
Unzipping the Secrets of Amyloid Disassembly by the Human Disaggregase

 , , , and
, , , and 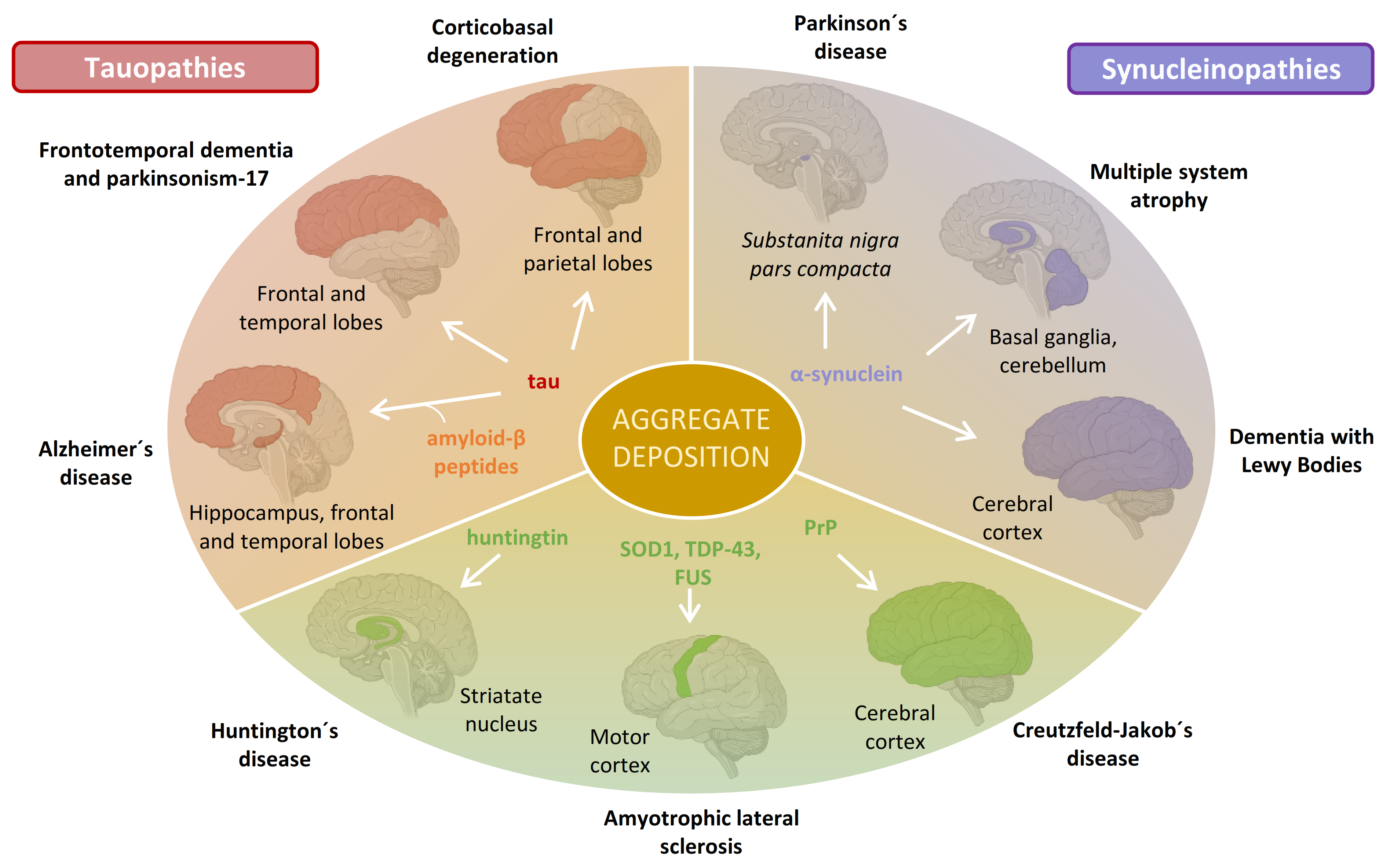{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Neurodegeneration and Amyloid Deposition
2. Synucleinopathies and α-Synuclein Amyloid Formation

3. Disaggregation of α-Synuclein Amyloids by Molecular Chaperones
3.1. Hsp104 Disaggregase
3.2. The Human Hsp70-Based Disaggregase System
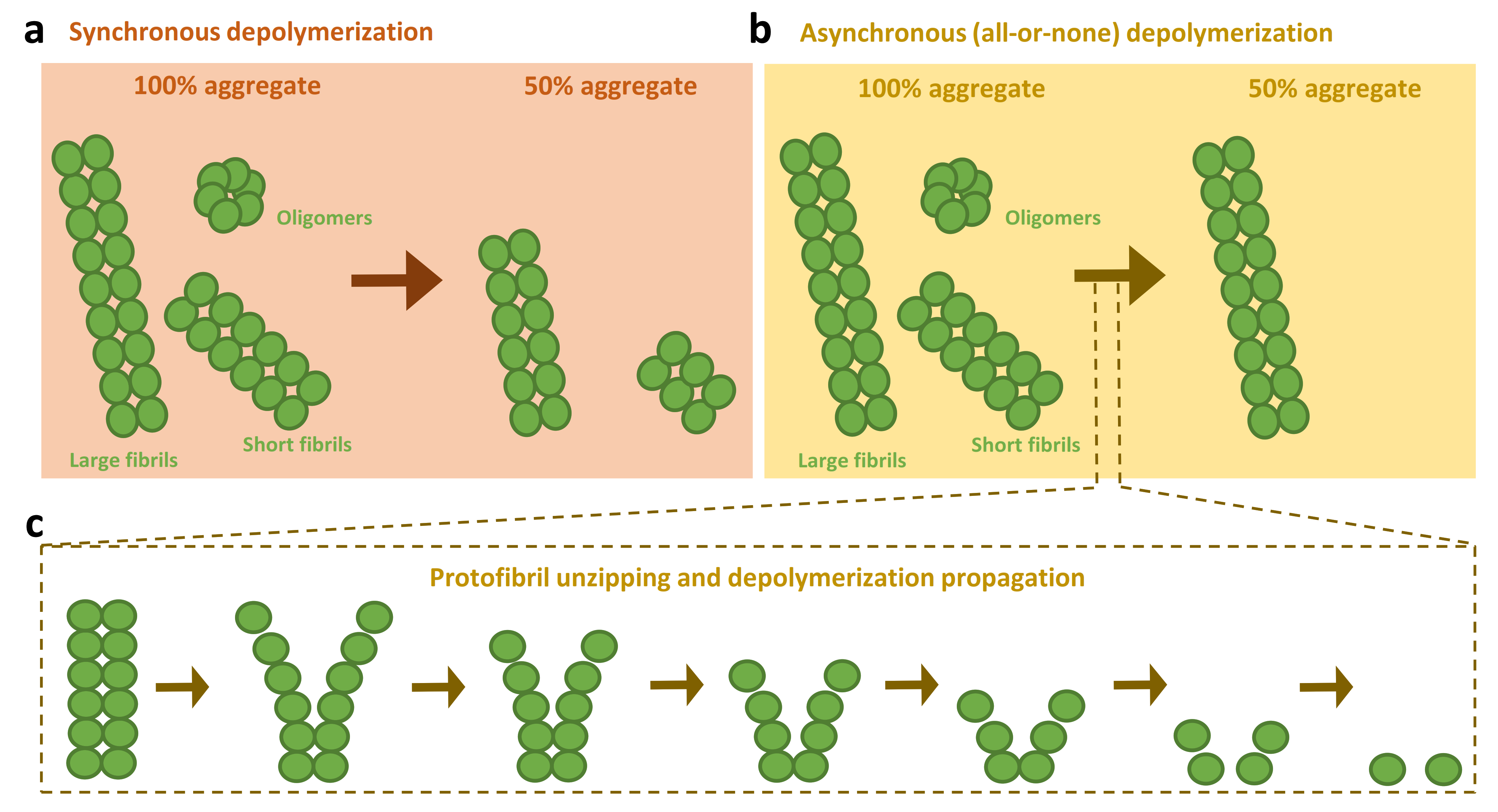
3.3. Models for the Disassembly of Amyloids by the Human Disaggregase

4. Expanding the Human Disaggregase Activity
4.1. Chaperone-Mediated Disassembly of Other Amyloidogenic Proteins
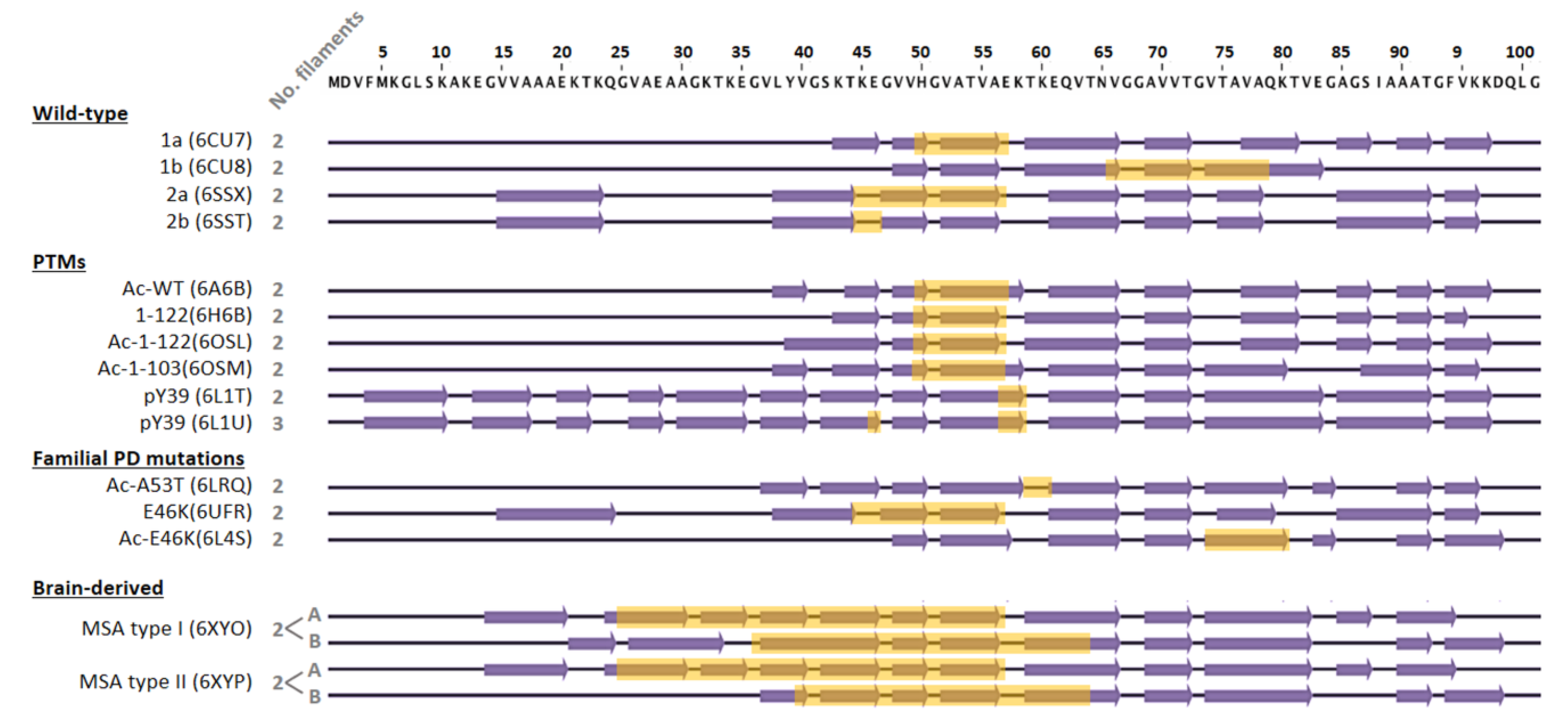
4.2. Impact of Amyloid Polymorphism in Chaperone Activity

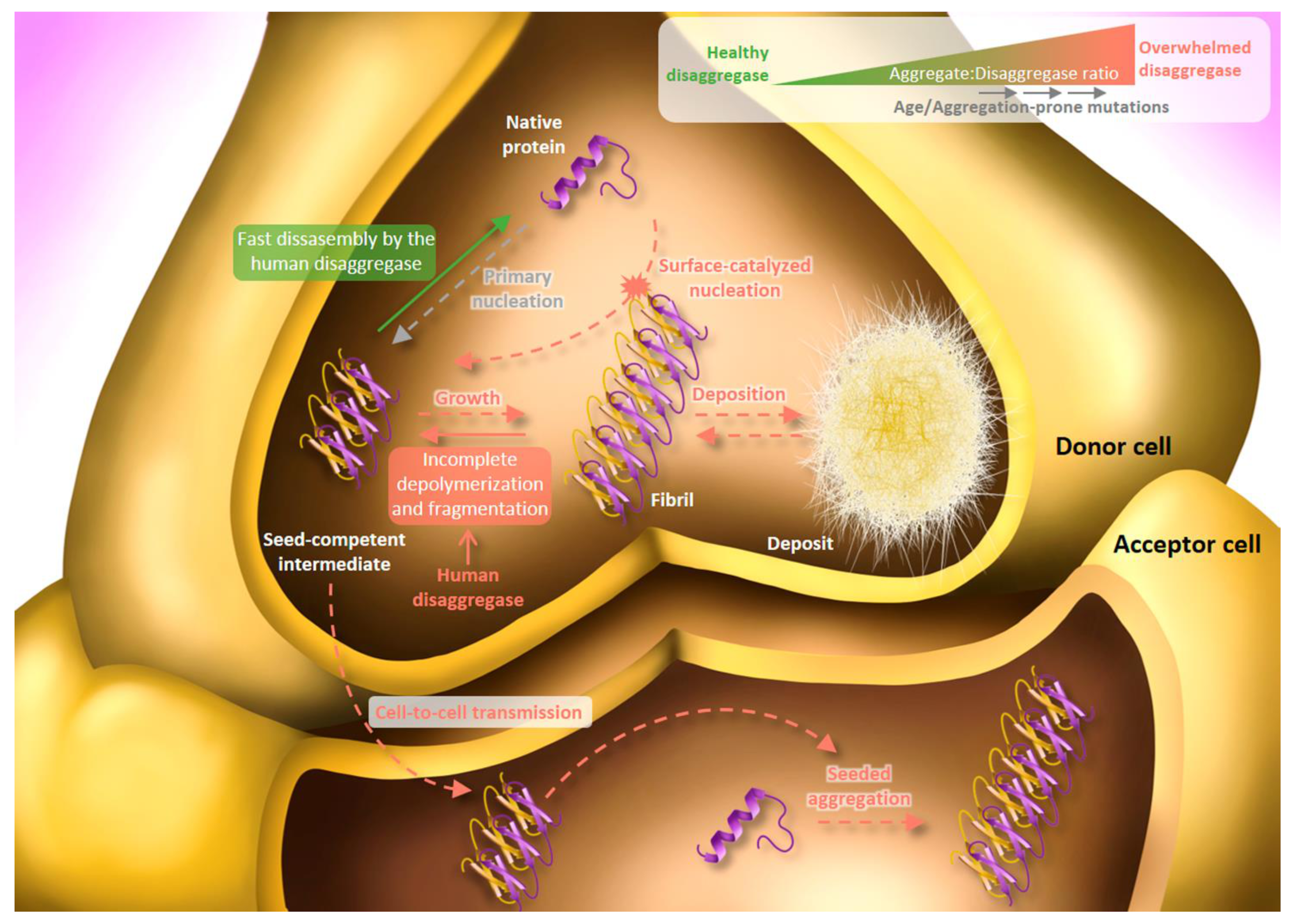
5. Human Disaggregase in Amyloid Toxicity and Propagation: Friend and Foe

6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Sheikh, S.; Haque, E.; Mir, S.S. Neurodegenerative diseases: Multifactorial conformational diseases and their therapeutic interventions. J. Neurodegener. Dis. 2013, 2013, 563481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Doré, V.; Burnham, S.C.; Masters, C.L.; Rowe, C.C. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 2018, 14, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Ozansoy, M.; Başak, A.N. The central theme of Parkinson’s disease: α-synuclein. Mol. Neurobiol. 2013, 47, 460–465. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in amyotrophic lateral sclerosis: “Ambivalent” behavior connected to the disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [Green Version]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Aksoy, Y.A.; Deng, W.; Stoddart, J.; Chung, R.; Guillemin, G.; Cole, N.J.; Neely, G.G.; Hesselson, D. “STRESSED OUT”: The role of FUS and TDP-43 in amyotrophic lateral sclerosis. Int. J. Biochem. Cell. Biol. 2020, 126, 105821. [Google Scholar] [CrossRef]
- Caterino, M.; Squillaro, T.; Montesarchio, D.; Giordano, A.; Giancola, C.; Melone, M. Huntingtin protein: A new option for fixing the Huntington’s disease countdown clock. Neuropharmacology 2018, 135, 126–138. [Google Scholar] [CrossRef]
- Gill, A.C.; Castle, A.R. The cellular and pathologic prion protein. Handb. Clin. Neurol. 2018, 153, 21–44. [Google Scholar]
- Winklhofer, K.F.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 Amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Li, J.-J.; Liu, S.; Zhao, J.; Jiang, Y.-J.; Song, A.-X.; Hu, H.-Y. Aggregation of polyglutamine-expanded Ataxin-3 sequesters its specific interacting partners into inclusions: Implication in a loss-of-function pathology. Sci. Rep. 2014, 4, 6410. [Google Scholar] [CrossRef] [Green Version]
- Budini, M.; Romano, V.; Quadri, Z.; Buratti, E.; Baralle, F.E. TDP-43 Loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N Prion-like domain. Hum. Mol. Genet. 2015, 24, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Surguchov, A. Intracellular dynamics of synucleins: “Here, There and Everywhere”. Int. Rev. Cell Mol. Biol. 2015, 320, 103–169. [Google Scholar]
