
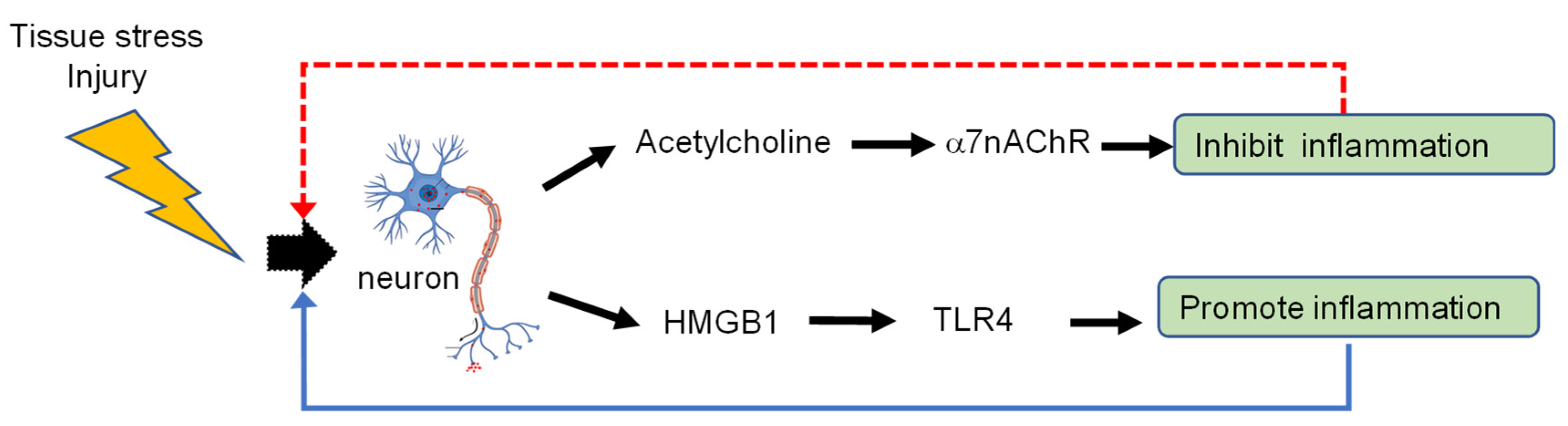
Neurons Are a Primary Driver of Inflammation via Release of HMGB1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Activation of Nociceptive Neurons Turns on Inflammation
2. Role of High Mobility Group Box 1 Protein (HMGB1) in the Inflammatory Response
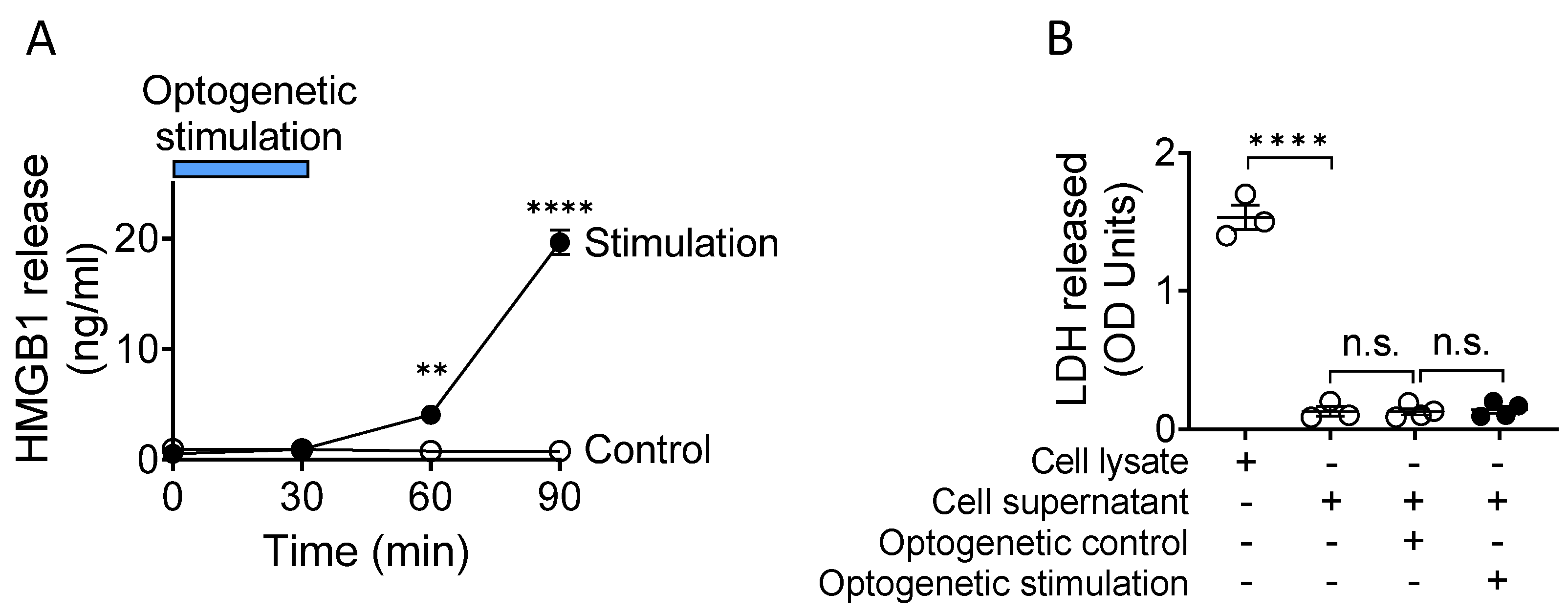
3. HMGB1 Is Actively Released by Nociceptive Neurons
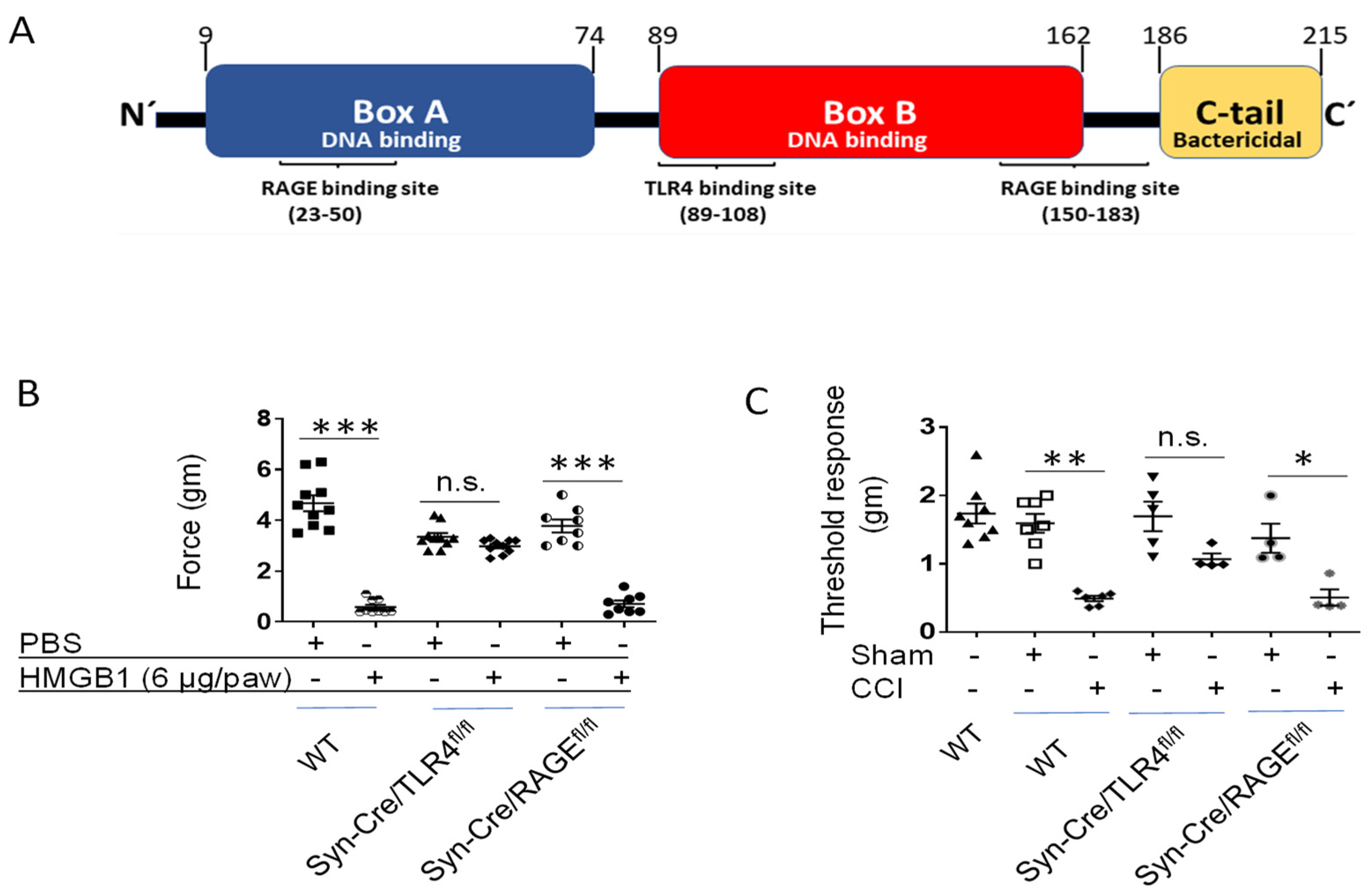
4. HMGB1 Induces Nociceptive Responses via Neuronal TLR4-Dependent Mechanisms
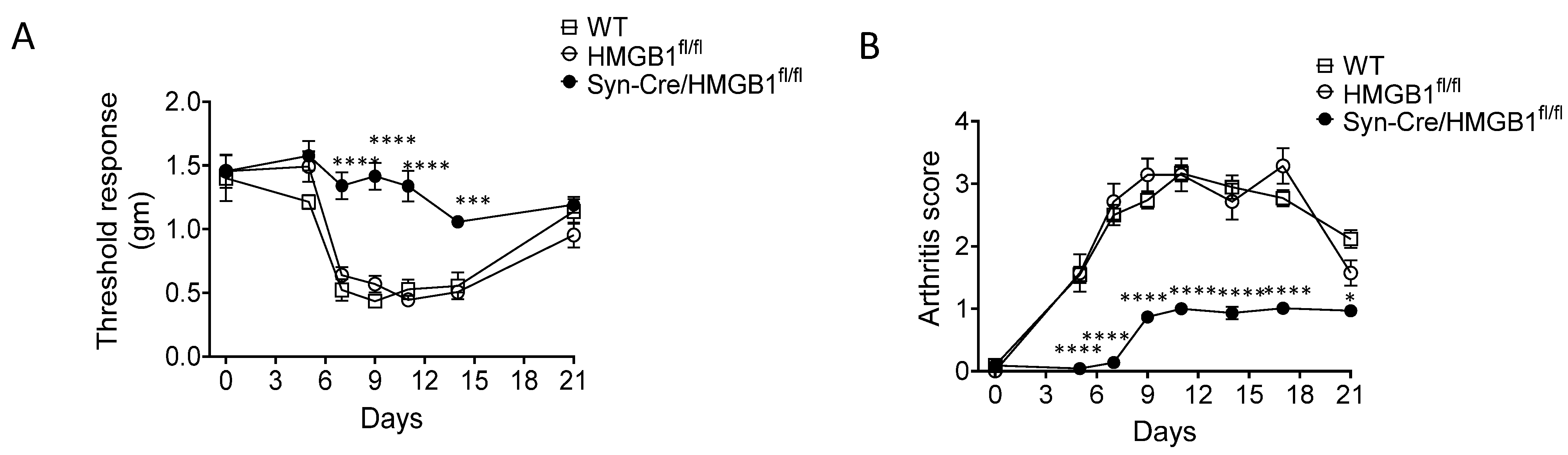
5. Neuronal HMGB1 Ablation/Neutralization Reduces Inflammation and Hyperalgesia
6. Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Cohen, J.A.; Edwards, T.N.; Liu, A.W.; Hirai, T.; Jones, M.R.; Wu, J.; Li, Y.; Zhang, S.; Ho, J.; Davis, B.M.; et al. Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 2019, 178, 919–932.e914. [Google Scholar] [CrossRef]
- Bersellini Farinotti, A.; Wigerblad, G.; Nascimento, D.; Bas, D.B.; Morado Urbina, C.; Nandakumar, K.S.; Sandor, K.; Xu, B.; Abdelmoaty, S.; Hunt, M.A.; et al. Cartilage-binding antibodies induce pain through immune complex-mediated activation of neurons. J. Exp. Med. 2019, 216, 1904–1924. [Google Scholar] [CrossRef] [Green Version]
- Borbély, É.; Kiss, T.; Szabadfi, K.; Pintér, E.; Szolcsányi, J.; Helyes, Z.; Botz, B. Complex Role of Capsaicin-Sensitive Afferents in the Collagen Antibody-Induced Autoimmune Arthritis of the Mouse. Sci. Rep. 2018, 8, 15916. [Google Scholar] [CrossRef] [Green Version]
- Kane, D.; Lockhart, J.C.; Balint, P.V.; Mann, C.; Ferrell, W.R.; McInnes, I.B. Protective effect of sensory denervation in inflammatory arthritis (evidence of regulatory neuroimmune pathways in the arthritic joint). Ann. Rheum. Dis. 2005, 64, 325–327. [Google Scholar] [CrossRef] [Green Version]
- Grace, P.M.; Tawfik, V.L.; Svensson, C.I.; Burton, M.D.; Loggia, M.L.; Hutchinson, M.R. The Neuroimmunology of Chronic Pain: From Rodents to Humans. J. Neurosci. Off. J. Soc. Neurosci. 2021, 41, 855–865. [Google Scholar] [CrossRef]
