattacharyya, S. Radicicol-Mediated Inhibition of Topoisomerase VIB-VIA Activity of the Human Malaria Parasite Plasmodium falciparum. mSphere 2016, 1, e00025-15. [Google Scholar] [CrossRef] [Green Version]
- Bansod, S.; Raj, N.; Nair, A.S.; Bhattacharyya, S. Molecular docking and molecular dynamics simulation identify a novel Radicicol derivative that predicts exclusive binding to Plasmodium falciparum Topoisomerase VIB. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, A.; Crowley, V.M.; Blagg, B.S.J. Resorcinol-Based Grp94-Selective Inhibitors. ACS Med. Chem. Lett. 2017, 8, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.J.; Ghosh, S.; Stothert, A.R.; Dickey, C.A.; Blagg, B.S.J. Transformation of the Non-Selective Aminocyclohexanol-Based Hsp90 Inhibitor into a Grp94-Seletive Scaffold. ACS Chem. Biol. 2016, 12, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, V.M.; Khandelwal, A.; Mishra, S.; Stothert, A.R.; Huard, D.J.E.; Zhao, J.; Muth, A.; Duerfeldt, A.S.; Kizziah, J.L.; Lieberman, R.L.; et al. Development of Glucose Regulated Protein 94-Selective Inhibitors Based on the BnIm and Radamide Scaffold. J. Med. Chem. 2016, 59, 3471–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerfeldt, A.S.; Peterson, L.B.; Maynard, J.C.; Ng, C.L.; Eletto, D.; Ostrovsky, O.; Shinogle, H.E.; Moore, D.S.; Argon, Y.; Nicchitta, C.V.; et al. Development of a Grp94 inhibitor. J. Am. Chem. Soc. 2012, 134, 9796–9804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magwenyane, A.M.; Mhlongo, N.N.; Lawal, M.M.; Amoako, D.G.; Somboro, A.M.; Sosibo, S.C.; Shunmugam, L.; Khan, R.B.; Kumalo, H.M. Understanding the Hsp90 N-Terminal Dynamics: Structural and Molecular Insights into the Therapeutic Activities of Anticancer Inhibitors Radicicol (RD) and Radicicol Derivative (NVP-YUA922). Molecules 2020, 25, 1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terracciano, S.; Foglia, A.; Chini, M.G.; Vaccaro, M.C.; Russo, A.; Piaz, F.D.; Saturnino, C.; Riccio, R.; Bifulco, G.; Bruno, I. New dihydropyrimidin-2(1H)-one based Hsp90 C-terminal inhibitors. RSC Adv. 2016, 6, 82330–82340. [Google Scholar] [CrossRef]
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and Related Coumarins and Depletion of Heat Shock Protein 90-Dependent Signaling Proteins. J. Natl. Cancer Inst. 2000, 92, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, C.; Lafitte, D.; Tsvetkov, P.O.; Barbier, P.; Leclerc-Devin, J.; Millot, J.-M.; Briand, C.; Makarov, A.A.; Catelli, M.G.; Peyrot, V. Binding of ATP to Heat Shock Protein 90. J. Biol. Chem. 2002, 277, 12208–12214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.M.; Shen, G.; Neckers, L.; Blake, H.; Holzbeierlein, J.; Cronk, B.; Blagg, B.S.J. Hsp90 Inhibitors Identified from a Library of Novobiocin Analogues. J. Am. Chem. Soc. 2005, 127, 12778–12779. [Google Scholar] [CrossRef] [PubMed]
- Burlison. J.A.; Neckers,.L.; Smith,.A.B.; Maxwell,.A.A.; Blagg, B.S.J. Novobiocin: Redesigning a DNA Gyrase Inhibitor for Selective Inhibition of Hsp90. J. Am. Chem. Soc. 2006, 128, 15529–15536. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, A.; Blagg, B.S.J. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 2008, 15, 2517–2702. [Google Scholar] [CrossRef] [Green Version]
- Strocchia, M.; Terracciano, S.; Chini, M.G.; Vassallo, A.; Vaccaro, M.C.; Piaz, F.D.; Leone, A.; Riccio, R.; Bruno, I.; Bifulco, G. Targeting the Hsp90 C-terminal domain by the chemically accessible dihydropyrimidinone scaffold. Chem. Commun. 2015, 51, 3850–3853. [Google Scholar] [CrossRef] [PubMed]
