
Ion Channel Impairment and Myofilament Ca2+ Sensitization: Two Parallel Mechanisms Underlying Arrhythmogenesis in Hypertrophic Cardiomyopathy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Hypertrophic Cardiomyopathy: A Brief Overview Identifying Sudden Cardiac Death as the Principal Clinical Burden of the Disease
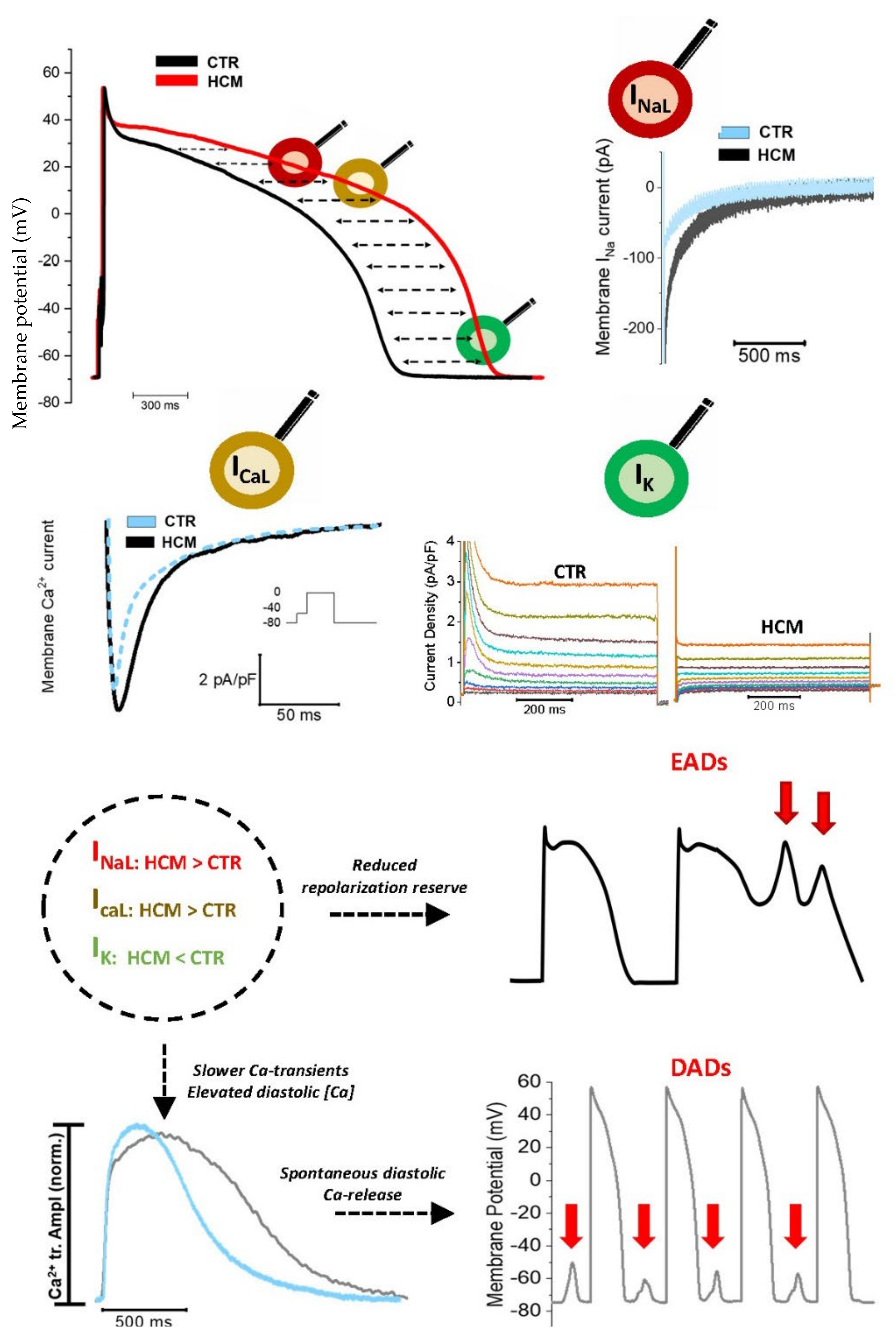
2. Changes in the Density of Ion Currents Result into Action Potential Prolongation in HCM Cardiomyocytes: Insights from Multiple Models
2.1. Human Surgical Samples
2.2. Animal Models
2.3. hiPSC
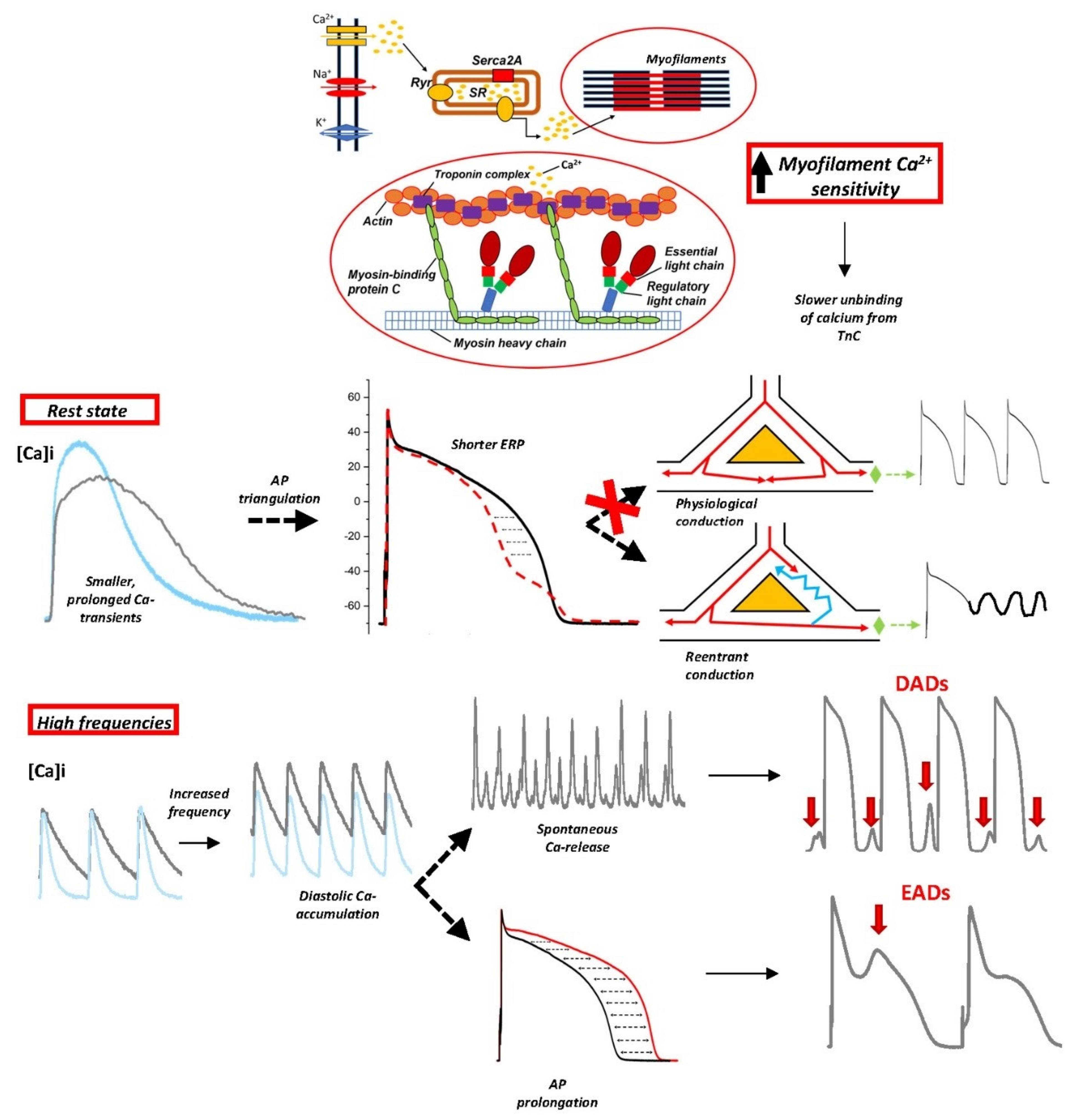
3. Myofilament Ca2+ Sensitization as a Determinant of Diastolic Dysfunction and Arrhythmogenesis in HCM Hearts
3.1. Increased Myofilament Ca2+ Sensitivity in HCM
3.2. Myofilament Ca2+ Sensitization as a Mechanism for Arrhythmias in HCM
4. Ion Channel Remodeling of the Cardiac Myocyte as a Major Trigger of Arrhythmogenesis in HCM Hearts
4.1. Role of CaMKII Activation in the Ion Channel Remodeling of HCM Myocardium
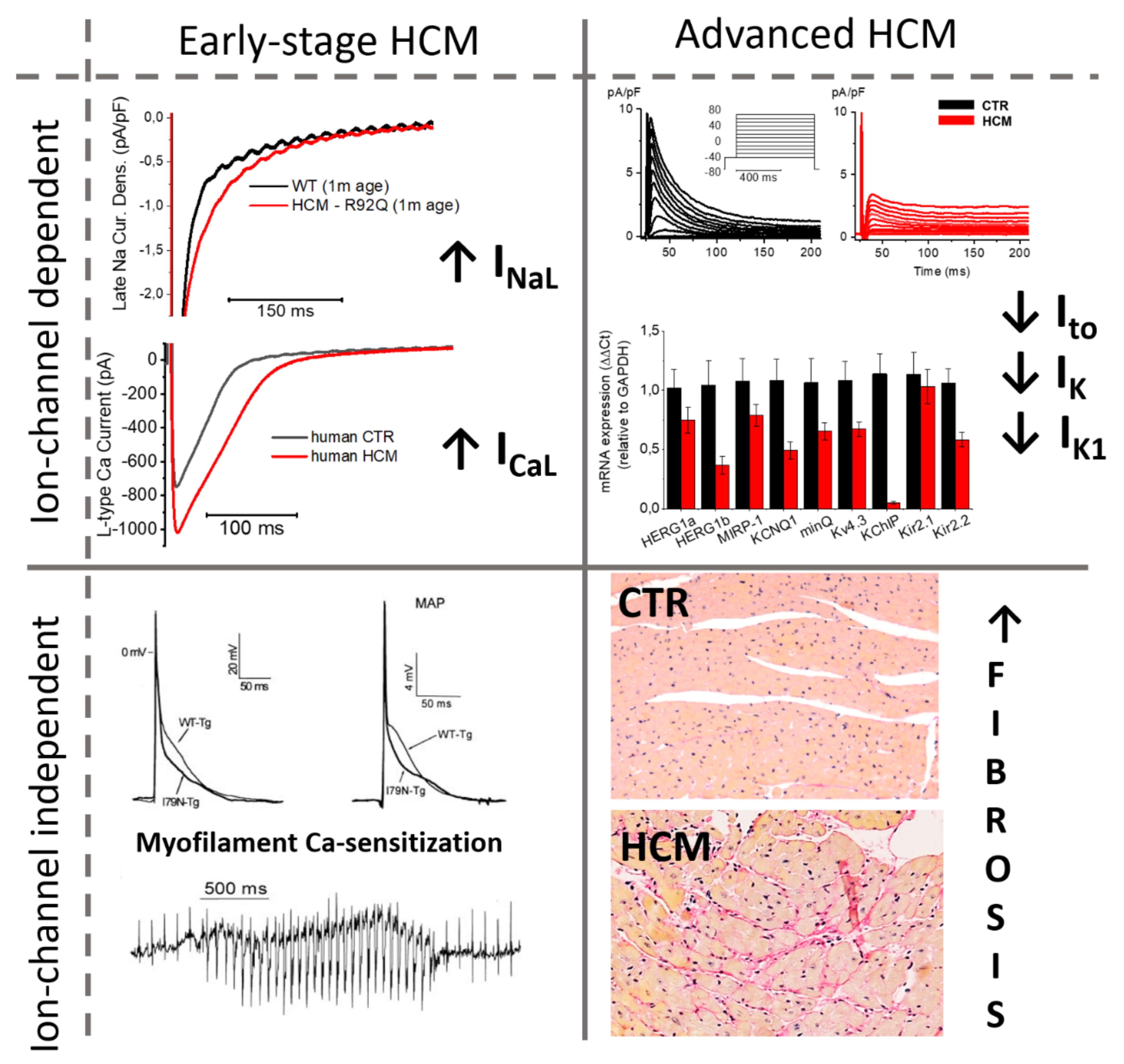
4.2. Timeline of Ion Current Remodeling in HCM
5. Conclusions: Different Electrophysiological Changes at Different Disease Stages
Author Contributions
Funding
Conflicts of Interest
References
- Maron, B.J. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002, 287, 1308–1320. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J. Hypertrophic cardiomyopathy: An important global disease. Am. J. Med. 2004, 116, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Alcalai, R.; Seidman, J.G.; Seidman, C.E. Genetic basis of hypertrophic cardiomyopathy: From bench to the clinics. J. Cardiovasc. Electrophysiol. 2008, 19, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; VanDriest, S.L.; Ommen, S.R.; Will, M.L.; Nishimura, R.A.; Tajik, A.J.; Gersh, B.J. Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin T genes in hypertrophic cardiomyopathy: A comprehensive outpatient perspective. J. Am. Coll. Cardiol. 2002, 39, 2042–2048. [Google Scholar] [CrossRef] [Green Version]
- Landstrom, A.P.; Ackerman, M.J. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation 2010, 122, 2441–2449. [Google Scholar] [CrossRef] [Green Version]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Kazmierczak, K.; Yun, H.; Szczesna-Cordary, D.; Kawai, M. Hypertrophic cardiomyopathy associated E22K mutation in myosin regulatory light chain decreases calcium-activated tension and stiffness and reduces myofilament Ca(2+) sensitivity. FEBS J. 2021, 288, 4596–4613. [Google Scholar] [CrossRef] [PubMed]
- Tadros, H.J.; Life, C.S.; Garcia, G.; Pirozzi, E.; Jones, E.G.; Datta, S.; Parvatiyar, M.S.; Chase, P.B.; Allen, H.D.; Kim, J.J.; et al. Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hot spots that are associated with worse clinical outcomes. J. Mol. Cell Cardiol. 2020, 142, 118–125. [Google Scholar] [CrossRef]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef]
- Maron, B.J.; Shen, W.K.; Link, M.S.; Epstein, A.E.; Almquist, A.K.; Daubert, J.P.; Bardy, G.H.; Favale, S.; Rea, R.F.; Boriani, G.; et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N. Engl. J. Med. 2000, 342, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Klues, H.G.; Schiffers, A.; Maron, B.J. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: Morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J. Am. Coll. Cardiol. 1995, 26, 1699–1708. [Google Scholar] [CrossRef] [Green Version]
- Maron, M.S. The current and emerging role of cardiovascular magnetic resonance imaging in hypertrophic cardiomyopathy. J. Cardiovasc. Transl. Res. 2009, 2, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Rickers, C.; Wilke, N.M.; Jerosch-Herold, M.; Casey, S.A.; Panse, P.; Panse, N.; Weil, J.; Zenovich, A.G.; Maron, B.J. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005, 112, 855–861. [Google Scholar] [CrossRef] [Green Version]
- Basso, C.; Thiene, G.; Corrado, D.; Buja, G.; Melacini, P.; Nava, A. Hypertrophic cardiomyopathy and sudden death in the young: Pathologic evidence of myocardial ischemia. Hum. Pathol. 2000, 31, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Camici, P.G.; Crea, F. Coronary microvascular dysfunction. N. Engl. J. Med. 2007, 356, 830–840. [Google Scholar] [CrossRef] [Green Version]
