
The Potential Role of Cytokines and Growth Factors in the Pathogenesis of Alzheimer’s Disease

 , , , , , , , and
, , , , , , , and 
Abstract
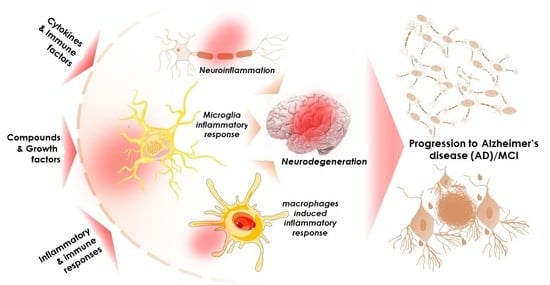
:
1. Introduction
2. Immune Response in AD: Role of Cytokines
2.1. Immune System in AD and Cytokines
2.2. Roles of Cytokines in Autophagy
2.3. Cytokines and BBB
3. Role of Cytokines and Chemokines in Neuropsychiatry
4. Neuroinflammation
4.1. Pro-Inflammatory Cytokines
4.2. Anti-Inflammatory Cytokines
4.3. APP Protein
4.4. TAU
4.5. Glial Cells
4.6. Advanced Glycation End Products
4.7. Complement System
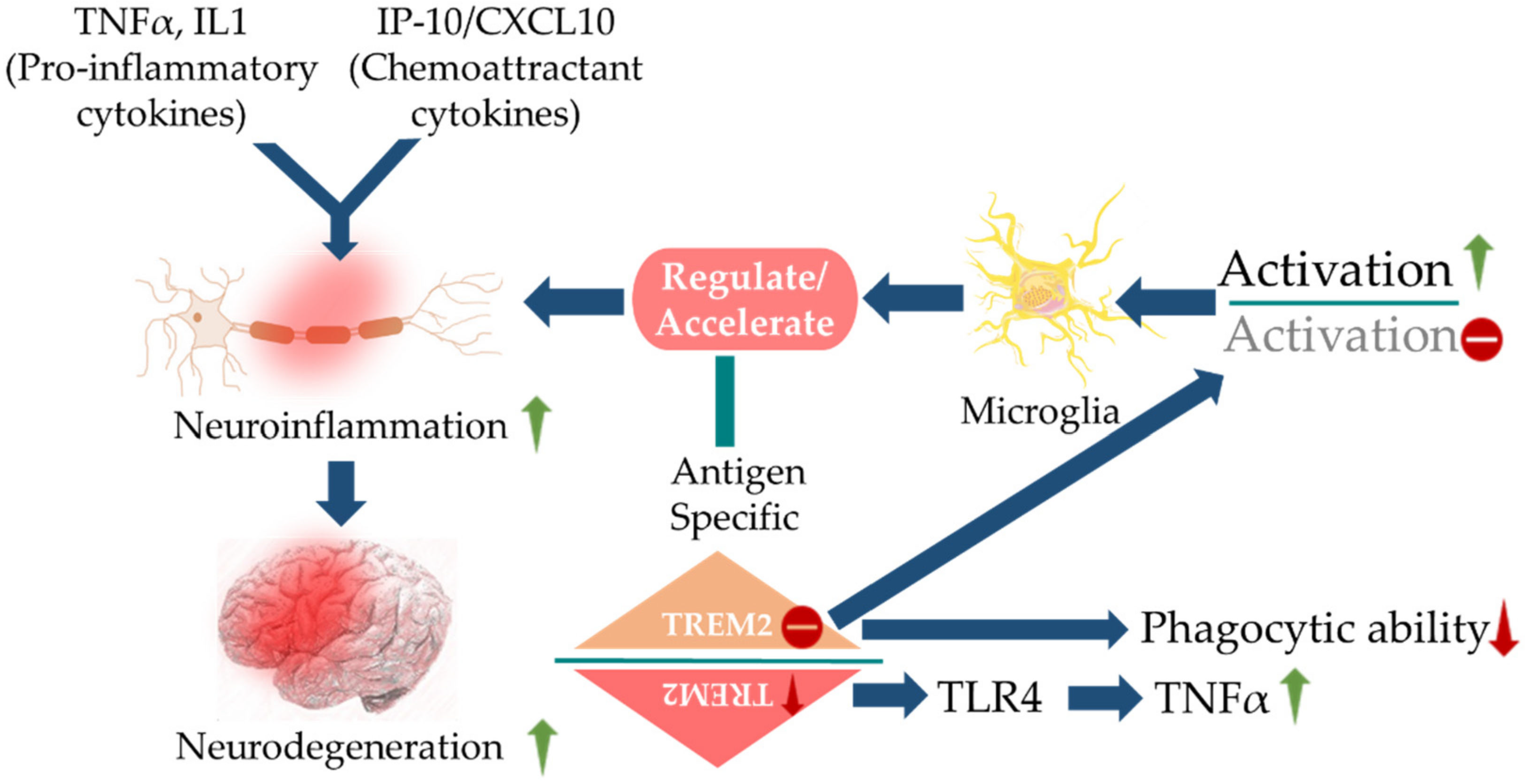
5. MMIFs in AD: Pathogenic or Protective?
6. Choroid Plexus Growth Factors and AD
6.1. Vascular Endothelial Growth Factors (VEGFs)
6.2. Fibroblast Growth Factors (FGF)
7. Neurotrophic Factors
8. Hematopoietic Growth Factors
9. Potential Strategies Involving Cytokines for Management of AD
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADAM | A disintegrin and metalloprotease |
| AICD | APP intracellular domain |
| APH-1 | Anterior pharynx defective-1 |
| ApoE | Apolipoprotein E |
| APP | Myloid precursor 69 protein |
| Aβ | Amyloid-beta |
| basic FGF/FGF2 | Basic fibroblast growth factor |
| BBB | Blood-brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| CDK5 | Cyclin-dependent kinase 5 |
| CNS | Central nervous system |
| CPLA2 | Cytosolic phospholipase A2 |
| CSF | Cerebrospinal fluid |
| DAMP | Danger-associated molecular pathways |
| GCSF | Granulocyte colony-stimulating factor |
| GDNF | Glial cell line-derived neurotrophic factor |
| GFAP | Glial fibrillary acidic protein |
| GMCSF | Granulocyte macrophage colony-stimulating factor |
| GSK-3β | Glycogen synthase kinase-3beta |
| IGF | Insulin-like growth factor |
| IL-1ra | IL-1 receptor antagonist |
| IL | Interleukin |
| INF | Interferon |
| LIFRb | Leukemia inhibitory factor receptor b |
| LPS | Lipopolysaccharide |
| MCI | Mild cognitive impairment |
| MCP1 | Monocyte chemoattractant protein 1 |
| MCSF | Macrophage colony-stimulating factor |
| MIIB | Myosin IIB |
| MIP | Macrophage inflammatory protein |
| MMIF | Macrophage migration inhibitory factor |
| NGF | Nerve growth factors |
| p38-MAPK | Mitogen-activated protein kinases |
| p75NTR | p75 neurotrophin receptor |
| Pen-2 | Presenilin enhancer-2 |
| PNS | Peripheral nervous system |
| PSEN | Presenilin |
| ROS | Reactive oxygen species |
| TACE | TNF-α converting enzyme |
| TGFβ | Transforming growth factor beta |
| TNFs | Tumor necrosis factors |
| TrK | Tropomyosin Receptor Kinases |
| VEGF | Vascular endothelial growth factor |
References
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Niikura, T.; Tajima, H.; Kita, Y. Neuronal cell death in Alzheimer’s disease and a neuroprotective factor, humanin. Curr. Neuropharmacol. 2006, 4, 139–147. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Menon, P.K.; Koistinen, N.A.; Iverfeldt, K.; Ström, A.L. Phosphorylation of the amyloid precursor protein (APP) at Ser-675 promotes APP processing involving meprin β. J. Biol. Chem. 2019, 294, 17768–17776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergström, P.; Agholme, L.; Nazir, F.H.; Satir, T.M.; Toombs, J.; Wellington, H.; Strandberg, J.; Bontell, T.O.; Kvartsberg, H.; Holmström, M.; et al. Amyloid precursor protein expression and processing are differentially regulated during cortical neuron differentiation. Sci. Rep. 2016, 6, 29200. [Google Scholar] [CrossRef] [Green Version]
- Medala, V.K.; Gollapelli, B.; Dewanjee, S.; Ogunmokun, G.; Kandimalla, R.; Vallamkondu, J. Mitochondrial dysfunction, mitophagy, and role of dynamin-related protein 1 in Alzheimer’s disease. J. Neurosci. Res. 2021, 99, 1120–1135. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers. Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Roy, E.R.; Wang, B.; Wan, Y.W.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Wang, M.M.; Miao, D.; Cao, X.P.; Tan, L.; Tan, L. Innate immune activation in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 177. [Google Scholar] [CrossRef] [PubMed]
- Suresh, J.; Khor, I.W.; Kaur, P.; Heng, H.L.; Torta, F.; Dawe, G.S.; Tai, E.S.; Tolwinski, N.S. Shared signaling pathways in Alzheimer’s and metabolic disease may point to new treatment approaches. FEBS J. 2021, 288, 3855–3873. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkelman, F.D.; Holmes, J.; Katona, I.M.; Urban, J.F., Jr.; Beckmann, M.P.; Park, L.S.; Schooley, K.A.; Coffman, R.L.; Mosmann, T.R.; Paul, W.E. Lymphokine control of in vivo immunoglobulin isotype selection. Annu. Rev. Immunol. 1990, 8, 303–333. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Forstl, H.; Levy, R. On certain peculiar diseases of old age. Hist. Psychiatry 1991, 2, 71–101. [Google Scholar] [CrossRef]
- Kawas, C.; Gray, S.; Brookmeyer, R.; Fozard, J.; Zonderman, A. Age-specific incidence rates of Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology 2000, 54, 2072–2077. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Ahn, I.S.; Kim, J.H.; Kim, S.; Ahn, I.S.; Kim, J.H.; Kim, S.; Chung, J.W.; Kim, H.; Kang, H.S.; Kim, D.K. Impairment of instrumental activities of daily living in patients with mild cognitive impairment. Psychiatry Investig. 2009, 6, 180–184. [Google Scholar] [CrossRef] [Green Version]