- Yim, K.; Prince, T.L.; Qu, S.; Bai, F.; Jennings, P.A.; Onuchic, J.N.; Theodorakis, E.A.; Neckers, L. Gambogic acid identifies an isoform-specific druggable pocket in the middle domain of Hsp90β. Proc. Natl. Acad. Sci. USA 2016, 113, E4801–E4809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q. Triptolide and its expanding multiple pharmacological functions. Int. Immunopharmacol. 2011, 11, 377–383. [Google Scholar] [CrossRef]
- Yi, F.; Zhu, P.; Southall, N.; Inglese, J.; Austin, C.P.; Zheng, W.; Regan, L. An AlphaScreenTM-Based High-Throughput Screen to Identify Inhibitors of Hsp90-Cochaperone Interaction. J. Biomol. Screen. 2009, 14, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Horibe, T.; Kohno, M.; Haramoto, M.; Ohara, K.; Kawakami, K. Designed hybrid TPR peptide targeting Hsp90 as a novel anticancer agent. J. Transl. Med. 2011, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Pimienta, G.; Herbert, K.M.; Regan, L. A Compound That Inhibits the HOP–Hsp90 Complex Formation and Has Unique Killing Effects in Breast Cancer Cell Lines. Mol. Pharm. 2011, 8, 2252–2261. [Google Scholar] [CrossRef]
- Vasko, R.C.; Rodriguez, R.A.; Cunningham, C.N.; Ardi, V.C.; Agard, D.A.; McAlpine, S.R. Mechanistic Studies of Sansalvamide A-Amide: An Allosteric Modulator of Hsp90. ACS Med. Chem. Lett. 2010, 1, 4–8. [Google Scholar] [CrossRef]
- Chadli, A.; Felts, S.J.; Wang, Q.; Sullivan, W.P.; Botuyan, M.V.; Fauq, A.; Ramirez-Alvarado, M.; Mer, G. Celastrol Inhibits Hsp90 Chaperoning of Steroid Receptors by Inducing Fibrillization of the Co-chaperone p23. J. Biol. Chem. 2010, 285, 4224–4231. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.A.; Seedarala, S.; Rice, N.; Kopel, L.; Halaweish, F.; Blagg, B.S.J. Cucurbitacin D Is a Disruptor of the HSP90 Chaperone Machinery. J. Nat. Prod. 2015, 78, 873–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patwardhan, C.A.; Fauq, A.; Peterson, L.B.; Miller, C.; Blagg, B.S.J.; Chadli, A. Gedunin Inactivates the Co-chaperone p23 Protein Causing Cancer Cell Death by Apoptosis. J. Biol. Chem. 2013, 288, 7313–7325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panaretou, B.; Siligardi, G.; Meyer, P.R.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.; Clarke, P.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the ATPase Activity of Hsp90 by the Stress-Regulated Cochaperone Aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Stiegler, S.C.; Rübbelke, M.; Korotkov, V.S.; Weiwad, M.; John, C.; Fischer, G.; Sieber, S.A.; Sattler, M.; Buchner, J. A chemical compound inhibiting the Aha1–Hsp90 chaperone complex. J. Biol. Chem. 2017, 292, 17073–17083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, D.; Woodford, M.; Truman, A.; Jensen, S.M.; Schulman, J.; Caza, T.; Remillard, T.C.; Loiselle, D.; Wolfgeher, D.; Blagg, B.S.; et al. c-Abl Mediated Tyrosine Phosphorylation of Aha1 Activates Its Co-chaperone Function in Cancer Cells. Cell Rep. 2015, 12, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, A.; Rezaei-Tavirani, M.; Farhadihosseinabadi, B.; Taranejoo, S.; Zali, H. HSP90 and Co-chaperones: Impact on Tumor Progression and Prospects for Molecular-Targeted Cancer Therapy. Cancer Investig. 2020, 38, 310–328. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bansal, A.; Hashimoto-Torii, K. HSP70 and HSP90 in neurodegenerative diseases. Neurosci. Lett. 2019, 716, 134678. [Google Scholar] [CrossRef] [PubMed]