- Surguchev, A.A.; Surguchov, A. Synucleins and gene expression: Ramblers in a crowd or cops regulating traffic? Front. Mol. Neurosci. 2017, 10, 224. [Google Scholar] [CrossRef] [Green Version]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-a beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Soll, L.G.; Eisen, J.N.; Vargas, K.J.; Medeiros, A.T.; Hammar, K.M.; Morgan, J.R. α-Synuclein-112 impairs synaptic vesicle recycling consistent with its enhanced membrane binding properties. Front. Cell Dev. Biol. 2020, 8, 405. [Google Scholar] [CrossRef]
- Cremades, N.; Chen, S.W.; Dobson, C.M. Structural characteristics of α-synuclein oligomers. Int. Rev. Cell Mol. Biol. 2017, 329, 79–143. [Google Scholar]
- Brás, I.C.; Xylaki, M.; Outeiro, T.F. Mechanisms of alpha-synuclein toxicity: An update and outlook. Prog. Brain Res. 2020, 252, 91–129. [Google Scholar]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Papp, M.I.; Kahn, J.E.; Lantos, P.L. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (Striatonigral degeneration, olivopontocerebellar atrophy and shy-drager syndrome). J. Neurol. Sci. 1989, 94, 79–100. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Trinkaus, V.A.; Riera-Tur, I.; Martínez-Sánchez, A.; Bäuerlein, F.J.B.; Guo, Q.; Arzberger, T.; Baumeister, W.; Dudanova, I.; Hipp, M.S.; Hartl, F.U.; et al. In situ architecture of neuronal α-synuclein inclusions. Nat. Commun. 2021, 12, 2110. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-synuclein aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [Green Version]
- Antonschmidt, L.; Dervişoğlu, R.; Sant, V.; Movellan, K.T.; Mey, I.; Riedel, D.; Steinem, C.; Becker, S.; Andreas, L.B.; Griesinger, C. Insights into the molecular mechanism of amyloid filament formation: Segmental folding of α-synuclein on lipid membranes/molecular mechanism of αs filament folding on membranes. Sci. Adv. 2021, 7, eabg2174. [Google Scholar] [CrossRef]
- Iljina, M.; Garcia, G.A.; Horrocks, M.H.; Tosatto, L.; Choi, M.L.; Ganzinger, K.A.; Abramov, A.Y.; Gandhi, S.; Wood, N.W.; Cremades, N.; et al. Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc. Natl. Acad. Sci. USA 2016, 113, E1206–E1215. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Dong, C.; Hoff, M.; Garen, C.R.; Cort, L.M.; Petersen, N.O.; Woodside, M.T.; Hoffmann, M.; Garen, C.R.; Cortez, L.M.; et al. Early stages of aggregation of engineered α-synuclein monomers and oligomers in solution. Sci. Rep. 2019, 9, 1734. [Google Scholar] [CrossRef]
- Buell, A.K.; Dobson, C.M.; Knowles, T.P.J. The physical chemistry of the amyloid phenomenon: Thermodynamics and kinetics of filamentous protein aggregation. Essays Biochem. 2014, 56, 11–39. [Google Scholar]