- Cloutier, S.; Chertkow, H.; Kergoat, M.J.; Gauthier, S.; Belleville, S. Patterns of Cognitive Decline Prior to Dementia in Persons with Mild Cognitive Impairment. J. Alzheimers Dis. 2015, 47, 901–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistridis, P.; Krumm, S.; Monsch, A.U.; Berres, M.; Taylor, K.I. The 12 Years Preceding Mild Cognitive Impairment Due to Alzheimer’s Disease: The Temporal Emergence of Cognitive Decline. J. Alzheimers Dis. 2015, 48, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, V.; Pupi, A.; Mosconi, L. PET/CT in diagnosis of dementia. Ann. N. Y. Acad. Sci. 2011, 1228, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werman, A.; Werman-Venkert, R.; White, R.; Lee, J.K.; Werman, B.; Krelin, Y.; Voronov, E.; Dinarello, C.A.; Apte, R.N. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 2434–2439. [Google Scholar] [CrossRef] [Green Version]
- Sattler, S. The Role of the Immune System Beyond the Fight Against Infection. Adv. Exp. Med. Biol. 2017, 1003, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Hartley, D.M.; Cahill, C.M.; Lahiri, D.K.; Chattopadhyay, N.; Rogers, J.T. Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J. Neurosci. Res. 2006, 84, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.J. Effects of interleukin-1beta polymorphisms on brain function and behavior in healthy and psychiatric disease conditions. Cytokine Growth Factor Rev. 2017, 37, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, M.L.; Brosnan, C.F.; Lee, S.C. Expression of type II nitric oxide synthase in primary human astrocytes and microglia: Role of IL-1beta and IL-1 receptor antagonist. J. Immunol. 1996, 157, 3569–3576. [Google Scholar]
- Rivera-Escalera, F.; Pinney, J.J.; Owlett, L.; Ahmed, H.; Thakar, J.; Olschowka, J.A.; Elliott, M.R.; O’Banion, M.K. IL-1beta-driven amyloid plaque clearance is associated with an expansion of transcriptionally reprogrammed microglia. J. Neuroinflamm. 2019, 16, 261. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory Cytokines Induce Amyloid Beta Neurotoxicity through Modulating Amyloid Precursor Protein Levels/Metabolism. Biomed. Res. Int. 2018, 2018, 3087475. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, P.; Margenthaler, E.; Reed, J.; Crawford, F.; Mullan, M. Depletion of CXCR2 inhibits gamma-secretase activity and amyloid-beta production in a murine model of Alzheimer’s disease. Cytokine 2011, 53, 163–169. [Google Scholar] [CrossRef]
- Koper, O.M.; Kaminska, J.; Sawicki, K.; Kemona, H. CXCL9, CXCL10, CXCL11, and their receptor (CXCR3) in neuroinflammation and neurodegeneration. Adv. Clin. Exp. Med. 2018, 27, 849–856. [Google Scholar] [CrossRef]
- Magalhaes, C.A.; Carvalho, M.D.G.; Sousa, L.P.; Caramelli, P.; Gomes, K.B. Alzheimer’s disease and cytokine IL-10 gene polymorphisms: Is there an association? Arq. Neuropsiquiatr. 2017, 75, 649–656. [Google Scholar] [CrossRef] [Green Version]
- Culjak, M.; Perkovic, M.N.; Uzun, S.; Strac, D.S.; Erjavec, G.N.; Leko, M.B.; Simic, G.; Tudor, L.; Konjevod, M.; Kozumplik, O.; et al. The Association between TNF-alpha, IL-1 alpha and IL-10 with Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 972–984. [Google Scholar] [CrossRef]
- Ojala, J.O.; Sutinen, E.M. The Role of Interleukin-18, Oxidative Stress and Metabolic Syndrome in Alzheimer’s Disease. J. Clin. Med. 2017, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, C.; Chromek, M.; Ahrengart, L.; Brauner, A.; Schultzberg, M.; Garlind, A. Soluble interleukin-1 receptor type II, IL-18 and caspase-1 in mild cognitive impairment and severe Alzheimer’s disease. Neurochem. Int. 2005, 46, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem. Int. 2006, 49, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef]
- Liu, L.; Martin, R.; Chan, C. Palmitate-activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol. Aging 2013, 34, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, L.D.; Oliveira-Cruz, L.; Cabrera, D. Transforming Growth Factor Beta Type I Role in Neurodegeneration: Implications for Alzheimer s Disease. Curr. Protein Pept. Sci. 2018, 19, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Khedr, E.M.; Gomaa, A.; Ahmed, O.G.; Sayed, H.M.; Gamea, A. Cognitive Impairment, P300, and Transforming Growth Factor β1 in Different Forms of Dementia. J. Alzheimer Dis. 2020, 78, 837–845. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Hunter, J.; Quadri, Z.; Zamudio, F.; Rocha-Rangel, P.V.; Chan, D.; Kesarwani, A.; Nash, K.; Lee, D.C.; Morgan, D.; et al. CCL2 Overexpression in the Brain Promotes Glial Activation and Accelerates Tau Pathology in a Mouse Model of Tauopathy. Front. Immunol. 2020, 11, 997. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse strocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Park, J.; Baik, S.H.; Mook-Jung, I.; Irimia, D.; Cho, H. Mimicry of Central-Peripheral Immunity in Alzheimer’s Disease and Discovery of Neurodegenerative Roles in Neutrophil. Front. Immunol. 2019, 10, 2231. [Google Scholar] [CrossRef]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro models of neurodegenerative diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Naveed, M.; Zhou, Q.G.; Han, F. Cerebrovascular inflammation: A critical trigger for neurovascular injury? Neurochem. Int. 2019, 126, 165–177. [Google Scholar] [CrossRef]