- Tavella, T.A.; da Silva, N.S.M.; Spillman, N.; Kayano, A.C.A.V.; Cassiano, G.C.; Vasconcelos, A.A.; Camargo, A.P.; da Silva, D.C.B.; Fontinha, D.; Alvarez, L.C.S.; et al. Violacein-Induced Chaperone System Collapse Underlies Multistage Antiplasmodial Activity. ACS Infect. Dis. 2021, 7, 759–776. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.L.; Rashid, S.; Wajda, B.; Wolmarans, A.; LaPointe, P.; Spyracopoulos, L. The Hsp90 Chaperone: 1H and 19F Dynamic Nuclear Magnetic Resonance Spectroscopy Reveals a Perfect Enzyme. Biochemistry 2019, 58, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Pallares, I.; de Groot, N.S.; Iglesias, V.; Sant’Anna, R.; Biosca, A.; Fernàndez-Busquets, X.; Ventura, S. Discovering Putative Prion-Like Proteins in Plasmodium falciparum: A Computational and Experimental Analysis. Front. Microbiol. 2018, 9, 1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajapandi, T. Chaperoning of asparagine repeat-containing proteins in Plasmodium falciparum. J. Parasit. Dis. 2020, 44, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Mabonga, L.; Kappo, A.P. Peptidomimetics: A synthetic tool for inhibiting protein–protein interactions in cancer. Int. J. Pept. Res. Ther. 2020, 26, 225–241. [Google Scholar] [CrossRef] [Green Version]
- Helton, L.G.; Kennedy, E.J. Targeting Plasmodium with constrained peptides and peptidomimetics. IUBMB Life 2020, 72, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Eotvos, L.; Wade, J.D. Current challenges in peptide-based drug discovery. Front. Chem. 2014, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Basore, K.; Cheng, Y.; Kushwaha, A.K.; Nguyen, S.T.; Desai, S.A. How do antimalarial drugs reach their intracellular targets? Front. Pharmacol. 2015, 6, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, L.; Knölker, H.; Simons, K. Subcellular targeting strategies for drug design and delivery. Nat. Rev. Drug Discov. 2010, 9, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Shadrack, D.M.; Swai, H.S.; Hassanali, A. A computational study on the role of water and conformational fluctuations in Hsp90 in response to inhibitors. J. Mol. Graph. Model. 2019, 96, 107510. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Co-Chaperone | P. falciparum Homolog | Function | References |
|---|---|---|---|
| Hop | PfHop (PF3D7_1434300) | Adaptor for Hsp70 and Hsp90; inhibitor of ATPase function | [44,45,50,51] |
| Cdc37 | Kinase-specific co-chaperone | [52] | |
| TTC4 | Interaction with Cpr7/Cyp40 | [53] | |
| Tah1 | PfRPAP3/PfTah1 (PF3D7_0213500) | Component of Rvb1-Rvb2-Tah1-Pih1 (R2TP) complex | [54] |
| Pih1 | PfPih1 (PF3D7_1235000) | Component of (R2TP) complex | [37,54,55] |
| Cyp40 | PF3D7_1111800 | Peptidylprolyl-cis/trans-isomerase | [48] |
| P23 | Pfp23A (PF3D7_1453700) Pf23B (PF3D7_0927000) | Inhibits ATPase | [56,57] |
| Aha1 | PfAha1 (PF3D7_0306200) | ATPase activator | [48,58] |
| PfAha1 (PF3D7_1334200) | ATPase activator | ||
| PP5 | PfPP5 (PF3D7_1355500) | Phosphatase | [59,60] |
| Sgt1 | PfCBP (PF3D7_0933200) | kinetochore assembly | [61] |
| FKBP38 | PfFKBP35 (PF3D7_1247400) | Peptidylprolyl-cis/trans-isomerase | [62,63] |
| PfCns1 (PF3D7_1108900) | [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stofberg, M.L.; Caillet, C.; de Villiers, M.; Zininga, T. Inhibitors of the Plasmodium falciparum Hsp90 towards Selective Antimalarial Drug Design: The Past, Present and Future. Cells 2021, 10, 2849. https://doi.org/10.3390/cells10112849
Stofberg ML, Caillet C, de Villiers M, Zininga T. Inhibitors of the Plasmodium falciparum Hsp90 towards Selective Antimalarial Drug Design: The Past, Present and Future. Cells. 2021; 10(11):2849. https://doi.org/10.3390/cells10112849
Chicago/Turabian StyleStofberg, Melissa Louise, Celine Caillet, Marianne de Villiers, and Tawanda Zininga. 2021. "Inhibitors of the Plasmodium falciparum Hsp90 towards Selective Antimalarial Drug Design: The Past, Present and Future" Cells 10, no. 11: 2849. https://doi.org/10.3390/cells10112849