- Peirs, C.; Seal, R.P. Neural circuits for pain: Recent advances and current views. Science 2016, 354, 578–584. [Google Scholar] [CrossRef]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The crucial roles of inflammatory mediators in inflammation: A review. Vet. World 2018, 11, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Yang, H.; Harris, H. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin. Ther. Targets 2018, 22, 263–277. [Google Scholar] [CrossRef]
- Feldman, P.; Due, M.R.; Ripsch, M.S.; Khanna, R.; White, F.A. The persistent release of HMGB1 contributes to tactile hyperalgesia in a rodent model of neuropathic pain. J. Neuroinflamm. 2012, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Karatas, H.; Erdener, S.E.; Gursoy-Ozdemir, Y.; Lule, S.; Eren-Koçak, E.; Sen, Z.D.; Dalkara, T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013, 339, 1092–1095. [Google Scholar] [CrossRef]
- Faraco, G.; Fossati, S.; Bianchi, M.E.; Patrone, M.; Pedrazzi, M.; Sparatore, B.; Moroni, F.; Chiarugi, A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J. Neurochem. 2007, 103, 590–603. [Google Scholar] [CrossRef]
- Gao, S.Q.; Zhang, H.; He, J.G.; Zheng, H.L.; Zhang, P.W.; Xu, J.F.; Shen, Z.C.; Zhao, H.H.; Wang, F.; Hu, Z.L.; et al. Neuronal HMGB1 in nucleus accumbens regulates cocaine reward memory. Addict. Biol. 2019, 25, e12739. [Google Scholar] [CrossRef]
- Wang, X.; Chu, G.; Yang, Z.; Sun, Y.; Zhou, H.; Li, M.; Shi, J.; Tian, B.; Zhang, C.; Meng, X. Ethanol directly induced HMGB1 release through NOX2/NLRP1 inflammasome in neuronal cells. Toxicology 2015, 334, 104–110. [Google Scholar] [CrossRef]
- Qian, J.; Zhu, Y.; Bai, L.; Gao, Y.; Jiang, M.; Xing, F.; Zhang, J.; Zhao, W.; Gu, H.; Mi, Y.; et al. Chronic morphine-mediated upregulation of high mobility group box 1 in the spinal cord contributes to analgesic tolerance and hyperalgesia in rats. Neurotherapeutics 2019, 17, 722–742. [Google Scholar] [CrossRef]