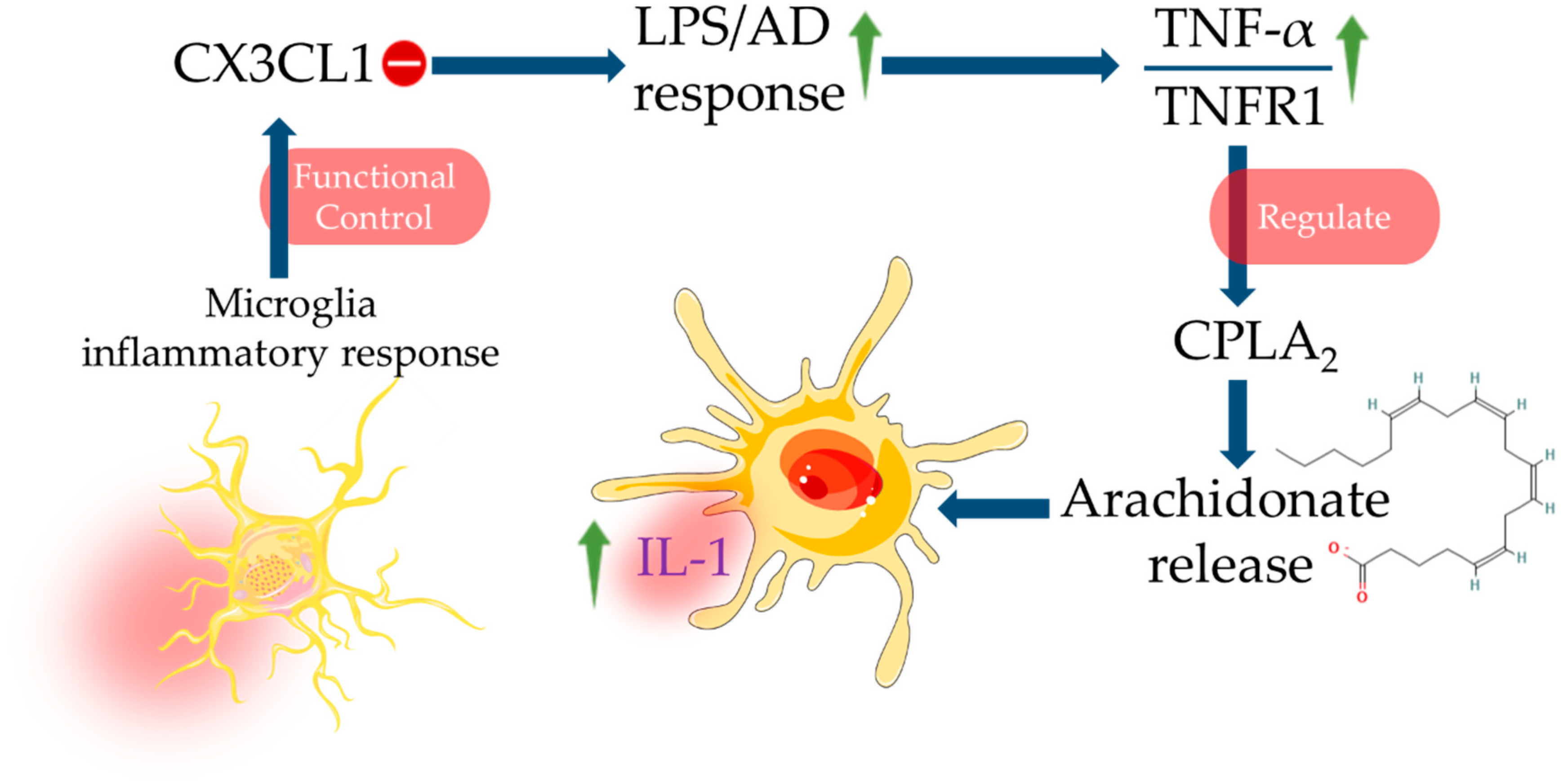
- Lee, S.; Xu, G.; Jay, T.R.; Bhatta, S.; Kim, K.W.; Jung, S.; Landreth, G.E.; Ransohoff, R.M.; Lamb, B.T. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J. Neurosci. 2014, 34, 12538–12546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, X.A.; Alvarez, I.; Aleixandre, M.; Linares, C.; Muresanu, D.; Winter, S.; Moessler, H. Severity-Related Increase and Cognitive Correlates of Serum VEGF Levels in Alzheimer’s Disease ApoE4 Carriers. J. Alzheimers Dis. 2018, 63, 1003–1013. [Google Scholar] [CrossRef]
- Cao, R.; Eriksson, A.; Kubo, H.; Alitalo, K.; Cao, Y.; Thyberg, J. Comparative evaluation of FGF-2-, VEGF-A-, and VEGF-C-induced angiogenesis, lymphangiogenesis, vascular fenestrations, and permeability. Circ. Res. 2004, 94, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Xie, Z.H.; Guo, Y.J.; Zhao, C.P.; Jiang, H.; Song, Y.; Zhu, Z.Y.; Lai, C.; Xu, S.L.; Bi, J.Z. VEGF-induced angiogenesis ameliorates the memory impairment in APP transgenic mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2011, 411, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Donnini, S.; Cantara, S.; Morbidelli, L.; Giachetti, A.; Ziche, M. FGF-2 overexpression opposes the beta amyloid toxic injuries to the vascular endothelium. Cell Death Differ. 2006, 13, 1088–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuello, A.C. The Involvement of NGF in the Alzheimer’s Pathology. Front. Neurosci. 2019, 13, 872. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Behbahani, H.; Eriksdotter, M. Innovative Therapy for Alzheimer’s Disease-With Focus on Biodelivery of NGF. Front. Neurosci. 2019, 13, 38. [Google Scholar] [CrossRef] [Green Version]
- Shabani, S.F.; Sarkaki, A.; Marda, S.A.; Ahangarpour, A.; Khorsandi, L. The effect of triiodothyronine on the hippocampal long-term potentiation in an animal model of the Alzheimer’s disease: The role of BDNF and reelin. Neurol. Psychiatry Brain Res. 2019, 33, 82–88. [Google Scholar] [CrossRef]
- Friedman, W.J.; Black, I.B.; Persson, H.; Ibanez, C.F. Synergistic trophic actions on rat basal forebrain neurons revealed by a synthetic NGF/BDNF chimaeric molecule. Eur. J. Neurosci. 1995, 7, 656–662. [Google Scholar] [CrossRef]
- Sharif, M.; Noroozian, M.; Hashemian, F. Do serum GDNF levels correlate with severity of Alzheimer’s disease? Neurol. Sci. 2021, 42, 2865–2872. [Google Scholar] [CrossRef]
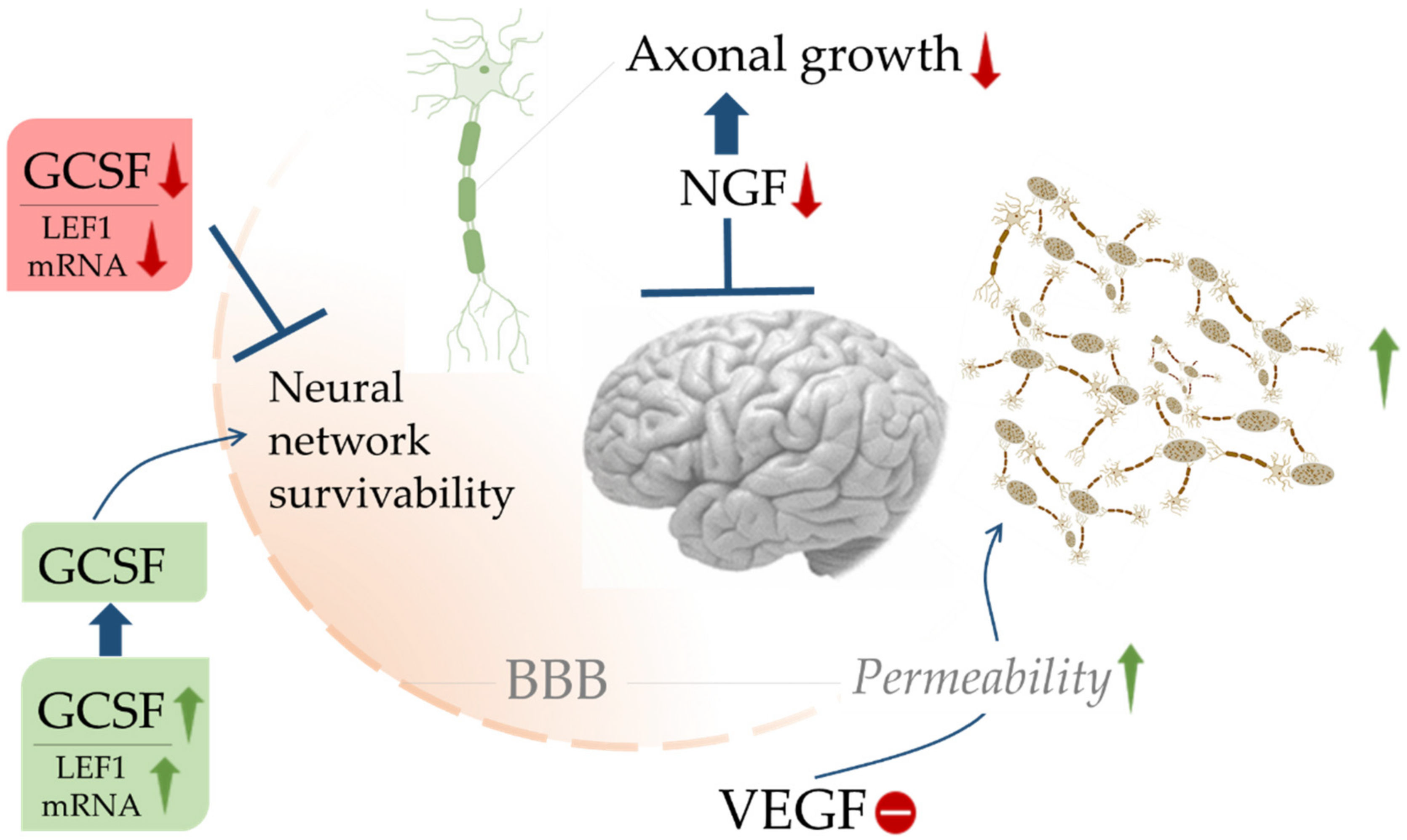
- Rahi, V.; Jamwal, S.; Kumar, P. Neuroprotection through G-CSF: Recent advances and future viewpoints. Pharmacol. Rep. 2021, 73, 372–385. [Google Scholar] [CrossRef]
- Laske, C.; Stellos, K.; Stransky, E.; Leyhe, T.; Gawaz, M. Decreased plasma levels of granulocyte-colony stimulating factor (G-CSF) in patients with early Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 115–123. [Google Scholar] [CrossRef]
- Laske, C.; Stellos, K.; Stransky, E.; Seizer, P.; Akcay, O.; Eschweiler, G.W.; Leyhe, T.; Gawaz, M. Decreased plasma and cerebrospinal fluid levels of stem cell factor in patients with early Alzheimer’s disease. J. Alzheimers Dis. 2008, 15, 451–460. [Google Scholar] [CrossRef]
- Boese, A.C.; Hamblin, M.H.; Lee, J.P. Neural stem cell therapy for neurovascular injury in Alzheimer’s disease. Exp. Neurol. 2020, 324, 113112. [Google Scholar] [CrossRef]
- Dalan, B.; Timirci-Kahraman, O.; Gulec-Yilmaz, S.; Altinkilic, E.M.; Duman, S.; Ayhan, H.; Isbir, T. Potential Protective Role of SDF-1 and CXCR4 Gene Variants in the Development of Dementia. Psychiatr. Danub. 2020, 32, 92–96. [Google Scholar] [CrossRef]
- Karaca, I.; Wagner, H.; Ramirez, A. Search for risk genes in Alzheimer’s disease. Der Nervenarzt 2017, 88, 744–750. [Google Scholar] [CrossRef]
- Karakus, S.; Bagci, B.; Bagci, G.; Sancakdar, E.; Yildiz, C.; Akkar, O.; Cetin, A. SDF-1/CXCL12 and CXCR4 gene variants, and elevated serum SDF-1 levels are associated with preeclampsia. Hypertens. Pregnancy 2017, 36, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.I.; Rakugi, H.; Morishita, R. Insight into the Role of Angiopoietins in Ageing-Associated Diseases. Cells 2020, 9, 2636. [Google Scholar] [CrossRef]
- Eklund, L.; Saharinen, P. Angiopoietin signaling in the vasculature. Exp. Cell Res. 2013, 319, 1271–1280. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Zhang, L.; Croll, S.D.; Chopp, M. Angiopoietin-1 reduces cerebral blood vessel leakage and ischemic lesion volume after focal cerebral embolic ischemia in mice. Neuroscience 2002, 113, 683–687. [Google Scholar] [CrossRef]
- Schindowski, K.; Belarbi, K.; Buee, L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav. 2008, 7, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L.; Li, W.W.; Xu, Y.L.; Gao, S.H.; Xu, M.Y.; Bu, X.L.; Liu, Y.H.; Wang, J.; Zhu, J.; Zeng, F.; et al. Neurotrophin receptor p75 mediates amyloid beta-induced tau pathology. Neurobiol. Dis. 2019, 132, 104567. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Skaper, S.D.; Dal Toso, R.; Petrelli, L.; Leon, A. Nerve growth factor: From neurotrophin to neurokine. Trends Neurosci. 1996, 19, 514–520. [Google Scholar] [CrossRef]