- Gaspar, R.; Meisl, G.; Buell, A.K.; Young, L.; Kaminski, C.F.; Knowles, T.P.J.; Sparr, E.; Linse, S. Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q. Rev. Biophys. 2017, 50, e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.J.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisl, G.; Knowles, T.P.J.; Klenerman, D. The molecular processes underpinning prion-like spreading and seed amplification in protein aggregation. Curr. Opin. Neurobiol. 2020, 61, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.; Brennan, S.; Keon, M.; Ooi, L. The role of amyloid oligomers in neurodegenerative pathologies. Int. J. Biol. Macromol. 2021, 181, 582–604. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Aguado, A.; Fernández-Higuero, J.A.; Moro, F.; Muga, A. Chaperone-assisted protein aggregate reactivation: Different solutions for the same problem. Arch. Biochem. Biophys. 2015, 580, 121–134. [Google Scholar] [CrossRef]
- Wentink, A.; Nussbaum-Krammer, C.; Bukau, B. Modulation of amyloid states by molecular chaperones. Cold Spring Harb. Perspect. Biol. 2019, 11, a033969. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Wentink, A.S.; Bukau, B. Protein disaggregation in multicellular organisms. Trends Biochem. Sci. 2018, 43, 285–300. [Google Scholar] [CrossRef]
- Avellaneda, M.J.; Franke, K.B.; Sunderlikova, V.; Bukau, B.; Mogk, A.; Tans, S.J. processive extrusion of polypeptide loops by a hsp100 disaggregase. Nature 2020, 578, 317–320. [Google Scholar] [CrossRef]
- Gates, S.N.; Gates, S.N.; Yokom, A.L.; Lin, J.; Jackrel, M.E.; Rizo, A.N.; Kendsersky, N.M.; Buell, C.E.; Sweeny, E.A.; Mack, K.L.; et al. Ratchet-like polypeptide translocation mechanism of the AAA + disaggregase Hsp104. Science 2017, 357, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogk, A.; Kummer, E.; Bukau, B. Cooperation of Hsp70 and Hsp100 Chaperone Machines in Protein Disaggregation. Front. Mol. Biosci. 2015, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef] [Green Version]
- Desantis, M.E.; Leung, E.H.; Sweeny, E.A.; Jackrel, M.E.; Cushman-Nick, M.; Neuhaus-Follini, A.; Vashist, S.; Sochor, M.A.; Knight, M.N.; Shorter, J. Operational plasticity enables Hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 2012, 151, 778–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Bianco, C.; Shorter, J.; Régulier, E.; Lashuel, H.; Iwatsubo, T.; Lindquist, S.; Aebischer, P. Hsp104 antagonizes α-synuclein aggregation and reduces dopaminergic degeneration in a rat model of parkinson disease. J. Clin. Investig. 2008, 118, 3087–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, A.; Jackrel, M.E.; Lin, J.; Hesketh, C.D.; Carman, P.J.; Mack, K.L.; Weitzman, R.; Gambogi, C.; Murillo, O.A.H.; Sweeny, E.A.; et al. Mining disaggregase sequence space to counter TDP-43, FUS, and a-synuclein proteotoxicity. Cell Rep. 2019, 28, 2080–2095.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampelt, H.; Kirstein-Miles, J.; Nillegoda, N.B.; Chi, K.; Scholz, S.R.; Morimoto, R.I.; Bukau, B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012, 31, 4221–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Kirstein, J.; Arnsburg, K.; Scior, A.; Szlachcic, A.; Guilbride, D.L.; Morimoto, R.I.; Bukau, B.; Nillegoda, N.B. In vivo properties of the disaggregase function of j-proteins and Hsc70 in caenorhabditis elegans stress and aging. Aging Cell 2017, 16, 1414–1424. