
Hormonally Induced Hepatocellular Carcinoma in Diabetic Wild Type and Carbohydrate Responsive Element Binding Protein Knockout Mice

 , ,
, ,
Abstract
:
1. Background
1.1. Preneoplastic and Neoplastic Hepatocellular Lesions in Rodent Models and Humans
1.2. Role of Carbohydrate-Response Element-Binding Protein in Hepatocellular Metabolism and Carcinogenesis
2. Methods
2.1. Immunohistochemistry
2.2. Morphologic and Proliferation Kinetic Investigations
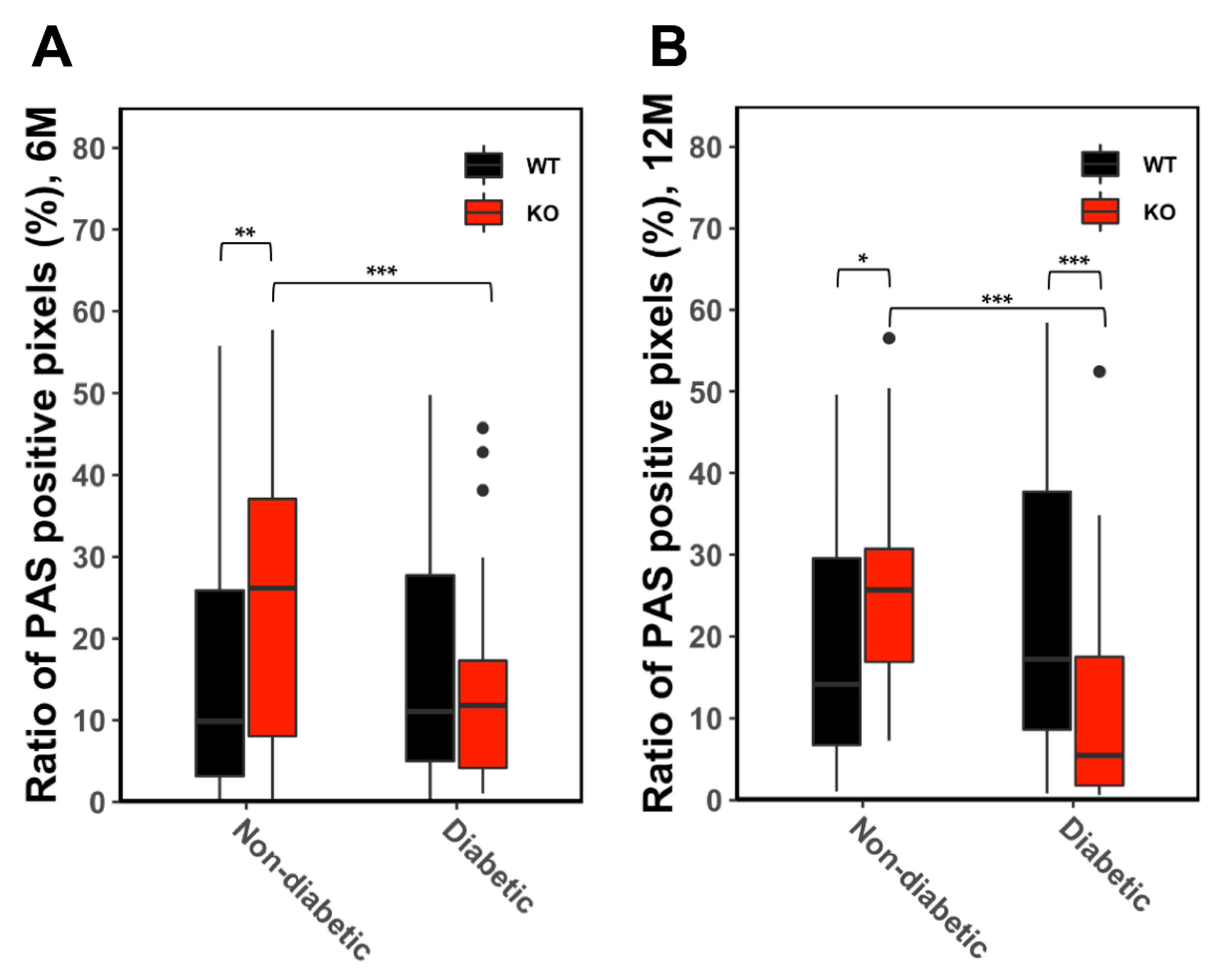
2.3. Glycogen Quantification (Automated PAS-Quantification)