- Savaskan, E.; Muller-Spahn, F.; Olivieri, G.; Bruttel, S.; Otten, U.; Rosenberg, C.; Hulette, C.; Hock, C. Alterations in trk A, trk B and trk C receptor immunoreactivities in parietal cortex and cerebellum in Alzheimer’s disease. Eur. Neurol. 2000, 44, 172–180. [Google Scholar] [CrossRef]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Nabeshima, T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharmacol. Sci. 2003, 91, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, G.J.; Chauhan, N.B. Neurotrophic factors in Alzheimer’s and Parkinson’s disease brain. Brain Res. Brain. Res. Rev. 2000, 33, 199–227. [Google Scholar] [CrossRef]
- Ohm, D.T.; Fought, A.J.; Martersteck, A.; Coventry, C.; Sridhar, J.; Gefen, T.; Weintraub, S.; Bigio, E.; Mesulam, M.M.; Rogalski, E.; et al. Accumulation of neurofibrillary tangles and activated microglia is associated with lower neuron densities in the aphasic variant of Alzheimer’s disease. Brain Pathol. 2021, 31, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.B.A.; Cusack, C.A.; Joshi, S.G. Alzheimer’s Disease: Intracellular Beta Amyloid Completes the Irreversible Pathway from Spirochetes to Biofilms to Beta Amyloid to Hyperphosphorylated Tau Protein. J. Neuroinfect. Dis. 2018, 9, 276. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Basile, M.S.; Lenzo, V.; Quattropani, M.C.; Di Nuovo, S.; Bendtzen, K.; Nicoletti, F. The cytokine network in the pathogenesis of major depressive disorder. Close to translation? Autoimmun. Rev. 2020, 19, 102504. [Google Scholar] [CrossRef]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular Senescence in Brain Aging. Front. Aging Neurosci. 2021, 13, 646924. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov AGAtanasov, A.G. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Lambelet, M.; Terra, L.F.; Fukaya, M.; Meyerovich, K.; Labriola, L.; Cardozo, A.K.; Allagnat, F. Dysfunctional autophagy following exposure to pro-inflammatory cytokines contributes to pancreatic β-cell apoptosis. Cell Death Dis. 2018, 9, 96. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- François, A.; Terro, F.; Janet, T.; RiouxBilan, A.; Paccalin, M.; Page, G. Involvement of interleukin-1β in the autophagic process of microglia: Relevance to Alzheimer’s disease. J. Neuroinflamm. 2013, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Yarlagadda, A.; Alfson, E.; Clayton, A.H. The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry 2009, 6, 18–22. [Google Scholar]
- Dantzer, R.; Kelley, K.W. Twenty years of research on cytokine-induced sickness behavior. Brain Behav. Immun. 2007, 21, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, E.G.; Banks, W.A.; Kastin, A.J. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J. Neuroimmunol. 1993, 47, 169–176. [Google Scholar] [CrossRef]
- Wang, K.; Wang, H.; Lou, W.; Ma, L.; Li, Y.; Zhang, N.; Wang, C.; Li, F.; Awais, M.; Cao, S.; et al. IP-10 Promotes Blood-Brain Barrier Damage by Inducing Tumor Necrosis Factor Alpha Production in Japanese Encephalitis. Front. Immunol. 2018, 9, 1148. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Rahman, M.A.; Kabir, M.T.; Behl, T.; Mathew, B.; Perveen, A.; Barreto, G.E.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Multifarious roles of mTOR signaling in cognitive aging and cerebrovascular dysfunction of Alzheimer’s disease. IUBMB Life 2020, 72, 1843–1855. [Google Scholar] [CrossRef]
- Maes, M. A review on the acute phase response in major depression. Rev. Neurosci. 1993, 4, 407–416. [Google Scholar] [CrossRef]
- Bahniwal, M.; Little, J.P.; Klegeris, A. High Glucose Enhances Neurotoxicity and Inflammatory Cytokine Secretion by Stimulated Human Astrocytes. Curr. Alzheimer Res. 2017, 14, 731–741. [Google Scholar] [CrossRef]
- Griffin, W.S.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflamm. 2006, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachida, Y.; Nakagawa, K.; Saito, T.; Saido, T.C.; Honda, T.; Saito, Y.; Murayama, S.; Endo, T.; Sakaguchi, G.; Kato, A.; et al. Interleukin-1 beta up-regulates TACE to enhance alpha-cleavage of APP in neurons: Resulting decrease in Abeta production. J. Neurochem. 2008, 104, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Lue, L.F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Tredici, K.D.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimers Dis. 2019, 6, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Olloquequi, J.; Cornejo-Cordova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef]
- Chen, S.; Yang, J.; Wei, Y.; Wei, X. Epigenetic regulation of macrophages: From homeostasis maintenance to host defense. Cell Mol. Immunol. 2020, 17, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.Z.; Yang, C.; Zhuang, X.X.; Yuan, N.N.; Wu, M.Y.; Tan, J.Q.; Song, J.X.; Cheung, K.H.; Su, H.; Wang, Y.T.; et al. NRBF2 is a RAB7 effector required for autophagosome maturation and mediates the association of APP-CTFs with active form of RAB7 for degradation. Autophagy 2021, 17, 1112–1130. [Google Scholar] [CrossRef]
- Yang, C.; Cai, C.Z.; Song, J.X.; Tan, J.Q.; Durairajan, S.S.K.; Iyaswamy, A.; Wu, M.Y.; Chen, L.L.; Yue, Z.; Li, M.; et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 2017, 13, 2028–2040. [Google Scholar] [CrossRef] [PubMed]