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Echeverria, A.; Shorter, J. Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol. 2012, 10, e1001346. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 Disaggregase Reverses Parkinson’s-Linked α-Synuclein Amyloid Fibrils. Mol. Cell 2015, 59, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, Y.; Dublang, L.; Fernández-Higuero, J.A.; Albesa-Jové, D.; Lucas, M.; Viguera, A.R.; Guerin, M.E.; Vilar, J.M.G.; Muga, A.; Moro, F. Regulation of human Hsc70 ATPase and chaperone activities by Apg2: Role of the acidic subdomain. J. Mol. Biol. 2019, 431, 444–461. [Google Scholar] [CrossRef]
- Zhuravleva, A.; Clerico, E.M.; Gierasch, L.M. An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 2012, 151, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef] [Green Version]
- Faust, O.; Abayev-Avraham, M.; Wentink, A.S.; Maurer, M.; Nillegoda, N.B.; London, N.; Bukau, B.; Rosenzweig, R. Hsp40s employ class-specific regulation to drive Hsp70 functional diversity. Nature 2020, 587, 489–494. [Google Scholar] [CrossRef]
- Wentink, A.S.; Nillegoda, N.B.; Feufel, J.; Ubartaitė, G.; Schneider, C.P.; De Los Rios, P.; Hennig, J.; Barducci, A.; Bukau, B. Molecular Dissection of Amyloid Disaggregation by Human HSP70. Nature 2020, 587, 483–488. [Google Scholar] [CrossRef]
- De Los Rios, P.; Ben-Zvi, A.; Slutsky, O.; Azem, A.; Goloubinoff, P. Hsp70 chaperones accelerate protein translocation and the unfolding of stable protein aggregates by entropic pulling. Proc. Natl. Acad. Sci. USA 2006, 103, 6166–6171. [Google Scholar] [CrossRef] [Green Version]
- Sousa, R.; Liao, H.; Cuéllar, J.; Jin, S.; Valpuesta, J.M.; Jin, A.J.; Lafer, E.M. Clathrin-coat disassembly illuminates the mechanisms of Hsp70 force generation. Nat. Struct. Mol. Biol. 2016, 23, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Bracher, A.; Verghese, J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2015, 2, 10. [Google Scholar] [CrossRef]
- Franco, A.; Gracia, P.; Colom, A.; Camino, J.D.; Fernández-Higuero, J.A.; Orozco, N.; Dulebo, A.; Saiz, L.; Cremades, N.; Vilar, J.M.; et al. All-or-none amyloid disassembly via chaperone-triggered fibril unzipping favors clearance of α-synuclein toxic species. Proc. Natl. Acad. Sci. USA 2021, 118, e2105548118. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.M.; Gautam, S.; Herling, T.W.; Andrzejewska, E.; Vendruscolo, M.; Bracher, A.; Dobson, C.M.; Hartl, F.U. The Hsc70 disaggregation machinery removes monomer units directly from α-synuclein fibril ends. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ferrari, L.; Geerts, W.J.C.; Van Wezel, M.; Kos, R.; Van Bezouwen, L.S.; Förster, F.G.; Stefan, G.D. Human chaperones untangle fibrils of the Alzheimer protein Tau. bioRxiv 2018. [Google Scholar] [CrossRef]
