
Therapeutic Approaches for Patients with Cystic Fibrosis Not Eligible for Current CFTR Modulators

Abstract
:1. Introduction
2. Broadening the Numbers of Mutations Eligible for CFTR Modulators
3. Readthrough Agents for Nonsense Mutations
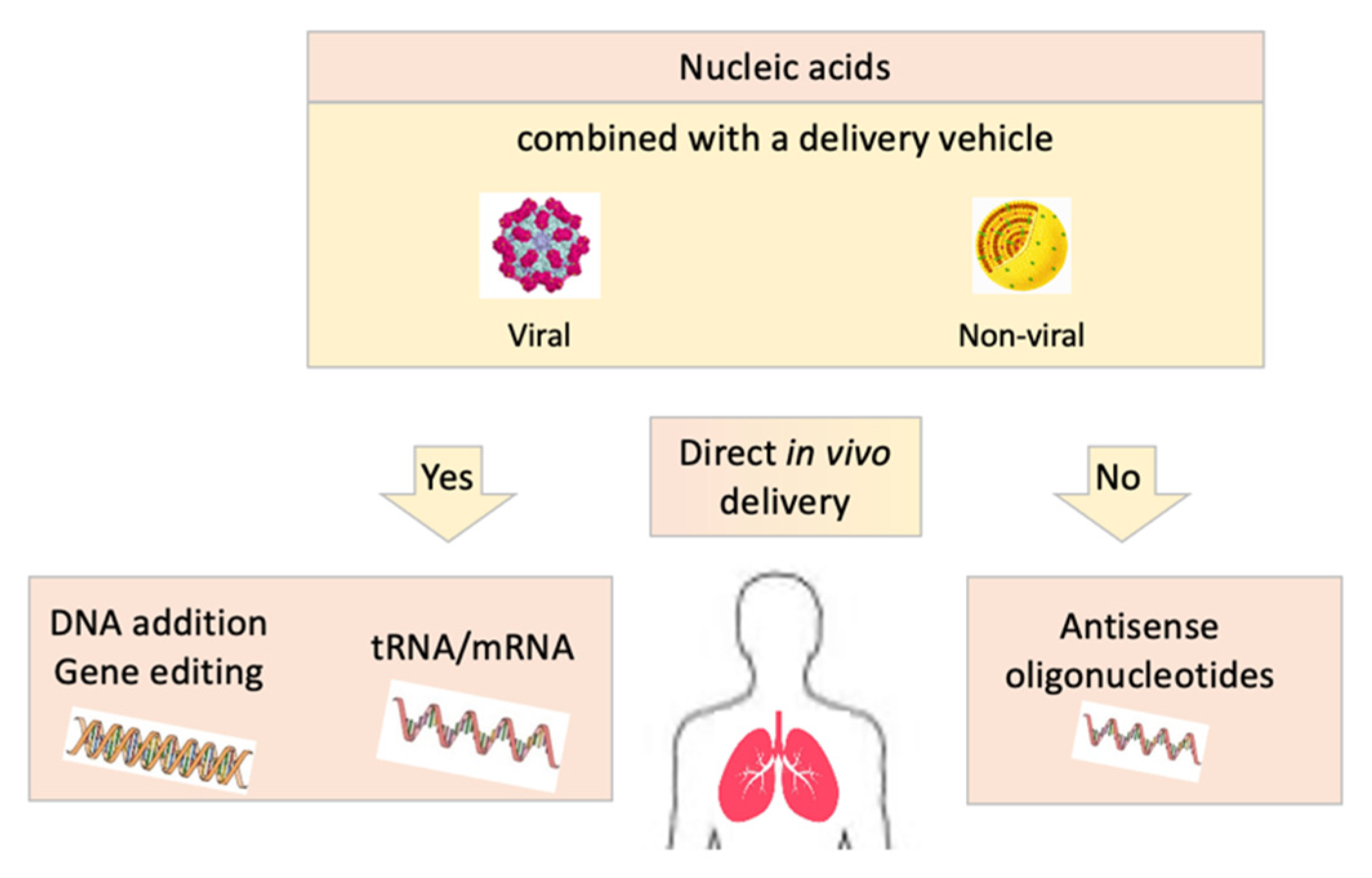
4. RNA-Based Therapies
5. DNA-Based Therapies: Gene Therapy and Gene Editing
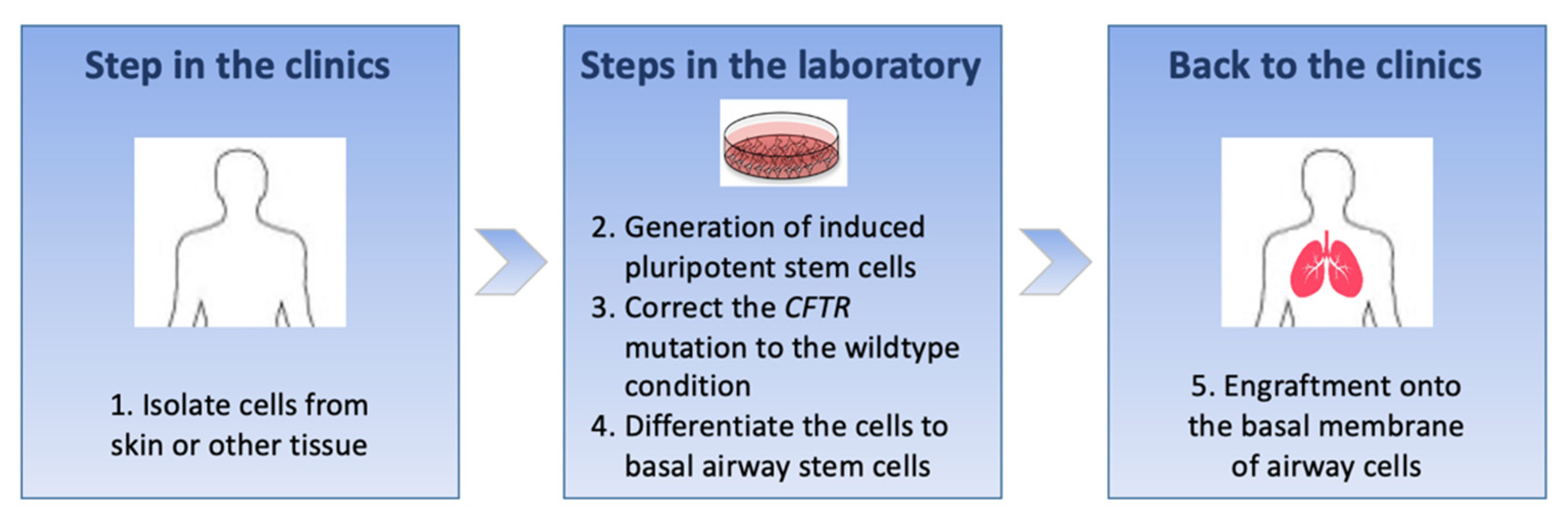
6. Cell-Based Therapies
7. Delivery Vectors
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E. Managing Cystic Fibrosis: Strategies That Increase Life Expectancy and Improve Quality of Life. Am. J. Respir. Crit. Care Med. 2011, 183, 1463–1471. [Google Scholar] [CrossRef] [Green Version]
- Bardin, E.; Pastor, A.; Semeraro, M.; Golec, A.; Hayes, K.; Chevalier, B.; Berhal, F.; Prestat, G.; Hinzpeter, A.; Gravier-Pelletier, C.; et al. Modulators of CFTR. Updates on Clinical Development and Future Directions. Eur. J. Med. Chem. 2021, 213, 113195. [Google Scholar] [CrossRef] [PubMed]
- Gramegna, A.; Contarini, M.; Bindo, F.; Aliberti, S.; Blasi, F. Elexacaftor–Tezacaftor–Ivacaftor: The New Paradigm to Treat People with Cystic Fibrosis with at Least One p.Phe508del Mutation. Curr. Opin. Pharmacol. 2021, 57, 81–88. [Google Scholar] [CrossRef]
- Amaral, M.D.; de Boeck, K. ECFS Strategic Planning Task Force on ‘Speeding up access to new drugs for CF’ Theranostics by Testing CFTR Modulators in Patient-Derived Materials: The Current Status and a Proposal for Subjects with Rare CFTR Mutations. J. Cyst. Fibros. 2019, 18, 685–692. [Google Scholar] [CrossRef]
- Durmowicz, A.G.; Lim, R.; Rogers, H.; Rosebraugh, C.J.; Chowdhury, B.A. The U.S. Food and Drug Administration’s Experience with Ivacaftor in Cystic Fibrosis. Establishing Efficacy Using In Vitro Data in Lieu of a Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef] [Green Version]
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might Brushed Nasal Cells Be a Surrogate for CFTR Modulator Clinical Response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126. [Google Scholar] [CrossRef]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A Meta-Analysis of Nonsense Mutations Causing Human Genetic Disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef]
- Mendell, J.T.; Dietz, H.C. When the Message Goes Awry. Cell 2001, 107, 411–414. [Google Scholar] [CrossRef] [Green Version]
- Frischmeyer, P.A. Nonsense-Mediated MRNA Decay in Health and Disease. Hum. Mol. Genet. 1999, 8, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- McCague, A.F.; Raraigh, K.S.; Pellicore, M.J.; Davis-Marcisak, E.F.; Evans, T.A.; Han, S.T.; Lu, Z.; Joynt, A.T.; Sharma, N.; Castellani, C.; et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1116–1126. [Google Scholar] [CrossRef]