- Ennerfelt, H.E.; Lukens, J.R. The role of innate immunity in Alzheimer’s disease. Immunol. Rev. 2020, 297, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Popp, J.; Oikonomidi, A.; Tautvydaite, D.; Dayon, L.; Bacher, M.; Migliavacca, E.; Henry, H.; Kirkland, R.; Severin, I.; Wojcik, J.; et al. Markers of neuroinflammation associated with Alzheimer’s disease pathology in older adults. Brain Behav. Immun. 2017, 62, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, Y.; Singh, R.; Parhar, I.; Kuhad, A.; Soga, T. Quinolinic Acid and Nuclear Factor Erythroid 2-Related Factor 2 in Depression: Role in Neuroprogression. Front. Pharmacol. 2019, 10, 452. [Google Scholar] [CrossRef]
- Khemka, V.K.; Ganguly, A.; Bagchi, D.; Ghosh, A.; Bir, A.; Biswas, A.; Chattopadhyay, S.; Chakrabarti, S. Raised serum proinflammatory cytokines in Alzheimer’s disease with depression. Aging Dis. 2014, 5, 170–176. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm. 2005, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.D.; Iannotti, F.; Pringle, A.K.; Trikkas, C.; Clough, G.F.; Church, M.K. A microdialysis method for the recovery of IL-1beta, IL-6 and nerve growth factor from human brain in vivo. J. Neurosci. Methods 2002, 119, 45–50. [Google Scholar] [CrossRef]
- Chong, Y. Effect of a carboxy-terminal fragment of the Alzheimer’s amyloid precursor protein on expression of proinflammatory cytokines in rat glial cells. Life Sci. 1997, 61, 2323–2333. [Google Scholar] [CrossRef]
- Agbo, D.B.; Neff, F.; Seitz, F.; Binder, C.; Oertel, W.H.; Bacher, M.; Dodel, R. Immunization as treatment for Parkinson’s disease. J. Neural. Transm. Suppl. 2009, 73, 311–315. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Lin, C.; von Euw, D.; Masliah, E.; Mucke, L.; Lacombe, P. Alzheimer’s disease-like cerebrovascular pathology in transforming growth factor-beta 1 transgenic mice and functional metabolic correlates. Ann. N. Y. Acad. Sci. 2000, 903, 317–323. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Frey, W.H., 2nd; Ala, T.A.; Tourtellotte, W.W.; Peterson, P.K. Transforming growth factor beta in Alzheimer’s disease. Clin. Diagn. Lab. Immunol. 1994, 1, 109–110. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Prachayasittikul, V.; Choomwattana, S.; Wongchitrat, P.; Phopin, K.; Suwanjang, W.; Malik, A.A.; Vincent, B.; Nantasenamat, C. Multidisciplinary approaches for targeting the secretase protein family as a therapeutic route for Alzheimer’s disease. Med. Res. Rev. 2019, 39, 1730–1778. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid beta oligomers (AbetaOs) in Alzheimer’s disease. J. Neural. Transm. 2018, 125, 177–191. [Google Scholar] [CrossRef]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates with Changes in Its Function Beyond Microtubule Stability. Front. Cell. Neurosci. 2018, 12, 338. [Google Scholar] [CrossRef] [Green Version]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3beta: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery? J. Med. Chem. 2021, 64, 26–41. [Google Scholar] [CrossRef]
- Gotz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Busciglio, J.; Lorenzo, A.; Yeh, J.; Yankner, B.A. beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995, 14, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2007, 2, e536. [Google Scholar] [CrossRef]
- Dzamba, D.; Harantova, L.; Butenko, O.; Anderova, M. Glial Cells—The Key Elements of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 894–911. [Google Scholar] [CrossRef] [PubMed]
- Baaklini, C.S.; Rawji, K.S.; Duncan, G.J.; Ho, M.F.S.; Plemel, J.R. Central Nervous System Remyelination: Roles of Glia and Innate Immune Cells. Front. Mol. Neurosci. 2019, 12, 225. [Google Scholar] [CrossRef] [PubMed]
- Vesce, S.; Rossi, D.; Brambilla, L.; Volterra, A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int. Rev. Neurobiol. 2007, 82, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Rossner, S.; Lange-Dohna, C.; Zeitschel, U.; Perez-Polo, J.R. Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J. Neurochem. 2005, 92, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Yang, Y.; Ju, W.N.; Wang, X.; Zhang, H.L. Emerging Roles of Astrocytes in Neuro-Vascular Unit and the Tripartite Synapse with Emphasis on Reactive Gliosis in the Context of Alzheimer’s Disease. Front. Cell Neurosci. 2018, 12, 193. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintela-Lopez, T.; Ortiz-Sanz, C.; Serrano-Regal, M.P.; Gaminde-Blasco, A.; Valero, J.; Baleriola, J.; Sanchez-Gomez, M.V.; Matute, C.; Alberdi, E. Abeta oligomers promote oligodendrocyte differentiation and maturation via integrin beta1 and Fyn kinase signaling. Cell Death Dis. 2019, 10, 445. [Google Scholar] [CrossRef]
- Li, Y.M.; Dickson, D.W. Enhanced binding of advanced glycation endproducts (AGE) by the ApoE4 isoform links the mechanism of plaque deposition in Alzheimer’s disease. Neurosci Lett. 1997, 226, 155–158. [Google Scholar] [CrossRef]