- Olivotto, I.; Cecchi, F.; Gistri, R.; Lorenzoni, R.; Chiriatti, G.; Girolami, F.; Torricelli, F.; Camici, P.G. Relevance of coronary microvascular flow impairment to long-term remodeling and systolic dysfunction in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 47, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Olivotto, I.; Girolami, F.; Nistri, S.; Rossi, A.; Rega, L.; Garbini, F.; Grifoni, C.; Cecchi, F.; Yacoub, M.H. The many faces of hypertrophic cardiomyopathy: From developmental biology to clinical practice. J. Cardiovasc. Transl. Res. 2009, 2, 349–367. [Google Scholar] [CrossRef]
- Maron, B.J.; Spirito, P.; Shen, W.K.; Haas, T.S.; Formisano, F.; Link, M.S.; Epstein, A.E.; Almquist, A.K.; Daubert, J.P.; Lawrenz, T.; et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA 2007, 298, 405–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, M.; Atkins, D.L.; Triedman, J.K. Sudden Cardiac Death in the Young. Circulation 2016, 133, 1006–1026. [Google Scholar] [CrossRef] [Green Version]
- Webster, G.; Olson, R.; Schoppen, Z.J.; Giancola, N.; Balmert, L.C.; Cherny, S.; George, A.L., Jr. Cardiac Evaluation of Children With a Family History of Sudden Death. J. Am. Coll. Cardiol. 2019, 74, 759–770. [Google Scholar] [CrossRef]
- Ulus, T.; Kudaiberdieva, G.; Gorenek, B. The onset mechanisms of ventricular tachycardia. Int. J. Cardiol. 2013, 167, 619–623. [Google Scholar] [CrossRef]
- Maron, B.J.; Casey, S.A.; Hauser, R.G.; Aeppli, D.M. Clinical course of hypertrophic cardiomyopathy with survival to advanced age. J. Am. Coll. Cardiol. 2003, 42, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Ashrafian, H.; McKenna, W.J.; Watkins, H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 2011, 109, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733. [Google Scholar] [CrossRef] [PubMed]
- Coppini, R.; Ferrantini, C.; Yao, L.; Fan, P.; Del Lungo, M.; Stillitano, F.; Sartiani, L.; Tosi, B.; Suffredini, S.; Tesi, C.; et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 2013, 127, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Barajas-Martinez, H.; Hu, D.; Goodrow, R.J., Jr.; Joyce, F.; Antzelevitch, C. Electrophysiologic characteristics and pharmacologic response of human cardiomyocytes isolated from a patient with hypertrophic cardiomyopathy. Pacing Clin. Electrophysiol. 2013, 36, 1512–1515. [Google Scholar] [CrossRef]
- Toib, A.; Zhang, C.; Borghetti, G.; Zhang, X.; Wallner, M.; Yang, Y.; Troupes, C.D.; Kubo, H.; Sharp, T.E.; Feldsott, E.; et al. Remodeling of repolarization and arrhythmia susceptibility in a myosin-binding protein C knockout mouse model. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H620–H630. [Google Scholar] [CrossRef] [Green Version]
- Flenner, F.; Jungen, C.; Kupker, N.; Ibel, A.; Kruse, M.; Koivumaki, J.T.; Rinas, A.; Zech, A.T.L.; Rhoden, A.; Wijnker, P.J.M.; et al. Translational investigation of electrophysiology in hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2021, 157, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Mitchell, S.; Vainoriene, M.; Lou, Q.; Xie, L.H.; Ren, S.; Goldhaber, J.I.; Wang, Y. Gi alpha 1-mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathy. Circulation 2007, 116, 596–605. [Google Scholar] [CrossRef] [Green Version]
- Hueneke, R.; Adenwala, A.; Mellor, R.L.; Seidman, J.G.; Seidman, C.E.; Nerbonne, J.M. Early remodeling of repolarizing K(+) currents in the alphaMHC(403/+) mouse model of familial hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2017, 103, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horvath, A.; Di Mauro, V.; Koivumaki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Kramer, E.; et al. Disease modeling of a mutation in alpha-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm 2018, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem. Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Haim, T.E.; Dowell, C.; Diamanti, T.; Scheuer, J.; Tardiff, J.C. Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J. Mol. Cell Cardiol. 2007, 42, 1098–1110. [Google Scholar] [CrossRef]
- Knollmann, B.C.; Kirchhof, P.; Sirenko, S.G.; Degen, H.; Greene, A.E.; Schober, T.; Mackow, J.C.; Fabritz, L.; Potter, J.D.; Morad, M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ. Res. 2003, 92, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Fraysse, B.; Weinberger, F.; Bardswell, S.C.; Cuello, F.; Vignier, N.; Geertz, B.; Starbatty, J.; Kramer, E.; Coirault, C.; Eschenhagen, T.; et al. Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J. Mol. Cell Cardiol. 2012, 52, 1299–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, L.; Palandri, C.; Nediani, C.; Cerbai, E.; Coppini, R. Modelling genetic diseases for drug development: Hypertrophic cardiomyopathy. Pharmacol. Res. 2020, 160, 105176. [Google Scholar] [CrossRef]