- Nachman, E.; Wentink, A.; Madiona, K.; Bousset, L.; Katsinelos, T.; Kampinga, H.; Mcewan, W.A.; Jahn, T.R.; Melki, R.; Mogk, A.; et al. Disassembly of tau fibrils by the human Hsp70 disaggregation machinery generates small seeding-competent species. J. Biol. Chem. 2020, 295, 9676–9690. [Google Scholar] [CrossRef] [PubMed]
- Scior, A.; Buntru, A.; Arnsburg, K.; Ast, A.; Iburg, M.; Juenemann, K.; Pigazzini, M.L.; Mlody, B.; Puchkov, D.; Priller, J.; et al. Complete suppression of htt fibrilization and disaggregation of htt fibrils by a trimeric chaperone complex. EMBO J. 2018, 37, 282–299. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Rossi, M.; Baiardi, S.; Parchi, P. Understanding prion strains: Evidence from studies of the disease forms affecting humans. Viruses 2019, 11, 309. [Google Scholar] [CrossRef] [Green Version]
- Adamcik, J.; Mezzenga, R. Amyloid polymorphism in the protein folding and aggregation energy landscape. Angew. Chem. Int. Ed. Engl. 2018, 57, 8370–8382. [Google Scholar] [CrossRef]
- Tycko, R. Amyloid polymorphism: Structural basis and neurobiological relevance. Neuron 2015, 86, 632–645. [Google Scholar] [CrossRef] [Green Version]
- Gracia, P.; Camino, J.D.; Volpicelli-Daley, L.; Cremades, N. Multiplicity of α-synuclein aggregated species and their possible roles in disease. Int. J. Mol. Sci. 2020, 21, 8043. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Kovacik, L.; Ni, D.; Stahlberg, H. New insights on the structure of alpha-synuclein fibrils using cryo-electron microscopy. Curr. Opin. Neurobiol. 2020, 61, 89–95. [Google Scholar] [CrossRef]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Mi Taylor, N.; Arteni, A.-A.; Kumari, P.; Mona, D.; Ringler, P.; Britschgi, M.; Lauer, M.E.; Makky, A.; Verasdonck, J.; et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife 2019, 8, e48907. [Google Scholar] [CrossRef]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. Structures of fibrils formed by α-synuclein hereditary disease mutant h50q reveal new polymorphs. Nat. Struct. Mol. Biol. 2019, 26, 1044–1052. [Google Scholar] [CrossRef]
- Zhao, K.; Li, Y.; Liu, Z.; Long, H.; Zhao, C.; Luo, F.; Sun, Y.; Tao, Y.; Su, X.; Li, D.; et al. Parkinson’s disease associated mutation e46k of α-synuclein triggers the formation of a distinct fibril structure. Nat. Commun. 2020, 11, 2643. [Google Scholar] [CrossRef]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Zhou, K.; Hughes, M.P.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. The α-synuclein hereditary mutation e46k unlocks a more stable, pathogenic fibril structure. Proc. Natl. Acad. Sci. USA 2020, 4, 75–84. [Google Scholar] [CrossRef]