- Prasad, K. AGE-RAGE stress: A changing landscape in pathology and treatment of Alzheimer’s disease. Mol. Cell Biochem. 2019, 459, 95–112. [Google Scholar] [CrossRef]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palimeri, S.; Palioura, E.; Diamanti-Kandarakis, E. Current perspectives on the health risks associated with the consumption of advanced glycation end products: Recommendations for dietary management. Diabetes Metab. Syndr. Obes. 2015, 8, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Puyvelde, K.; Mets, T.; Njemini, R.; Beyer, I.; Bautmans, I. Effect of advanced glycation end product intake on inflammation and aging: A systematic review. Nutr. Rev. 2014, 72, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Fatoba, O.; Itokazu, T.; Yamashita, T. Complement cascade functions during brain development and neurodegeneration. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tenner, A.J. Complement-Mediated Events in Alzheimer’s Disease: Mechanisms and Potential Therapeutic Targets. J. Immunol. 2020, 204, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Tenner, A.J. Complement in Alzheimer’s disease: Opportunities for modulating protective and pathogenic events. Neurobiol. Aging 2001, 22, 849–861. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Yan, F.; Lin, A.H.; Lambris, J.D.; Alexander, J.J.; Quigg, R.J.; Masliah, E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. USA 2002, 99, 10837–10842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Hou, W.T.; Zeng, L.; Li, Z.P.; Ge, W.; Yi, C.; Kang, J.P.; Li, W.M.; Wang, F.; Wu, D.B.; et al. Progress in the study of markers related to glioma prognosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7690–7697. [Google Scholar] [CrossRef]
- Munaut, C.; Boniver, J.; Foidart, J.M.; Deprez, M. Macrophage migration inhibitory factor (MIF) expression in human glioblastomas correlates with vascular endothelial growth factor (VEGF) expression. Neuropathol. Appl. Neurobiol. 2002, 28, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Nie, K.; Zhang, Q.; Guo, M.; Qiu, Y.; Li, Y.; Gao, Y.; Wang, L. Macrophage Migration Inhibitory Factor Mediates Neuroprotective Effects by Regulating Inflammation, Apoptosis and Autophagy in Parkinson’s Disease. Neuroscience 2019, 416, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Leyton-Jaimes, M.F.; Kahn, J.; Israelson, A. AAV2/9-mediated overexpression of MIF inhibits SOD1 misfolding, delays disease onset, and extends survival in mouse models of ALS. Proc. Natl. Acad. Sci. USA 2019, 116, 14755–14760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyton-Jaimes, M.F.; Benaim, C.; Abu-Hamad, S.; Kahn, J.; Guetta, A.; Bucala, R.; Israelson, A. Endogenous macrophage migration inhibitory factor reduces the accumulation and toxicity of misfolded SOD1 in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2016, 113, 10198–10203. [Google Scholar] [CrossRef] [Green Version]
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; John, A.; Reddy, P.H.; Kandimalla, R. Autophagy in the diabetic heart: A potential pharmacotherapeutic target in diabetic cardiomyopathy. Ageing Res. Rev. 2021, 68, 101338. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; Chakraborty, P.; Gangopadhyay, M.; Sahu, R.; Medala, V.; John, A.; Reddy, P.H.; De Feo, V.; et al. The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus. Cells 2021, 10, 1340. [Google Scholar] [CrossRef]
- Stosic-Grujicic, S.; Saksida, T.; Miljkovic, D.; Stojanovic, I. MIF and insulin: Lifetime companions from common genesis to common pathogenesis. Cytokine 2020, 125, 154792. [Google Scholar] [CrossRef]
- Liang, C.J.; Li, J.H.; Zhang, Z.; Zhang, J.Y.; Liu, S.Q.; Yang, J. Suppression of MIF-induced neuronal apoptosis may underlie the therapeutic effects of effective components of Fufang Danshen in the treatment of Alzheimer’s disease. Acta Pharmacol. Sin. 2018, 39, 1421–1438. [Google Scholar] [CrossRef]
- Bacher, M.; Deuster, O.; Aljabari, B.; Egensperger, R.; Neff, F.; Jessen, F.; Popp, J.; Noelker, C.; Reese, J.P.; Al-Abed, Y.; et al. The role of macrophage migration inhibitory factor in Alzheimer’s disease. Mol. Med. 2010, 16, 116–121. [Google Scholar] [CrossRef]
- Makhouri, F.R.; Ghasemi, J.B. In Silico Studies in Drug Research Against Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 664–725. [Google Scholar] [CrossRef]
- Popp, J.; Bacher, M.; Kolsch, H.; Noelker, C.; Deuster, O.; Dodel, R.; Jessen, F. Macrophage migration inhibitory factor in mild cognitive impairment and Alzheimer’s disease. J. Psychiatr. Res. 2009, 43, 749–753. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Li, J.; Schultz, L.; McAllister, J.P., 2nd. Intraventricular infusion of hyperosmolar dextran induces hydrocephalus: A novel animal model of hydrocephalus. Cereb. Fluid Res. 2009, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Zachary, I. Signaling mechanisms mediating vascular protective actions of vascular endothelial growth factor. Am. J. Physiol. Cell Physiol. 2001, 280, C1375–C1386. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Ramini, D.; Giuliani, A.; Recchioni, R.; Spazzafumo, L.; Olivieri, F. Connecting vascular aging and frailty in Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111444. [Google Scholar] [CrossRef]
- Price, B.R.; Johnson, L.A.; Norris, C.M. Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101335. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Cohen, D.L.; Premkumar, D.R.; Nag, S.; LaManna, J.C.; Lust, W.D. Vascular endothelial growth factor in Alzheimer’s disease and experimental cerebral ischemia. Brain Res. Mol. Brain Res. 1998, 62, 101–105. [Google Scholar] [CrossRef]