- Ferrantini, C.; Pioner, J.M.; Mazzoni, L.; Gentile, F.; Tosi, B.; Rossi, A.; Belardinelli, L.; Tesi, C.; Palandri, C.; Matucci, R.; et al. Late sodium current inhibitors to treat exercise-induced obstruction in hypertrophic cardiomyopathy: An in vitro study in human myocardium. Br. J. Pharmacol. 2018, 175, 2635–2652. [Google Scholar] [CrossRef] [Green Version]
- Coppini, R.; Ferrantini, C.; Pioner, J.M.; Santini, L.; Wang, Z.J.; Palandri, C.; Scardigli, M.; Vitale, G.; Sacconi, L.; Stefano, P.; et al. Electrophysiological and Contractile Effects of Disopyramide in Patients With Obstructive Hypertrophic Cardiomyopathy: A Translational Study. JACC Basic Transl. Sci. 2019, 4, 795–813. [Google Scholar] [CrossRef]
- Fischer, T.H.; Herting, J.; Tirilomis, T.; Renner, A.; Neef, S.; Toischer, K.; Ellenberger, D.; Forster, A.; Schmitto, J.D.; Gummert, J.; et al. Ca2+/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 2013, 128, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Toischer, K.; Rokita, A.G.; Unsold, B.; Zhu, W.; Kararigas, G.; Sossalla, S.; Reuter, S.P.; Becker, A.; Teucher, N.; Seidler, T.; et al. Differential cardiac remodeling in preload versus afterload. Circulation 2010, 122, 993–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Zhang, T.; Pereira, L.; Means, C.K.; Cheng, H.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Chen, J.; Bers, D.; et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Investig. 2009, 119, 1230–1240. [Google Scholar] [CrossRef] [Green Version]
- Lehman, S.J.; Tal-Grinspan, L.; Lynn, M.L.; Strom, J.; Benitez, G.E.; Anderson, M.E.; Tardiff, J.C. Chronic Calmodulin-Kinase II Activation Drives Disease Progression in Mutation-Specific Hypertrophic Cardiomyopathy. Circulation 2019, 139, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Hudmon, A.; Schulman, H.; Kim, J.; Maltez, J.M.; Tsien, R.W.; Pitt, G.S. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 2005, 171, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Dybkova, N.; Rasenack, E.C.; Jacobshagen, C.; Fabritz, L.; Kirchhof, P.; Maier, S.K.; Zhang, T.; Hasenfuss, G.; Brown, J.H.; et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Investig. 2006, 116, 3127–3138. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Lee, H.C.; Kabat, J.A.; Shibata, E.F. Modulation of rat cardiac sodium channel by the stimulatory G protein alpha subunit. J. Physiol. 1999, 518, 371–384. [Google Scholar] [CrossRef]
- Coppini, R.; Ferrantini, C.; Mugelli, A.; Poggesi, C.; Cerbai, E. Altered Ca(2+) and Na(+) Homeostasis in Human Hypertrophic Cardiomyopathy: Implications for Arrhythmogenesis. Front Physiol. 2018, 9, 1391. [Google Scholar] [CrossRef]
- Ferrantini, C.; Coppini, R.; Pioner, J.M.; Gentile, F.; Tosi, B.; Mazzoni, L.; Scellini, B.; Piroddi, N.; Laurino, A.; Santini, L.; et al. Pathogenesis of Hypertrophic Cardiomyopathy is Mutation Rather Than Disease Specific: A Comparison of the Cardiac Troponin T E163R and R92Q Mouse Models. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.Y.; Charron, P.; Richard, P.; Girolami, F.; Van Spaendonck-Zwarts, K.Y.; Pinto, Y. Genetic advances in sarcomeric cardiomyopathies: State of the art. Cardiovasc. Res. 2015, 105, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.P.; Bartley, C.R.; Hacker, T.A.; McDonald, K.S.; Douglas, P.S.; Greaser, M.L.; Powers, P.A.; Moss, R.L. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 2002, 90, 594–601. [Google Scholar] [CrossRef] [Green Version]
- Kensler, R.W.; Harris, S.P. The structure of isolated cardiac Myosin thick filaments from cardiac Myosin binding protein-C knockout mice. Biophys. J. 2008, 94, 1707–1718. [Google Scholar] [CrossRef] [Green Version]