- Fan-Minogue, H.; Bedwell, D.M. Eukaryotic Ribosomal RNA Determinants of Aminoglycoside Resistance and Their Role in Translational Fidelity. RNA 2007, 14, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside Antibiotics Restore CFTR Function by Overcoming Premature Stop Mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I.; Renouil, M.; Fajac, A.; Bidou, L.; Parbaille, B.; Pierrot, S.; Davy, N.; Bismuth, E.; Reinert, P.; Lenoir, G.; et al. Correction to: In Vitro Prediction of Stop-Codon Suppression by Intravenous Gentamicin in Patients with Cystic Fibrosis: A Pilot Study. BMC Med. 2018, 16, 159. [Google Scholar] [CrossRef]
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Aviram, M.; Bdolah-Abram, T.; Bebok, Z.; Shushi, L.; et al. Gentamicin-Induced Correction of CFTR Function in Patients with Cystic Fibrosis and CFTR Stop Mutations. N. Engl. J. Med. 2003, 349, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the Treatment of Nonsense-Mutation Cystic Fibrosis: A Randomised, Double-Blind, Placebo-Controlled Phase 3 Trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Kerem, E. ELX-02: An Investigational Read-through Agent for the Treatment of Nonsense Mutation-Related Genetic Disease. Expert Opin. Investig. Drugs 2020, 29, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Floquet, C.; Hatin, I.; Rousset, J.-P.; Bidou, L. Statistical Analysis of Readthrough Levels for Nonsense Mutations in Mammalian Cells Reveals a Major Determinant of Response to Gentamicin. PLoS Genet. 2012, 8, e1002608. [Google Scholar] [CrossRef]
- Yeh, J.-T.; Hwang, T.-C. Positional Effects of Premature Termination Codons on the Biochemical and Biophysical Properties of CFTR. J. Physiol. 2020, 598, 517–541. [Google Scholar] [CrossRef]
- Keenan, M.M.; Huang, L.; Jordan, N.J.; Wong, E.; Cheng, Y.; Valley, H.C.; Mahiou, J.; Liang, F.; Bihler, H.; Mense, M.; et al. Nonsense-Mediated RNA Decay Pathway Inhibition Restores Expression and Function of W1282X CFTR. Am. J. Respir. Cell Mol. Biol. 2019, 61, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.; Bidou, L.; Martin, N.; Blanchet, S.; Hatton, A.; Karri, S.; Cornu, D.; Costes, B.; Chevalier, B.; Tondelier, D.; et al. Factors Influencing Readthrough Therapy for Frequent Cystic Fibrosis Premature Termination Codons. ERJ Open Res. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.; Keeling, K.M.; Rowe, S.M. Pharmacological Approaches for Targeting Cystic Fibrosis Nonsense Mutations. Eur. J. Med. Chem. 2020, 200, 112436. [Google Scholar] [CrossRef]