- Edmonston, D.; Wolf, M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat. Rev. Nephrol. 2020, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Govindpani, K.; McNamara, L.G.; Smith, N.R.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J. Clin. Med. 2019, 8, 651. [Google Scholar] [CrossRef] [Green Version]
- Belluardo, N.; Mudo, G.; Blum, M.; Itoh, N.; Agnati, L.; Fuxe, K. Nicotine-induced FGF-2 mRNA in rat brain is preserved during aging. Neurobiol. Aging 2004, 25, 1333–1342. [Google Scholar] [CrossRef]
- Chen, S.; Chen, S.T.; Sun, Y.; Xu, Z.; Wang, Y.; Yao, S.Y.; Yao, W.B.; Gao, X.D. Fibroblast growth factor 21 ameliorates neurodegeneration in rat and cellular models of Alzheimer’s disease. Redox Biol. 2019, 22, 101133. [Google Scholar] [CrossRef]
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Mol. Neurosci. 2018, 65, 289–300. [Google Scholar] [CrossRef]
- Shizuru, J.A.; Negrin, R.S.; Weissman, I.L. Hematopoietic stem and progenitor cells: Clinical and preclinical regeneration of the hematolymphoid system. Annu. Rev. Med. 2005, 56, 509–538. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Peel, A.L.; Mao, X.O.; Xie, L.; Cottrell, B.A.; Henshall, D.C.; Greenberg, D.A. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Sopova, K.; Gatsiou, K.; Stellos, K.; Laske, C. Dysregulation of neurotrophic and haematopoietic growth factors in Alzheimer’s disease: From pathophysiology to novel treatment strategies. Curr. Alzheimer Res. 2014, 11, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Boekhoorn, K.; Joels, M.; Lucassen, P.J. Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol. Dis. 2006, 24, 1–14. [Google Scholar] [CrossRef]
- Peng, Z.; Luo, Y.; Xiao, Z.Y. Angiopoietin-1 accelerates Alzheimer’s disease via FOXA2/PEN2/APP pathway in APP/PS1 mice. Life Sci. 2020, 246, 117430. [Google Scholar] [CrossRef]
- Wimo, A.; Handels, R.; Winblad, B.; Black, C.M.; Johansson, G.; Salomonsson, S.; Eriksdotter, M.; Khandker, R.K. Quantifying and Describing the Natural History and Costs of Alzheimer’s Disease and Effects of Hypothetical Interventions. J. Alzheimers Dis. 2020, 75, 891–902. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophic Factor and Glucocorticoid Stress in Neurogenesis. Int. J. Mol. Sci. 2017, 18, 2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Permanne, B.; Adessi, C.; Saborio, G.P.; Fraga, S.; Frossard, M.J.; Van Dorpe, J.; Dewachter, I.; Banks, W.A.; Van Leuven, F.; Soto, C. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer’s disease by treatment with a beta-sheet breaker peptide. FASEB J. 2002, 16, 860–862. [Google Scholar] [CrossRef]
- Bortolotto, V.; Grilli, M. Every Cloud Has a Silver Lining: Proneurogenic Effects of Abeta Oligomers and HMGB-1 via Activation of the RAGE-NF-kappaB Axis. CNS Neurol. Disord. Drug Targets 2017, 16, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Konar, A.; Kalra, R.S.; Chaudhary, A.; Nayak, A.; Guruprasad, K.P.; Satyamoorthy, K.; Ishida, Y.; Terao, K.; Kaul, S.C.; Wadhwa, R. Identification of Caffeic Acid Phenethyl Ester (CAPE) as a Potent Neurodifferentiating Natural Compound That Improves Cognitive and Physiological Functions in Animal Models of Neurodegenerative Diseases. Front. Aging Neurosci. 2020, 12, 561925. [Google Scholar] [CrossRef]
- Martin, I. Resveratrol for Alzheimer’s disease? Sci. Transl. Med. 2017, 9, eaam6055. [Google Scholar] [CrossRef]
- Delgado, A.; Cholevas, C.; Theoharides, T.C. Neuroinflammation in Alzheimer’s disease and beneficial action of luteolin. Biofactors 2021, 47, 207–217. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Paris, D.; Melck, D.; Angiolillo, A.; Corso, G.; Maniscalco, M.; Motta, A. Blood biomarkers indicate that the preclinical stages of Alzheimer’s disease present overlapping molecular features. Sci. Rep. 2020, 10, 15612. [Google Scholar] [CrossRef]
- Biringer, R.G. The Role of Eicosanoids in Alzheimer’s Disease. Int. J. Environ. Res. Public Health 2019, 16, 2560. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Letterio, J.J.; Roberts, A.B. TGF-beta: A critical modulator of immune cell function. Clin. Immunol. Immunopathol. 1997, 84, 244–250. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, R.; Kaelber, D.C.; Gurney, M.E. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS ONE 2020, 15, e0229819. [Google Scholar] [CrossRef] [Green Version]
- McGeer, P.L.; Schulzer, M.; McGeer, E.G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: A review of 17 epidemiologic studies. Neurology 1996, 47, 425–432. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Brites, D.; Fernandes, A. Neuroinflammation and Depression: Microglia Activation, Extracellular Microvesicles and microRNA Dysregulation. Front. Cell Neurosci. 2015, 9, 476. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Li, F.; Maiese, K. Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr. Neurovasc. Res. 2005, 2, 387–399. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Cisbani, G.; Rivest, S. Targeting innate immunity to protect and cure Alzheimer’s disease: Opportunities and pitfalls. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.L. Role of Microglia in Neurological Disorders and Their Potentials as a Therapeutic Target. Mol. Neurobiol. 2017, 54, 7567–7584. [Google Scholar] [CrossRef]