- Korte, F.S.; McDonald, K.S.; Harris, S.P.; Moss, R.L. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 2003, 93, 752–758. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, J.E.; Fitzsimons, D.P.; Moss, R.L. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys. J. 2006, 90, 4119–4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppini, R.; Mazzoni, L.; Ferrantini, C.; Gentile, F.; Pioner, J.M.; Laurino, A.; Santini, L.; Bargelli, V.; Rotellini, M.; Bartolucci, G.; et al. Ranolazine Prevents Phenotype Development in a Mouse Model of Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, A.J.; Watkins, H.; Daniels, M.J.; Redwood, C.; Robinson, P. Mavacamten rescues increased myofilament calcium sensitivity and dysregulation of Ca(2+) flux caused by thin filament hypertrophic cardiomyopathy mutations. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H715–H722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montag, J.; Petersen, B.; Flogel, A.K.; Becker, E.; Lucas-Hahn, A.; Cost, G.J.; Muhlfeld, C.; Kraft, T.; Niemann, H.; Brenner, B. Successful knock-in of Hypertrophic Cardiomyopathy-mutation R723G into the MYH7 gene mimics HCM pathology in pigs. Sci. Rep. 2018, 8, 4786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Rio, C.L.; Henze, M.P.; Wong, F.L.; Evanchik, M.J.; Divekar, A.; Gifford, L.M.; Ferhaan, A.; Green, E.M. A Novel Mini-Pig Genetic Model of Hypertrophic Cardiomyopathy: Altered Myofilament Dynamics, Hyper-Contractility, and Impaired Systolic/Diastolic Functional Reserve in vivo. Circulation 2017, 136, A20770. [Google Scholar]
- Del Rio, C.L.; Yadav, A.; Huang, N.; Geist, G.E.; Ueyama, Y.; Youngblood, B.L.; Evanchik, M.J.; Green, E.; Divekar, A.; Ahmad, F. Acute Effects of a Small-Molecule Direct Myosin-Attenuator (MYK-581) in a Mini-Pig Genetic Model of Non-Obstructed Hypertrophic Cardiomyopathy: In Vivo Evidence for Contractile Regulation With Improved Compliance and Functional Reserve. Circulation 2018, 138, A13204. [Google Scholar]
- Semsarian, C.; Healey, M.J.; Fatkin, D.; Giewat, M.; Duffy, C.; Seidman, C.E.; Seidman, J.G. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2001, 33, 2055–2060. [Google Scholar] [CrossRef]
- Semsarian, C.; Ahmad, I.; Giewat, M.; Georgakopoulos, D.; Schmitt, J.P.; McConnell, B.K.; Reiken, S.; Mende, U.; Marks, A.R.; Kass, D.A.; et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J. Clin. Investig. 2002, 109, 1013–1020. [Google Scholar] [CrossRef]
- Seidman, J.G.; Seidman, C. The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 2001, 104, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.B.; Fraser, S.T.; Semsarian, C. Induced pluripotent stem cells in the inherited cardiomyopathies: From disease mechanisms to novel therapies. Trends Cardiovasc. Med. 2016, 26, 663–672. [Google Scholar] [CrossRef]
- Clippinger, S.R.; Cloonan, P.E.; Wang, W.; Greenberg, L.; Stump, W.T.; Angsutararux, P.; Nerbonne, J.M.; Greenberg, M.J. Mechanical dysfunction of the sarcomere induced by a pathogenic mutation in troponin T drives cellular adaptation. J. Gen. Physiol. 2021, 153. [Google Scholar] [CrossRef] [PubMed]
- Kansakar, U.; Varzideh, F.; Jankauskas, S.S.; Gambardella, J.; Trimarco, B.; Santulli, G. Advances in the understanding of excitation-contraction coupling: The pulsing quest for drugs against heart failure and arrhythmias. Eur. Heart J. Cardiovasc. Pharmacother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, P.A.; Fuchs, F. Bound calcium and force development in skinned cardiac muscle bundles: Effect of sarcomere length. J. Mol. Cell Cardiol. 1988, 20, 667–677. [Google Scholar] [CrossRef]
- Lehman, W.; Craig, R. Tropomyosin and the steric mechanism of muscle regulation. Adv. Exp. Med. Biol. 2008, 644, 95–109. [Google Scholar] [CrossRef]
- Betocchi, S.; Hess, O.M.; Losi, M.A.; Nonogi, H.; Krayenbuehl, H.P. Regional left ventricular mechanics in hypertrophic cardiomyopathy. Circulation 1993, 88, 2206–2214. [Google Scholar] [CrossRef] [Green Version]
- Germans, T.; Russel, I.K.; Gotte, M.J.; Spreeuwenberg, M.D.; Doevendans, P.A.; Pinto, Y.M.; van der Geest, R.J.; van der Velden, J.; Wilde, A.A.; van Rossum, A.C. How do hypertrophic cardiomyopathy mutations affect myocardial function in carriers with normal wall thickness? Assessment with cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2010, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.Y.; Carlsen, C.; Thune, J.J.; Havndrup, O.; Bundgaard, H.; Farrohi, F.; Rivero, J.; Cirino, A.L.; Andersen, P.S.; Christiansen, M.; et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Maras, D.; Chung, R.; Duncan, A.; Li, W.; Thorp, C.; Morner, S.; Lindqvist, P.; Henein, M.Y. Patterns of cardiac dysfunction coinciding with exertional breathlessness in hypertrophic cardiomyopathy. Int. J. Cardiol. 2013, 170, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F.; Bachinski, L.L.; Meyer, D.; Hill, R.; Zoghbi, W.A.; Tam, J.W.; Quinones, M.A.; Roberts, R.; Marian, A.J. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation 2001, 104, 128–130. [Google Scholar] [CrossRef] [Green Version]