- Sun, Q.; Wu, W.; Hu, Y.C.; Li, H.; Zhang, D.; Li, S.; Li, W.; Li, W.D.; Ma, B.; Zhu, J.H.; et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2014, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; Cao, L.; Khanabdali, R.; Kalionis, B.; Tai, X.; Xia, S. The Emerging Role of HMGB1 in Neuropathic Pain: A Potential Therapeutic Target for Neuroinflammation. J. Immunol. Res. 2016, 2016, 6430423. [Google Scholar] [CrossRef] [Green Version]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Fan, M.; Wang, X.; Xu, J.; Wang, Y.; Tu, F.; Gill, P.S.; Ha, T.; Liu, L.; Williams, D.L.; et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2021, 1–14. [Google Scholar] [CrossRef]
- Merianda, T.T.; Coleman, J.; Kim, H.H.; Kumar Sahoo, P.; Gomes, C.; Brito-Vargas, P.; Rauvala, H.; Blesch, A.; Yoo, S.; Twiss, J.L. Axonal amphoterin mRNA is regulated by translational control and enhances axon outgrowth. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 5693–5706. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M.; et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Chen, H.; Dai, J.; Wan, Z.; Xiong, P.; Xu, Y.; Han, Z.; Chai, W.; Gong, F.; Zheng, F. Glycyrrhizin Protects Mice Against Experimental Autoimmune Encephalomyelitis by Inhibiting High-Mobility Group Box 1 (HMGB1) Expression and Neuronal HMGB1 Release. Front. Immunol. 2018, 9, 1518. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.Y.; Crews, F.T. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS ONE 2014, 9, e87915. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zeng, Q.; Silverman, H.A.; Gunasekaran, M.; George, S.J.; Devarajan, A.; Addorisio, M.E.; Li, J.; Tsaava, T.; Shah, V.; et al. HMGB1 released from nociceptors mediates inflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2102034118. [Google Scholar] [CrossRef] [PubMed]