- Chaudhary, A.; Kalra, R.S.; Huang, C.; Prakash, J.; Kaul, S.C.; Wadhwa, R. 2,3-Dihydro-3beta-methoxy Withaferin-A Protects Normal Cells against Stress: Molecular Evidence of Its Potent Cytoprotective Activity. J. Nat. Prod. 2017, 80, 2756–2760. [Google Scholar] [CrossRef]
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef]
- Zhang, F.; Zhong, R.J.; Cheng, C.; Li, S.; Le, W.D. New therapeutics beyond amyloid-beta and tau for the treatment of Alzheimer’s disease. Acta Pharmacol. Sin. 2021, 42, 1382–1389. [Google Scholar] [CrossRef]
- Casali, B.T.; Reed-Geaghan, E.G. Microglial Function and Regulation during Development, Homeostasis and Alzheimer’s Disease. Cells 2021, 10, 957. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial. No. | Stages | Pathological Symptoms |
|---|---|---|
| 1 | Early onset AD/MCI | Impairment of non-memory features of cognition, difficulty in word finding, decline in reasoning/judgement. |
| 2 | Mild AD | Loss of spontaneity, memory loss, anxiety, aggression, restlessness, altered personality, misplacing items. |
| 3 | Moderate AD | Confusion, attention deficit, continuous cognition problems, impulsive behavior, delusion, paranoia, hallucination, recognition problem. |
| 4 | Severe AD | Severe dementia, continued cognitive decline, seizures, functional limitations, lack of bowel/bladder control, weight loss, skin infection, swallowing difficulty, enhanced sleep time, brain atrophy. |
| Serial No. | Mediators | Functions | References |
|---|---|---|---|
| 1 | IL-1α | Increases α-secretase, decreases amyloidogenic processing, increases sAPPα | [24,26,27] |
| 2 | IL-1β | Increases APP mRNA, increases α-secretase and γ-secretase, downregulates β-secretase, upregulates TAU mRNA | [28,29,30] |
| 3 | IL-4 | Upregulates Aβ production, increases p-TAU | [30,31] |
| 4 | IL-6 | Upregulates APP mRNA, increases p-TAU | [10,32] |
| 5 | IL-8/CXCL8 | Upregulates γ-secretase activity by increasing substrates C83 and C99 | [33,34] |
| 6 | IL-10 | Favors Aβ deposition | [10,35,36] |
| 7 | IL-18 | Increases APP, upregulates both β-secretase and γ-secretase, increases Aβ formation | [10,37,38] |
| 8 | TNF-α | Upregulates APP mRNA, upregulates both β-secretase and γ-secretase, increases sAPPβ | [10,36,39] |
| 9 | IFN-γ | Upregulates APP intracellular domains, upregulates both β-secretase and γ-secretase, increases Aβ deposition | [40,41,42,43] |
| 10 | TGF-β1 | Increases APP mRNA, increases Aβ deposition | [10,42,43] |
| 11 | CCL2 | Increases Aβ formation and deposition | [44,45] |
| 12 | CCL3 | Upregulates β-secretase, increases C99, increases Aβ deposition | [45,46] |
| 13 | CCL5 | Upregulates β-secretase, increase C99, increases Aβ deposition | [46,47] |
| 14 | CXCL10 | Decreases Aβ deposition | [34,48] |
| 15 | CX3CL1 | Decreased Aβ deposition, upregulated p-TAU | [49,50] |
| 16 | VEGF | Upregulates expressions of monocytes and macrophages, increases proliferation of endothelial cells | [51,52,53] |
| 17 | FGF | Attenuates Aβ related pathologies | [52,54] |
| 18 | NGF | Increases degeneration leads to loss of cholinergic nerve endings in cortex and hippocampus | [55,56] |
| 19 | BDNF | Upregulates sAPPα, promotes non-amyloidogenic pathway, astrocyte activation, improved memory performance | [57,58] |
| 20 | GDNF | Neuroprotection | [55,59] |
| 21 | GCSF | Induces neurogenesis | [60,61] |
| 22 | Stem cell factor | Maintains hematopoietic brain support, neurogenesis | [62,63] |
| 23 | SDF | Neurogenesis, inflammatory disruption of BBB | [64,65] |
| 24 | CXCR4 | Ligand for SDF-1 | [64,66] |
| 25 | Angiopoeitins | Angiopoeitin-1 prevents neuronal apoptosis, Angiopoeitin-2 promotes neurogenesis via migration of neural progenitor cells | [67,68,69] |
| 26 | Neurotrophin-3 | Upregulates neuronal apoptosis inhibitory protein 1, limits cleavage of caspases 3, 8 and 9 | [70,71] |
| 27 | Neurotrophin-4 | Regulates TAU dephosphorylation | [70,72] |
| 28 | TrKA | Receptor protein for β-NGF | [73,74] |
| 29 | TrKB | Receptor protein for brain derived neurotrophic factor and neurotrophins | [73,75] |
| 30 | TrKC | Receptor protein for neurotrophin-3 | [73,76] |
| 31 | p75 | Neurotrophin receptor protein, regulates phosphorylation of TAU | [71,72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogunmokun, G.; Dewanjee, S.; Chakraborty, P.; Valupadas, C.; Chaudhary, A.; Kolli, V.; Anand, U.; Vallamkondu, J.; Goel, P.; Paluru, H.P.R.; et al. The Potential Role of Cytokines and Growth Factors in the Pathogenesis of Alzheimer’s Disease. Cells 2021, 10, 2790. https://doi.org/10.3390/cells10102790
Ogunmokun G, Dewanjee S, Chakraborty P, Valupadas C, Chaudhary A, Kolli V, Anand U, Vallamkondu J, Goel P, Paluru HPR, et al. The Potential Role of Cytokines and Growth Factors in the Pathogenesis of Alzheimer’s Disease. Cells. 2021; 10(10):2790. https://doi.org/10.3390/cells10102790
Chicago/Turabian StyleOgunmokun, Gilbert, Saikat Dewanjee, Pratik Chakraborty, Chandrasekhar Valupadas, Anupama Chaudhary, Viswakalyan Kolli, Uttpal Anand, Jayalakshmi Vallamkondu, Parul Goel, Hari Prasad Reddy Paluru, and et al. 2021. "The Potential Role of Cytokines and Growth Factors in the Pathogenesis of Alzheimer’s Disease" Cells 10, no. 10: 2790. https://doi.org/10.3390/cells10102790