- Chandra, M.; Rundell, V.L.; Tardiff, J.C.; Leinwand, L.A.; De Tombe, P.P.; Solaro, R.J. Ca(2+) activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H705–H713. [Google Scholar] [CrossRef]
- Kim, S.J.; Iizuka, K.; Kelly, R.A.; Geng, Y.J.; Bishop, S.P.; Yang, G.; Kudej, A.; McConnell, B.K.; Seidman, C.E.; Seidman, J.G.; et al. An alpha-cardiac myosin heavy chain gene mutation impairs contraction and relaxation function of cardiac myocytes. Am. J. Physiol. 1999, 276, H1780–H1787. [Google Scholar] [CrossRef]
- Knollmann, B.C.; Blatt, S.A.; Horton, K.; de Freitas, F.; Miller, T.; Bell, M.; Housmans, P.R.; Weissman, N.J.; Morad, M.; Potter, J.D. Inotropic stimulation induces cardiac dysfunction in transgenic mice expressing a troponin T (I79N) mutation linked to familial hypertrophic cardiomyopathy. J. Biol. Chem. 2001, 276, 10039–10048. [Google Scholar] [CrossRef] [Green Version]
- Nagueh, S.F.; Kopelen, H.A.; Lim, D.S.; Zoghbi, W.A.; Quinones, M.A.; Roberts, R.; Marian, A.J. Tissue Doppler imaging consistently detects myocardial contraction and relaxation abnormalities, irrespective of cardiac hypertrophy, in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2000, 102, 1346–1350. [Google Scholar] [CrossRef] [Green Version]
- Najafi, A.; Schlossarek, S.; van Deel, E.D.; van den Heuvel, N.; Guclu, A.; Goebel, M.; Kuster, D.W.; Carrier, L.; van der Velden, J. Sexual dimorphic response to exercise in hypertrophic cardiomyopathy-associated MYBPC3-targeted knock-in mice. Pflugers Arch. 2015, 467, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
- Schober, T.; Huke, S.; Venkataraman, R.; Gryshchenko, O.; Kryshtal, D.; Hwang, H.S.; Baudenbacher, F.J.; Knollmann, B.C. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ. Res. 2012, 111, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Tardiff, J.C.; Hewett, T.E.; Palmer, B.M.; Olsson, C.; Factor, S.M.; Moore, R.L.; Robbins, J.; Leinwand, L.A. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J. Clin. Investig. 1999, 104, 469–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, S.; Copeland, O.; Gehmlich, K.; Schlossarek, S.; Carrier, L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J. Muscle Res. Cell Motil. 2012, 33, 75–80. [Google Scholar] [CrossRef]
- Baudenbacher, F.; Schober, T.; Pinto, J.R.; Sidorov, V.Y.; Hilliard, F.; Solaro, R.J.; Potter, J.D.; Knollmann, B.C. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Investig. 2008, 118, 3893–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, T.; Witjas-Paalberends, E.R.; Boontje, N.M.; Tripathi, S.; Brandis, A.; Montag, J.; Hodgkinson, J.L.; Francino, A.; Navarro-Lopez, F.; Brenner, B.; et al. Familial hypertrophic cardiomyopathy: Functional effects of myosin mutation R723G in cardiomyocytes. J. Mol. Cell Cardiol. 2013, 57, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Najafi, A.; Sequeira, V.; Helmes, M.; Bollen, I.A.; Goebel, M.; Regan, J.A.; Carrier, L.; Kuster, D.W.; Van Der Velden, J. Selective phosphorylation of PKA targets after beta-adrenergic receptor stimulation impairs myofilament function in Mybpc3-targeted HCM mouse model. Cardiovasc. Res. 2016, 110, 200–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, R.; Boivin, G.P.; Grupp, I.L.; Hoit, B.; Arteaga, G.; Solaro, R.J.; Wieczorek, D.F. A familial hypertrophic cardiomyopathy alpha-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J. Mol. Cell Cardiol. 2001, 33, 1815–1828. [Google Scholar] [CrossRef]
- Sequeira, V.; Wijnker, P.J.; Nijenkamp, L.L.; Kuster, D.W.; Najafi, A.; Witjas-Paalberends, E.R.; Regan, J.A.; Boontje, N.; Ten Cate, F.J.; Germans, T.; et al. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 2013, 112, 1491–1505. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.; et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009, 119, 1473–1483. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, S.J.; Paalberends, E.R.; Najafi, A.; Michels, M.; Sadayappan, S.; Carrier, L.; Boontje, N.M.; Kuster, D.W.; van Slegtenhorst, M.; Dooijes, D.; et al. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 2012, 5, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Mijailovich, S.M.; Prodanovic, M.; Poggesi, C.; Powers, J.D.; Davis, J.; Geeves, M.A.; Regnier, M. The effect of variable troponin C mutation thin filament incorporation on cardiac muscle twitch contractions. J. Mol. Cell Cardiol. 2021, 155, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; McKenna, W.J.; Thierfelder, L.; Suk, H.J.; Anan, R.; O’Donoghue, A.; Spirito, P.; Matsumori, A.; Moravec, C.S.; Seidman, J.G.; et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N. Engl. J. Med. 1995, 332, 1058–1064. [Google Scholar] [CrossRef]