- Daou, I.; Tuttle, A.H.; Longo, G.; Wieskopf, J.S.; Bonin, R.P.; Ase, A.R.; Wood, J.N.; De Koninck, Y.; Ribeiro-da-Silva, A.; Mogil, J.S.; et al. Remote optogenetic activation and sensitization of pain pathways in freely moving mice. J. Neurosci. 2013, 33, 18631–18640. [Google Scholar] [CrossRef] [PubMed]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundback, P.; Long, W.; Valdes-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Bianchi, M.E.; Coleman, T.R.; Tracey, K.J.; Al-Abed, Y. Correction to: Exploring the biological functional mechanism of the HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Mol. Med. 2018, 24, 31. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Roncarati, P.; Demoulin, S.; Pilard, C.; Ancion, M.; Reynders, C.; Lerho, T.; Bruyere, D.; Lebeau, A.; Radermecker, C.; et al. Extracellular HMGB1 blockade inhibits tumor growth through profoundly remodeling immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. J. Immunother. Cancer 2021, 9, e001966. [Google Scholar] [CrossRef]
- Agalave, N.M.; Larsson, M.; Abdelmoaty, S.; Su, J.; Baharpoor, A.; Lundback, P.; Palmblad, K.; Andersson, U.; Harris, H.; Svensson, C.I. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 2014, 155, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Agalave, N.M.; Svensson, C.I. Extracellular high-mobility group box 1 protein (HMGB1) as a mediator of persistent pain. Mol. Med. 2015, 20, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Gao, T.; Shi, T.; Xiang, Q.; Xu, X.; Wiesenfeld-Hallin, Z.; Hökfelt, T.; Svensson, C.I. Phenotypic changes in dorsal root ganglion and spinal cord in the collagen antibody-induced arthritis mouse model. J. Comp. Neurol. 2015, 523, 1505–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakthiswary, R.; Singh, R. Has the median nerve involvement in rheumatoid arthritis been overemphasized? Rev. Bras. Reumatol. 2017, 57, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Takahashi, H.K.; Liu, K.; Wake, H.; Liu, R.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Mori, S.; et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 2011, 42, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, K.; Wake, H.; Teshigawara, K.; Mori, S.; Nishibori, M. Anti-high mobility group box-1 (HMGB1) antibody inhibits hemorrhage-induced brain injury and improved neurological deficits in rats. Sci. Rep. 2017, 7, 46243. [Google Scholar] [CrossRef] [Green Version]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.K.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef]