- Sun, Y.; Hou, S.; Zhao, K.; Long, H.; Liu, Z.; Gao, J.; Zhang, Y.; Su, X.-D.; Li, D.; Liu, C. Cryo-EM structure of full-length α-synuclein amyloid fibril with parkinson’s disease familial a53t mutation. Cell Res. 2020, 30, 360–362. [Google Scholar] [CrossRef]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Seyfried, N.T.; Petrucelli, L.; Fitzpatrick, A.W.P.; Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; et al. Post-translational modifications mediate the structural diversity of tauopathy strains. Cell 2020, 180, 633–644.e12. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; De Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic lewy. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, P.J.; Timothy Greenamyre, J. Post-translational modification of α-synuclein in Parkinson’s disease. Brain Res. 2015, 1628, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Giasson, B.I. The emerging role of α-synuclein truncation in aggregation and disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; McGlinchey, R.P.; Jiang, J.; Lee, J.C. Structural insights into α-synuclein fibril polymorphism: Effects of Parkinson’s disease-related C-terminal truncations. J. Mol. Biol. 2019, 431, 3913–3919. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [Green Version]
- González, N.; Arcos-López, T.; König, A.; Quintanar, L.; Menacho Márquez, M.; Outeiro, T.F.; Fernández, C.O. Effects of alpha-synuclein post-translational modifications on metal binding. J. Neurochem. 2019, 150, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Lim, Y.-J.; Liu, Z.; Long, H.; Sun, Y.; Hu, J.-J. Parkinson’s disease-related phosphorylation at tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proc. Natl. Acad. Sci. USA 2020, 117, 20305–20315. [Google Scholar] [CrossRef]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of alpha-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.G.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration, a four-repeat tauopathy. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Saibil, H.R. Cryo-EM of amyloid fibrils and cellular aggregates. Curr. Opin. Struct. Biol. 2019, 58, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Vats, A.; Taneja, V. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138–145. [Google Scholar]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Kukushkin, Y.; Gupta, R.; Chen, T.; Konagai, A.; Hipp, M.S.; Hayer-Hartl, M.; Hartl, F.U. PolyQ Proteins interfere with nuclear degradation of cytosolic proteins by sequestering the sis1p chaperone. Cell 2013, 154, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Hipp, M.S.; Park, S.H.; Hartl, U.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef]
- Yu, A.; Shibata, Y.; Shah, B.; Calamini, B.; Lo, D.C.; Morimoto, R.I. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc. Natl. Acad. Sci. USA 2014, 111, E1481–E1490. [Google Scholar] [CrossRef] [Green Version]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [Green Version]
- Choe, Y.J.; Park, S.H.; Hassemer, T.; Körner, R.; Vincenz-Donnelly, L.; Hayer-Hartl, M.; Hartl, F.U. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature 2016, 531, 191–195. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Lehmer, C.; Martínez-Sánchez, A.; Rudack, T.; Beck, F.; Hartmann, H.; Pérez-Berlanga, M.; Frottin, F.; Hipp, M.S.; Hartl, F.U.; et al. In situ structure of neuronal C9orf72 Poly-GA aggregates reveals proteasome recruitment. Cell 2018, 172, 696–705.e12. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Carulla, N.; Caddy, G.L.; Hall, D.R.; Gairı, M.; Feliz, M.; Giralt, E.; Robinson, C.V.; Dobson, C.M. Molecular recycling within amyloid fibrils. Nature 2005, 436, 554–558. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.A.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef] [Green Version]