- Laselva, O.; Eckford, P.D.; Bartlett, C.; Ouyang, H.; Gunawardena, T.N.; Gonska, T.; Moraes, T.J.; Bear, C.E. Functional Rescue of c.3846G>A (W1282X) in Patient-Derived Nasal Cultures Achieved by Inhibition of Nonsense Mediated Decay and Protein Modulators with Complementary Mechanisms of Action. J. Cyst. Fibros. 2020, 19, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Porter, J.J.; Heil, C.S.; Lueck, J.D. Therapeutic Promise of Engineered Nonsense Suppressor tRNAs. WIREs RNA 2021. [Google Scholar] [CrossRef] [PubMed]
- Lueck, J.D.; Yoon, J.S.; Perales-Puchalt, A.; Mackey, A.L.; Infield, D.T.; Behlke, M.A.; Pope, M.R.; Weiner, D.B.; Skach, W.R.; McCray, P.B.; et al. Engineered Transfer RNAs for Suppression of Premature Termination Codons. Nat. Commun. 2019, 10, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva Sanchez, A.; Paunovska, K.; Cristian, A.; Dahlman, J.E. Treating Cystic Fibrosis with MRNA and CRISPR. Hum. Gene Ther. 2020, 31, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.N.; Beiβert, T.; Simon, P.; Vallazza, B.; Buck, J.; Davies, B.P.; Tureci, O.; Sahin, U. MRNA as a Versatile Tool for Exogenous Protein Expression. Curr. Gene 2012, 12, 347–361. [Google Scholar] [CrossRef]
- Hajj, K.A.; Whitehead, K.A. Tools for Translation: Non-Viral Materials for Therapeutic MRNA Delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Zhang, H.; Leal, J.; Soto, M.R.; Smyth, H.D.C.; Ghosh, D. Aerosolizable Lipid Nanoparticles for Pulmonary Delivery of MRNA through Design of Experiments. Pharmaceutics 2020, 12, 1042. [Google Scholar] [CrossRef]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified MRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beumer, W.; Swildens, J.; Leal, T.; Noel, S.; Anthonijsz, H.; van der Horst, G.; Kuiperij-Boersma, H.; Potman, M.; van Putten, C.; Biasutto, P.; et al. Evaluation of Eluforsen, a Novel RNA Oligonucleotide for Restoration of CFTR Function in in Vitro and Murine Models of p.Phe508del Cystic Fibrosis. PLoS ONE 2019, 14, e0219182. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I.; Clancy, J.P.; Nichols, D.P.; Nick, J.A.; De Boeck, K.; Solomon, G.M.; Mall, M.A.; Bolognese, J.; Bouisset, F.; den Hollander, W.; et al. Antisense Oligonucleotide Eluforsen Improves CFTR Function in F508del Cystic Fibrosis. J. Cyst. Fibros. 2019, 18, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drevinek, P.; Pressler, T.; Cipolli, M.; De Boeck, K.; Schwarz, C.; Bouisset, F.; Boff, M.; Henig, N.; Paquette-Lamontagne, N.; Montgomery, S.; et al. Antisense Oligonucleotide Eluforsen Is Safe and Improves Respiratory Symptoms in F508del Cystic Fibrosis. J. Cyst. Fibros. 2020, 19, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, Y.S.; Irony-Tur Sinai, M.; Golec, A.; Barchad-Avitzur, O.; Mutyam, V.; Li, Y.; Hong, J.; Ozeri-Galai, E.; Hatton, A.; Leibson, C.; et al. Antisense Oligonucleotide-Based Drug Development for Cystic Fibrosis Patients Carrying the 3849+10 Kb C-to-T Splicing Mutation. J. Cyst. Fibros. 2021, 20, 865–875. [Google Scholar] [CrossRef]