- Okuma, Y.; Liu, K.; Wake, H.; Zhang, J.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Otani, N.; Tomura, S.; et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann. Neurol. 2012, 72, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Liu, K.; Agari, T.; Yasuhara, T.; Morimoto, J.; Okazaki, M.; Takeuchi, H.; Toyoshima, A.; Sasada, S.; Shinko, A.; et al. Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson’s disease. Exp. Neurol. 2016, 275 Pt 1, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Liu, K.; Wake, H.; Teshigawara, K.; Yoshino, T.; Takahashi, H.; Mori, S.; Nishibori, M. Therapeutic effects of anti-HMGB1 monoclonal antibody on pilocarpine-induced status epilepticus in mice. Sci. Rep. 2017, 7, 1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Wang, Y.; Xu, C.; Liu, K.; Wang, Y.; Chen, L.; Wu, X.; Gao, F.; Guo, Y.; Zhu, J.; et al. Therapeutic potential of an anti-high mobility group box-1 monoclonal antibody in epilepsy. Brain Behav. Immun. 2017, 64, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Uzawa, A.; Mori, M.; Taniguchi, J.; Masuda, S.; Muto, M.; Kuwabara, S. Anti-high mobility group box 1 monoclonal antibody ameliorates experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2013, 172, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.P.; Caldis, M.W.; Harp, C.T.; Goings, G.E.; Miller, S.D. High-mobility group box 1 protein (HMGB1) neutralization ameliorates experimental autoimmune encephalomyelitis. J. Autoimmun. 2013, 43, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otoshi, K.; Kikuchi, S.; Kato, K.; Sekiguchi, M.; Konno, S. Anti-HMGB1 neutralization antibody improves pain-related behavior induced by application of autologous nucleus pulposus onto nerve roots in rats. Spine 2011, 36, E692–E698. [Google Scholar] [CrossRef]