- Dweck, D.; Sanchez-Gonzalez, M.A.; Chang, A.N.; Dulce, R.A.; Badger, C.D.; Koutnik, A.P.; Ruiz, E.L.; Griffin, B.; Liang, J.; Kabbaj, M.; et al. Long term ablation of protein kinase A (PKA)-mediated cardiac troponin I phosphorylation leads to excitation-contraction uncoupling and diastolic dysfunction in a knock-in mouse model of hypertrophic cardiomyopathy. J. Biol. Chem. 2014, 289, 23097–23111. [Google Scholar] [CrossRef] [Green Version]
- Wolff, M.R.; Buck, S.H.; Stoker, S.W.; Greaser, M.L.; Mentzer, R.M. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: Role of altered beta-adrenergically mediated protein phosphorylation. J. Clin. Investig. 1996, 98, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Van der Velden, J.; Merkus, D.; Klarenbeek, B.R.; James, A.T.; Boontje, N.M.; Dekkers, D.H.; Stienen, G.J.; Lamers, J.M.; Duncker, D.J. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ. Res. 2004, 95, e85–e95. [Google Scholar] [CrossRef] [Green Version]
- Varnava, A.M.; Elliott, P.M.; Baboonian, C.; Davison, F.; Davies, M.J.; McKenna, W.J. Hypertrophic cardiomyopathy: Histopathological features of sudden death in cardiac troponin T disease. Circulation 2001, 104, 1380–1384. [Google Scholar] [CrossRef] [Green Version]
- Knollmann, B.C.; Katchman, A.N.; Franz, M.R. Monophasic action potential recordings from intact mouse heart: Validation, regional heterogeneity, and relation to refractoriness. J. Cardiovasc. Electrophysiol. 2001, 12, 1286–1294. [Google Scholar] [CrossRef]
- Dou, Y.; Arlock, P.; Arner, A. Blebbistatin specifically inhibits actin-myosin interaction in mouse cardiac muscle. Am. J. Physiol. Cell. Physiol. 2007, 293, C1148–C1153. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Kryshtal, D.O.; Kim, K.; Parikh, S.; Cadar, A.G.; Bersell, K.R.; He, H.; Pinto, J.R.; Knollmann, B.C. Myofilament Calcium-Buffering Dependent Action Potential Triangulation in Human-Induced Pluripotent Stem Cell Model of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 2600–2602. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kim, K.; Parikh, S.; Cadar, A.G.; Bersell, K.R.; He, H.; Pinto, J.R.; Kryshtal, D.O.; Knollmann, B.C. Hypertrophic cardiomyopathy-linked mutation in troponin T causes myofibrillar disarray and pro-arrhythmic action potential changes in human iPSC cardiomyocytes. J. Mol. Cell Cardiol. 2018, 114, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Parvatiyar, M.S.; Landstrom, A.P.; Figueiredo-Freitas, C.; Potter, J.D.; Ackerman, M.J.; Pinto, J.R. A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J. Biol. Chem. 2012, 287, 31845–31855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppini, R.; Santini, L.; Olivotto, I.; Ackerman, M.J.; Cerbai, E. Abnormalities in sodium current and calcium homoeostasis as drivers of arrhythmogenesis in hypertrophic cardiomyopathy. Cardiovasc. Res. 2020, 116, 1585–1599. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, T.; Virag, L.; Varro, A.; Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput. Biol. 2011, 7, e1002061. [Google Scholar] [CrossRef] [Green Version]
- Passini, E.; Minchole, A.; Coppini, R.; Cerbai, E.; Rodriguez, B.; Severi, S.; Bueno-Orovio, A. Mechanisms of pro-arrhythmic abnormalities in ventricular repolarisation and anti-arrhythmic therapies in human hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2016, 96, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.A.K.; Warren, C.M.; Simon, J.N.; Ryba, D.M.; Batra, A.; Varga, P.; Kranias, E.G.; Tardiff, J.C.; Solaro, R.J.; Wolska, B.M. Modifications of Sarcoplasmic Reticulum Function Prevent Progression of Sarcomere-Linked Hypertrophic Cardiomyopathy Despite a Persistent Increase in Myofilament Calcium Response. Front Physiol. 2020, 11, 107. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, W.J.; Li, Y.; Wu, F.; Bai, R.; Ma, S.; Dong, T.; Zhang, H.; Lee, A.S.; Wang, Y.; et al. MLP-deficient human pluripotent stem cell derived cardiomyocytes develop hypertrophic cardiomyopathy and heart failure phenotypes due to abnormal calcium handling. Cell Death Dis. 2019, 10, 610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chang, Y.; Li, X.; Li, X.; Gao, J.; Zhou, Y.; Wu, F.; Bai, R.; Dong, T.; Ma, S.; et al. RAD-Deficient Human Cardiomyocytes Develop Hypertrophic Cardiomyopathy Phenotypes Due to Calcium Dysregulation. Front Cell Dev. Biol. 2020, 8, 585879. [Google Scholar] [CrossRef]