- Scheidt, T.; Łapińska, U.; Kumita, J.R.; Whiten, D.R.; Klenerman, D.; Wilson, M.R.; Cohen, S.I.A.; Linse, S.; Vendruscolo, M.; Dobson, C.M.; et al. Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates. Sci. Adv. 2019, 5, eaau3112. [Google Scholar] [CrossRef] [Green Version]
- Tittelmeier, J.; Sandhof, C.A.; Ries, H.M.; Druffel-augustin, S.; Mogk, A.; Bukau, B.; Nussbaum-Krammer, C. The HSP 110/HSP 70 disaggregation system generates spreading-competent toxic α-synuclein species. EMBO J. 2020, 39, e103954. [Google Scholar] [CrossRef]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 targets hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Sweeny, E.A.; Shorter, J. Mechanistic and structural insights into the prion-disaggregase activity of Hsp104. J. Mol. Biol. 2016, 428, 1870–1885. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.K.; Sindi, S.S. A mathematical model of the dynamics of prion aggregates with chaperone-mediated fragmentation. J. Math. Biol. 2016, 72, 1555–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [Psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.V.; Gorenberg, E.L.; Nagy, M.; Thrasher, D.; Fenton, W.A.; Volpicelli-Daley, L.; Horwich, A.L.; Chandra, S.S. Hsp110 mitigates α-synuclein pathology in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 24310–24316. [Google Scholar] [CrossRef] [PubMed]
- Nagy, M.; Fenton, W.A.; Li, D.; Furtak, K.; Horwich, A.L. Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc. Natl. Acad. Sci. USA 2016, 113, 5424–5428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanenko, O.V.; Sulatsky, M.I.; Mikhailova, E.V.; Stepanenko, O.V.; Povarova, O.I.; Kuznetsova, I.M.; Turoverov, K.K.; Sulatskaya, A.I. Alpha-b-crystallin effect on mature amyloid fibrils: Different degradation mechanisms and changes in cytotoxicity. Int. J. Mol. Sci. 2020, 21, 7659. [Google Scholar] [CrossRef]
- Binger, K.J.; Ecroyd, H.; Yang, S.; Carver, J.A.; Howlett, G.J.; Griffin, M.D.W. Avoiding the oligomeric state: αB-crystallin inhibits fragmentation and induces dissociation of apolipoprotein C-II Amyloid fibrils. FASEB J. 2013, 27, 1214–1222. [Google Scholar] [CrossRef]
- Kandasamy, G.; Andréasson, C. Hsp70–Hsp110 chaperones deliver ubiquitin-dependent and -independent substrates to the 26s proteasome for proteolysis in yeast. J. Cell Sci. 2018, 131, jcs210948. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Klenerman, D.; Finley, D. N-Terminal ubiquitination of amyloidogenic proteins triggers removal of their oligomers by the proteasome holoenzyme. J. Mol. Biol. 2019, 432, 585–596. [Google Scholar] [CrossRef]