- Chavan, S.S.; Huerta, P.T.; Robbiati, S.; Valdes-Ferrer, S.I.; Ochani, M.; Dancho, M.; Frankfurt, M.; Volpe, B.T.; Tracey, K.J.; Diamond, B. HMGB1 mediates cognitive impairment in sepsis survivors. Mol. Med. 2012, 18, 930–937. [Google Scholar] [CrossRef]
- Terrando, N.; Yang, T.; Wang, X.; Fang, J.; Cao, M.; Andersson, U.; Erlandsson, H.H.; Ouyang, W.; Tong, J. Systemic HMGB1 Neutralization Prevents Postoperative Neurocognitive Dysfunction in Aged Rats. Front. Immunol. 2016, 7, 441. [Google Scholar] [CrossRef] [Green Version]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. HMGB1-Mediated Neuroinflammatory Responses in Brain Injuries: Potential Mechanisms and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 4609. [Google Scholar] [CrossRef] [PubMed]
- Nishibori, M.; Wang, D.; Ousaka, D.; Wake, H. High Mobility Group Box-1 and Blood-Brain Barrier Disruption. Cells 2020, 9, 2650. [Google Scholar] [CrossRef] [PubMed]
- Nishibori, M.; Mori, S.; Takahashi, H.K. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J. Pharm. Sci. 2019, 140, 94–101. [Google Scholar] [CrossRef]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49, 740–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Morioka, N.; Abe, H.; Zhang, F.F.; Hisaoka-Nakashima, K.; Liu, K.; Nishibori, M.; Nakata, Y. Neuropathic pain in rats with a partial sciatic nerve ligation is alleviated by intravenous injection of monoclonal antibody to high mobility group box-1. PLoS ONE 2013, 8, e73640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, W.; Harada, S.; Liu, K.; Nishibori, M.; Tokuyama, S. Evidence of a role for spinal HMGB1 in ischemic stress-induced mechanical allodynia in mice. Brain Res. 2018, 1687, 1–10. [Google Scholar] [CrossRef]