- Hamdani, N.; Krysiak, J.; Kreusser, M.M.; Neef, S.; Dos Remedios, C.G.; Maier, L.S.; Kruger, M.; Backs, J.; Linke, W.A. Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ. Res. 2013, 112, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, C.W.; Gaffin, R.D.; Zawieja, D.C.; Muthuchamy, M. Roles of phosphorylation of myosin binding protein-C and troponin I in mouse cardiac muscle twitch dynamics. J. Physiol. 2004, 558, 927–941. [Google Scholar] [CrossRef] [Green Version]
- Guilbert, A.; Lim, H.J.; Cheng, J.; Wang, Y. CaMKII-dependent myofilament Ca2+ desensitization contributes to the frequency-dependent acceleration of relaxation. Cell Calcium. 2015, 58, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Stanbury, D.M.; Bounds, P.L. Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radic. Biol. Med. 2010, 49, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Lin, C.Y.; Farmand, F.; Sindhu, R.K. Superoxide dismutase, catalase, glutathione peroxidase and NADPH oxidase in lead-induced hypertension. Kidney Int. 2003, 63, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Gasparetto, C.; Malinverno, A.; Culacciati, D.; Gritti, D.; Prosperini, P.G.; Specchia, G.; Ricevuti, G. Antioxidant vitamins reduce oxidative stress and ventricular remodeling in patients with acute myocardial infarction. Int. J. Immunopathol. Pharmacol. 2005, 18, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Cesselli, D.; Jakoniuk, I.; Barlucchi, L.; Beltrami, A.P.; Hintze, T.H.; Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ. Res. 2001, 89, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, D.B.; Siwik, D.A.; Xiao, L.; Pimentel, D.R.; Singh, K.; Colucci, W.S. Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell Cardiol. 2002, 34, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Kono, Y.; Nakamura, K.; Kimura, H.; Nishii, N.; Watanabe, A.; Banba, K.; Miura, A.; Nagase, S.; Sakuragi, S.; Kusano, K.F.; et al. Elevated levels of oxidative DNA damage in serum and myocardium of patients with heart failure. Circ. J. 2006, 70, 1001–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.F.; Singal, P.K. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am. J. Pathol. 1996, 148, 291–300. [Google Scholar]
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: A potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998, 97, 1536–1539. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, Z.; Hu, F.; Yang, W.; Yuan, J.; Cui, J.; Hao, S.; Hu, J.; Zhou, Y.; Qiao, S. 17beta-estradiol prevents cardiac diastolic dysfunction by stimulating mitochondrial function: A preclinical study in a mouse model of a human hypertrophic cardiomyopathy mutation. J. Steroid. Biochem. Mol. Biol. 2015, 147, 92–102. [Google Scholar] [CrossRef]
- Christiansen, L.B.; Dela, F.; Koch, J.; Hansen, C.N.; Leifsson, P.S.; Yokota, T. Impaired cardiac mitochondrial oxidative phosphorylation and enhanced mitochondrial oxidative stress in feline hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1237–H1247. [Google Scholar] [CrossRef] [Green Version]
- Flenner, F.; Friedrich, F.W.; Ungeheuer, N.; Christ, T.; Geertz, B.; Reischmann, S.; Wagner, S.; Stathopoulou, K.; Sohren, K.D.; Weinberger, F.; et al. Ranolazine antagonizes catecholamine-induced dysfunction in isolated cardiomyocytes, but lacks long-term therapeutic effects in vivo in a mouse model of hypertrophic cardiomyopathy. Cardiovasc. Res. 2016, 109, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, R.; Rodriguez, G.; Chen, S.N.; Ripplinger, C.M.; Li, W.; Chen, J.; Willerson, J.T.; Betocchi, S.; Wickline, S.A.; Efimov, I.R.; et al. Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol-sensitive mechanisms. Circulation 2009, 119, 1398–1407. [Google Scholar] [CrossRef]