- Igreja, S.; Clarke, L.A.; Botelho, H.M.; Marques, L.; Amaral, M.D. Correction of a Cystic Fibrosis Splicing Mutation by Antisense Oligonucleotides. Hum. Mutat. 2016, 37, 209–215. [Google Scholar] [CrossRef]
- Michaels, W.E.; Bridges, R.J.; Hastings, M.L. Antisense Oligonucleotide-Mediated Correction of CFTR Splicing Improves Chloride Secretion in Cystic Fibrosis Patient-Derived Bronchial Epithelial Cells. Nucleic Acids Res. 2020, 48, 7454–7467. [Google Scholar] [CrossRef]
- Boyd, A.C.; Guo, S.; Huang, L.; Kerem, B.; Oren, Y.S.; Walker, A.J.; Hart, S.L. New Approaches to Genetic Therapies for Cystic Fibrosis. J. Cyst. Fibros. 2020, 19, S54–S59. [Google Scholar] [CrossRef] [Green Version]
- Cooney, A.; McCray, P.; Sinn, P. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 2018, 9, 538. [Google Scholar] [CrossRef]
- Vu, A.; McCray, P.B. New Directions in Pulmonary Gene Therapy. Hum. Gene Ther. 2020, 31, 921–939. [Google Scholar] [CrossRef]
- Hodges, C.A.; Conlon, R.A. Delivering on the Promise of Gene Editing for Cystic Fibrosis. Genes Dis. 2019, 6, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, J.; Hirai, H.; Yang, D.; Ma, L.; Hou, X.; Jiang, H.; Wei, H.; Rajagopalan, C.; Mou, H.; Wang, G.; et al. Efficient Gene Editing at Major CFTR Mutation Loci. Mol. Ther.-Nucleic Acids 2019, 16, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, M.P.T.; Broeders, M.; Herrero-Hernandez, P.; Oussoren, E.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Ready for Repair? Gene Editing Enters the Clinic for the Treatment of Human Disease. Mol. Methods Clin. Dev. 2020, 18, 532–557. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S.; et al. Targeted Correction and Restored Function of the CFTR Gene in Cystic Fibrosis Induced Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A Revised Airway Epithelial Hierarchy Includes CFTR-Expressing Ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Allan, K.M.; Farrow, N.; Donnelley, M.; Jaffe, A.; Waters, S.A. Treatment of Cystic Fibrosis: From Gene- to Cell-Based Therapies. Front. Pharmacol. 2021, 12, 639475. [Google Scholar] [CrossRef]
- Berical, A.; Lee, R.E.; Randell, S.H.; Hawkins, F. Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis. Front. Pharmacol. 2019, 10, 74. [Google Scholar] [CrossRef]