- Morioka, N.; Miyauchi, K.; Miyashita, K.; Kochi, T.; Zhang, F.F.; Nakamura, Y.; Liu, K.; Wake, H.; Hisaoka-Nakashima, K.; Nishibori, M.; et al. Spinal high-mobility group box-1 induces long-lasting mechanical hypersensitivity through the toll-like receptor 4 and upregulation of interleukin-1beta in activated astrocytes. J. Neurochem. 2019, 150, 738–758. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Tracey, K.J. Neural regulation of immunity: Molecular mechanisms and clinical translation. Nat. Neurosci. 2017, 20, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Anti-inflammatory properties of the vagus nerve: Potential therapeutic implications of vagus nerve stimulation. J. Physiol. 2016, 594, 5781–5790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, F.A.; Chavan, S.S.; Miljko, S.; Grazio, S.; Sokolovic, S.; Schuurman, P.R.; Mehta, A.D.; Levine, Y.A.; Faltys, M.; Zitnik, R.; et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2016, 113, 8284–8289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlov, V.A.; Chavan, S.S.; Tracey, K.J. Molecular and Functional Neuroscience in Immunity. Annu. Rev. Immunol. 2018, 36, 783–812. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.S.; Pavlov, V.A.; Tracey, K.J. Mechanisms and Therapeutic Relevance of Neuro-immune Communication. Immunity 2017, 46, 927–942. [Google Scholar] [CrossRef] [Green Version]
- Zanos, T.P.; Silverman, H.A.; Levy, T.; Tsaava, T.; Battinelli, E.; Lorraine, P.W.; Ashe, J.M.; Chavan, S.S.; Tracey, K.J.; Bouton, C.E. Identification of cytokine-specific sensory neural signals by decoding murine vagus nerve activity. Proc. Natl. Acad. Sci. USA 2018, 115, E4843–E4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Andersson, U.; Brines, M. Neurons Are a Primary Driver of Inflammation via Release of HMGB1. Cells 2021, 10, 2791. https://doi.org/10.3390/cells10102791
Yang H, Andersson U, Brines M. Neurons Are a Primary Driver of Inflammation via Release of HMGB1. Cells. 2021; 10(10):2791. https://doi.org/10.3390/cells10102791
Chicago/Turabian StyleYang, Huan, Ulf Andersson, and Michael Brines. 2021. "Neurons Are a Primary Driver of Inflammation via Release of HMGB1" Cells 10, no. 10: 2791. https://doi.org/10.3390/cells10102791