- Senthil, V.; Chen, S.N.; Tsybouleva, N.; Halder, T.; Nagueh, S.F.; Willerson, J.T.; Roberts, R.; Marian, A.J. Prevention of cardiac hypertrophy by atorvastatin in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ. Res. 2005, 97, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Chimenti, C.; Scopelliti, F.; Vulpis, E.; Tafani, M.; Villanova, L.; Verardo, R.; De Paulis, R.; Russo, M.A.; Frustaci, A. Increased oxidative stress contributes to cardiomyocyte dysfunction and death in patients with Fabry disease cardiomyopathy. Hum. Pathol. 2015, 46, 1760–1768. [Google Scholar] [CrossRef]
- Dimitrow, P.P.; Undas, A.; Wolkow, P.; Tracz, W.; Dubiel, J.S. Enhanced oxidative stress in hypertrophic cardiomyopathy. Pharmacol. Rep. 2009, 61, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Koda, M.; Takemura, G.; Okada, H.; Kanoh, M.; Maruyama, R.; Esaki, M.; Li, Y.; Miyata, S.; Kanamori, H.; Li, L.; et al. Nuclear hypertrophy reflects increased biosynthetic activities in myocytes of human hypertrophic hearts. Circ. J. 2006, 70, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.L.; Dudley, S.C., Jr. Tandem promoters and developmentally regulated 5'- and 3'-mRNA untranslated regions of the mouse Scn5a cardiac sodium channel. J. Biol. Chem. 2005, 280, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Barth, A.S.; DiSilvestre, D.; Akar, F.G.; Tian, Y.; Tanskanen, A.; Kass, D.A.; Winslow, R.L.; Tomaselli, G.F. Key pathways associated with heart failure development revealed by gene networks correlated with cardiac remodeling. Physiol. Genomics 2008, 35, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Abramson, J.J.; Zable, A.C.; Pessah, I.N. Direct evidence for the existence and functional role of hyperreactive sulfhydryls on the ryanodine receptor-triadin complex selectively labeled by the coumarin maleimide 7-diethylamino-3-(4'-maleimidylphenyl)-4-methylcoumarin. Mol. Pharmacol. 1994, 45, 189–200. [Google Scholar]
- Xu, K.Y.; Zweier, J.L.; Becker, L.C. Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ. Res. 1997, 80, 76–81. [Google Scholar] [CrossRef]
- Wehrens, X.H.; Lehnart, S.E.; Marks, A.R. Intracellular calcium release and cardiac disease. Annu. Rev. Physiol. 2005, 67, 69–98. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, J.; Wang, H.; Luo, X.; Wang, J.; Villeneuve, L.R.; Zhang, H.; Bai, Y.; Yang, B.; Wang, Z. Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1446–H1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandra, C.J.A.; Kp, M.M.J.; Chua, J.; Hernandez-Resendiz, S.; Liehn, E.A.; Knoll, R.; Gan, L.M.; Michaelsson, E.; Jonsson, M.K.B.; Ryden-Markinhuhta, K.; et al. Inhibiting cardiac myeloperoxidase alleviates the relaxation defect in hypertrophic cardiomyocytes. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Coppini, R.; Ferrantini, C. NaV1.8: A novel contributor to cardiac arrhythmogenesis in heart failure. Cardiovasc. Res. 2018, 114, 1691–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.N.; Grifoni, C.; Bos, J.M.; Saber-Ayad, M.; Ommen, S.R.; Nistri, S.; Cecchi, F.; Olivotto, I.; Ackerman, M.J. Prevalence and clinical correlates of QT prolongation in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2011, 32, 1114–1120. [Google Scholar] [CrossRef] [Green Version]
- Coppini, R.; Ho, C.Y.; Ashley, E.; Day, S.; Ferrantini, C.; Girolami, F.; Tomberli, B.; Bardi, S.; Torricelli, F.; Cecchi, F.; et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J. Am. Coll. Cardiol. 2014, 64, 2589–2600. [Google Scholar] [CrossRef] [Green Version]
- Olivotto, I.; Coppini, R. Channelopathies, cardiac hypertrophy, and the theory of light. Eur. Heart J. 2018, 39, 2908–2910. [Google Scholar] [CrossRef]
- Solomon, T.; Filipovska, A.; Hool, L.; Viola, H. Preventative therapeutic approaches for hypertrophic cardiomyopathy. J. Physiol. 2021, 599, 3495–3512. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santini, L.; Coppini, R.; Cerbai, E. Ion Channel Impairment and Myofilament Ca2+ Sensitization: Two Parallel Mechanisms Underlying Arrhythmogenesis in Hypertrophic Cardiomyopathy. Cells 2021, 10, 2789. https://doi.org/10.3390/cells10102789
Santini L, Coppini R, Cerbai E. Ion Channel Impairment and Myofilament Ca2+ Sensitization: Two Parallel Mechanisms Underlying Arrhythmogenesis in Hypertrophic Cardiomyopathy. Cells. 2021; 10(10):2789. https://doi.org/10.3390/cells10102789
Chicago/Turabian StyleSantini, Lorenzo, Raffaele Coppini, and Elisabetta Cerbai. 2021. "Ion Channel Impairment and Myofilament Ca2+ Sensitization: Two Parallel Mechanisms Underlying Arrhythmogenesis in Hypertrophic Cardiomyopathy" Cells 10, no. 10: 2789. https://doi.org/10.3390/cells10102789