- Hayes, D.; Kopp, B.T.; Hill, C.L.; Lallier, S.W.; Schwartz, C.M.; Tadesse, M.; Alsudayri, A.; Reynolds, S.D. Cell Therapy for Cystic Fibrosis Lung Disease: Regenerative Basal Cell Amplification. Stem Cells Transl. Med. 2019, 8, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, A.S.; Beck, S.; Meyer, M.; Penque, D.; Cutting, G.R.; Amaral, M.D. Five Percent of Normal Cystic Fibrosis Transmembrane Conductance Regulator MRNA Ameliorates the Severity of Pulmonary Disease in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2002, 27, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Eisman, R.; Xu, J.; Harsch, A.D.; Mulberg, A.E.; Bevins, C.L.; Glick, M.C.; Scanlin, T.F. Turnover of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR): Slow Degradation of Wild-Type and Delta F508 CFTR in Surface Membrane Preparations of Immortalized Airway Epithelial Cells. J. Cell. Physiol. 1996, 168, 373–384. [Google Scholar] [CrossRef]
- Faustino, N.A. Pre-MRNA Splicing and Human Disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissim-Rafinia, M.; Kerem, B. Splicing Regulation as a Potential Genetic Modifier. Trends Genet. 2002, 18, 123–127. [Google Scholar] [CrossRef]
- Hawkins, F.J.; Suzuki, S.; Beermann, M.L.; Barillà, C.; Wang, R.; Villacorta-Martin, C.; Berical, A.; Jean, J.C.; Le Suer, J.; Matte, T.; et al. Derivation of Airway Basal Stem Cells from Human Pluripotent Stem Cells. Cell Stem Cell 2021, 28, 79–95.e8. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.L.; Green, M.D.; de Carvalho, A.T.; Mumau, M.; Chen, Y.-W.; D’Souza, S.L.; Snoeck, H.-W. The in Vitro Generation of Lung and Airway Progenitor Cells from Human Pluripotent Stem Cells. Nat. Protoc. 2015, 10, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alton, E.W.F.W.; Beekman, J.M.; Boyd, A.C.; Brand, J.; Carlon, M.S.; Connolly, M.M.; Chan, M.; Conlon, S.; Davidson, H.E.; Davies, J.C.; et al. Preparation for a First-in-Man Lentivirus Trial in Patients with Cystic Fibrosis. Thorax 2017, 72, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rollet-Cohen, V.; Bourderioux, M.; Lipecka, J.; Chhuon, C.; Jung, V.A.; Mesbahi, M.; Nguyen-Khoa, T.; Guérin-Pfyffer, S.; Schmitt, A.; Edelman, A.; et al. Comparative Proteomics of Respiratory Exosomes in Cystic Fibrosis, Primary Ciliary Dyskinesia and Asthma. J. Proteom. 2018, 185, 1–7. [Google Scholar] [CrossRef]
- Villamizar, O.; Waters, S.A.; Scott, T.; Grepo, N.; Jaffe, A.; Morris, K.V. Mesenchymal Stem Cell Exosome Delivered Zinc Finger Protein Activation of Cystic Fibrosis Transmembrane Conductance Regulator. J. Extracell. Vesicles 2021, 10, e12053. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Therapeutic Approaches | Mechanism of Action | Mutation Specificity | Issues to Overcome | Examples of References |
|---|---|---|---|---|