- Cliffe, R.; Sang, J.C.; Kundel, F.; Finley, D.; Klenerman, D.; Ye, Y. Filamentous aggregates are fragmented by the proteasome holoenzyme. Cell Rep. 2019, 26, 2140–2149. [Google Scholar] [CrossRef] [Green Version]
- Sang, J.C.; Hidari, E.; Klenerman, D.; Meisl, G.; Ranasinghe, R.T.; Spillantini, M.G. Super-resolution imaging reveals α-synuclein seeded aggregation in sh-sy5y cells. Commun. Biol. 2021, 4, 613. [Google Scholar] [CrossRef]
- Marrero-Winkens, C.; Sankaran, C.; Schätzl, H.M. from seeds to fibrils and back: Fragmentation as an overlooked step in the propagation of prions and prion-like proteins. Biomolecules 2020, 10, 1305. [Google Scholar] [CrossRef]
- Xu, Y.; Propson, N.E.; Du, S.; Xiong, W.; Zheng, H. Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proc. Natl. Acad. Sci. USA 2021, 118, e2023418118. [Google Scholar] [CrossRef]
- Sabath, N.; Levy-Adam, F.; Younis, A.; Rozales, K.; Meller, A.; Hadar, S.; Soueid-Baumgarten, S.; Shalgi, R. Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. USA 2020, 117, 31902–31913. [Google Scholar] [CrossRef]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [Green Version]
- Mckinnon, C.; Tabrizi, S.J. The ubiquitin-proteasome system in neurodegeneration. Antioxid. Redox Signal. 2014, 21, 2302–2321. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. Autophagy and aging. Adv. Exp. Med. Biol. 2015, 847, 73–87. [Google Scholar]
- Caldwell, K.A.; Caldwell, G.A.; Shorter, J. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2014, 156, 170–182. [Google Scholar]
- Fare, C.M.; Shorter, J. (Dis)solving the problem of aberrant protein states. Dis. Model Mech. 2021, 14, dmm048983. [Google Scholar] [CrossRef]
- Perni, M.; Flagmeier, P.; Limbocker, R.; Cascella, R.; Aprile, F.A.; Galvagnion, C.; Heller, G.T.; Meisl, G.; Chen, S.W.; Kumita, J.R.; et al. Multistep inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem. Biol. 2018, 13, 2308–2319. [Google Scholar] [CrossRef]
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 2020, 3, 435. [Google Scholar] [CrossRef]
- Santos, J.; Gracia, P.; Navarro, S.; Peña-Díaz, S.; Pujols, J.; Cremades, N.; Pallarès, I.; Ventura, S. α-Helical peptidic scaffolds to target α-synuclein toxic species with nanomolar affinity. Nat. Commun. 2021, 12, 3752. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, A.; Velasco-Carneros, L.; Alvarez, N.; Orozco, N.; Moro, F.; Prado, A.; Muga, A. Unzipping the Secrets of Amyloid Disassembly by the Human Disaggregase. Cells 2021, 10, 2745. https://doi.org/10.3390/cells10102745
Franco A, Velasco-Carneros L, Alvarez N, Orozco N, Moro F, Prado A, Muga A. Unzipping the Secrets of Amyloid Disassembly by the Human Disaggregase. Cells. 2021; 10(10):2745. https://doi.org/10.3390/cells10102745
Chicago/Turabian StyleFranco, Aitor, Lorea Velasco-Carneros, Naiara Alvarez, Natalia Orozco, Fernando Moro, Adelina Prado, and Arturo Muga. 2021. "Unzipping the Secrets of Amyloid Disassembly by the Human Disaggregase" Cells 10, no. 10: 2745. https://doi.org/10.3390/cells10102745