| Readthrough agents | Incorporation of an amino acid instead of a H2O molecule, leading to premature translation termination | Nonsense mutations | Nonsense-mediated decay reduces mRNA to act upon; amino acids altering the neoformed protein function can be incorporated; potential theoretical effect on the terminal stop codon; repeat administration needed | [13,14,15,16,17,18,19,20,21,22,23,24] |
| Engineered transfer RNA | Carries a nonsense suppressing anticodon to address the premature translation termination codon | Nonsense mutations | Need for an effective delivery vehicle to deliver to target cells and overcome natural barriers; repeat administration needed | [25,26] |
| mRNA addition | Addition of the correct CFTR-mRNA and synthesis of the CFTR protein | Mutation-agnostic | Need for an effective delivery vehicle to deliver to target cells and to overcome natural barriers; repeat administration needed | [27,28,29,30,31] |
| Antisense oligonucleotides | Oligonucleotides chemically modified to bind and restore target RNA | Mutation-specific | Need to deliver to target cells and to overcome natural barriers; repeat administration needed | [32,33,34,35,36,37] |
| DNA addition | Addition of the correct CFTR-encoding cDNA and synthesis of the CFTR protein | Mutation-agnostic | Need for an effective delivery vehicle to deliver to target cells and to overcome natural barriers, including the nuclear membrane | [38,39,40] |
| Gene editing | Repair of the mutation by the delivery of a nuclease and the correct CFTR cDNA sequence guide | Mutation-specific | Need for an effective delivery vehicle to deliver to target cells and to overcome natural barriers, including the nuclear membrane | [41,42,43,44] |
| Cell-based therapies | Gene editing of airway epithelial stem cells for later engraftment onto the airway basal membrane | Mutation-specific | Need to control the phenotype of corrected cells: type of cells, free of new mutation; safe and effective manner to engraft corrected cells into airways | [45,46,47,48,49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fajac, I.; Sermet, I. Therapeutic Approaches for Patients with Cystic Fibrosis Not Eligible for Current CFTR Modulators. Cells 2021, 10, 2793. https://doi.org/10.3390/cells10102793
Fajac I, Sermet I. Therapeutic Approaches for Patients with Cystic Fibrosis Not Eligible for Current CFTR Modulators. Cells. 2021; 10(10):2793. https://doi.org/10.3390/cells10102793
Chicago/Turabian StyleFajac, Isabelle, and Isabelle Sermet. 2021. "Therapeutic Approaches for Patients with Cystic Fibrosis Not Eligible for Current CFTR Modulators" Cells 10, no. 10: 2793. https://doi.org/10.3390/cells10102793