2.4. Ultrastructural Analysis
2.5. RNA Extraction, Quality Control and RNA-seq Transcriptomic Data Analysis
2.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.7. Statistical Analysis
3. Results
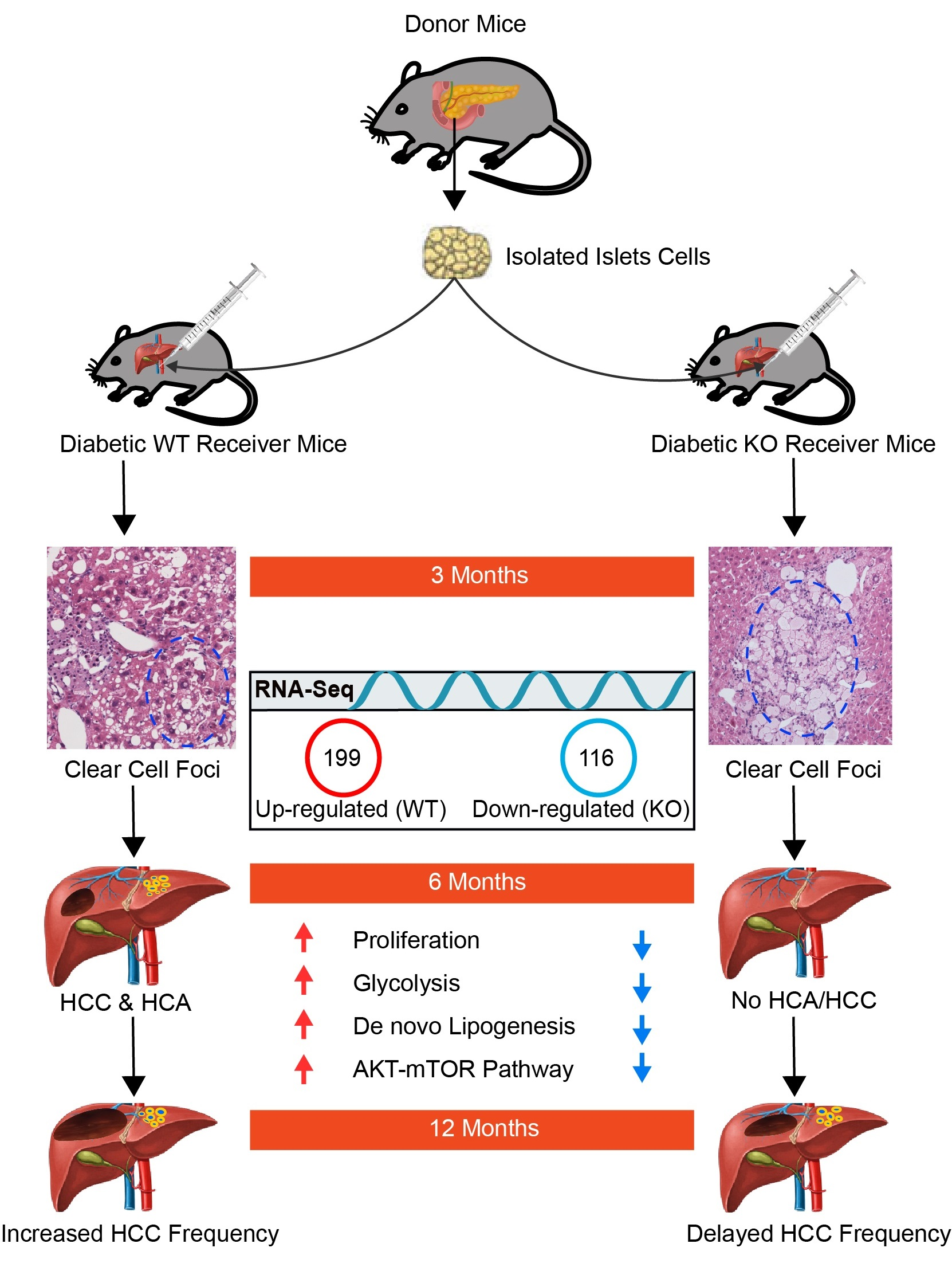
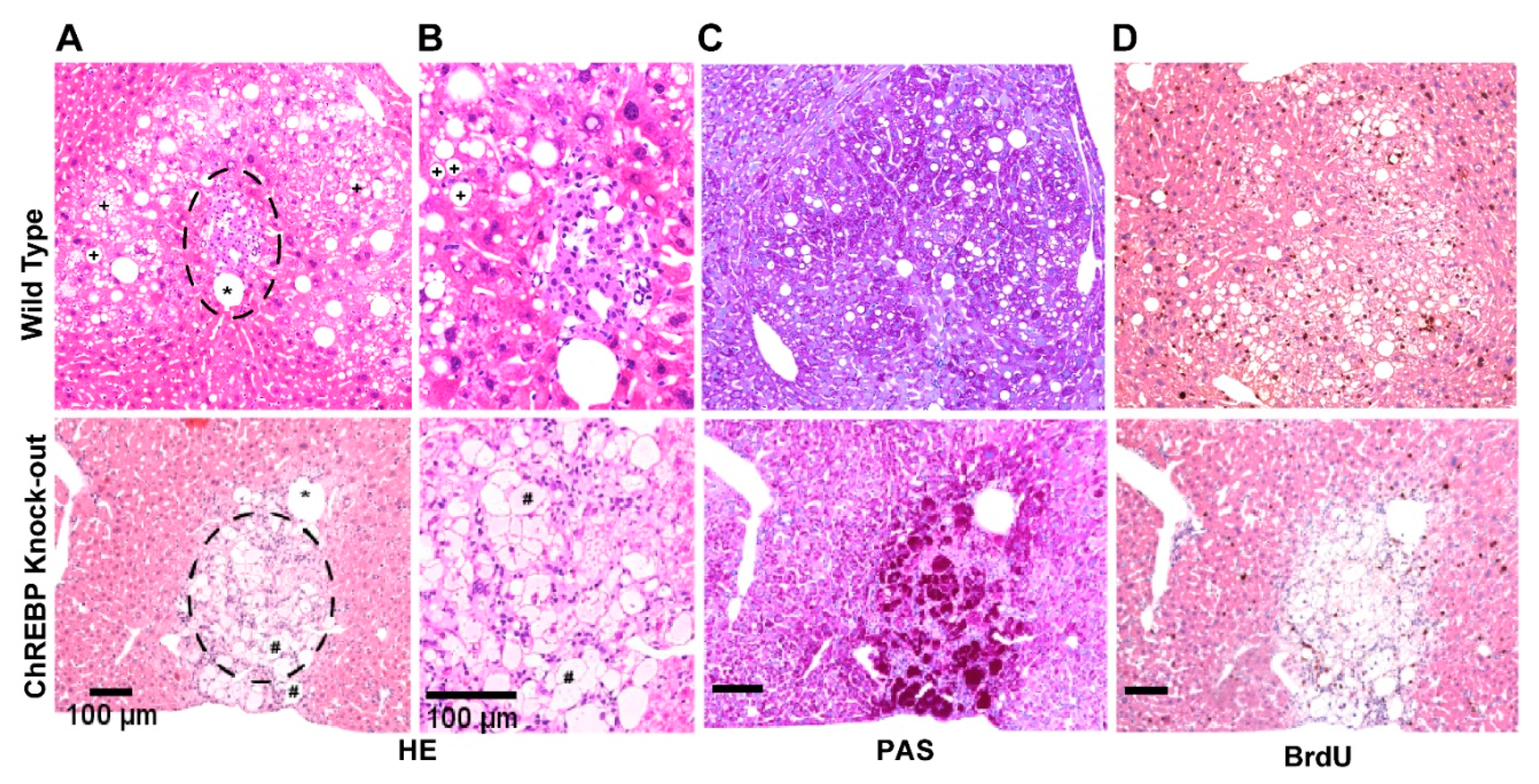
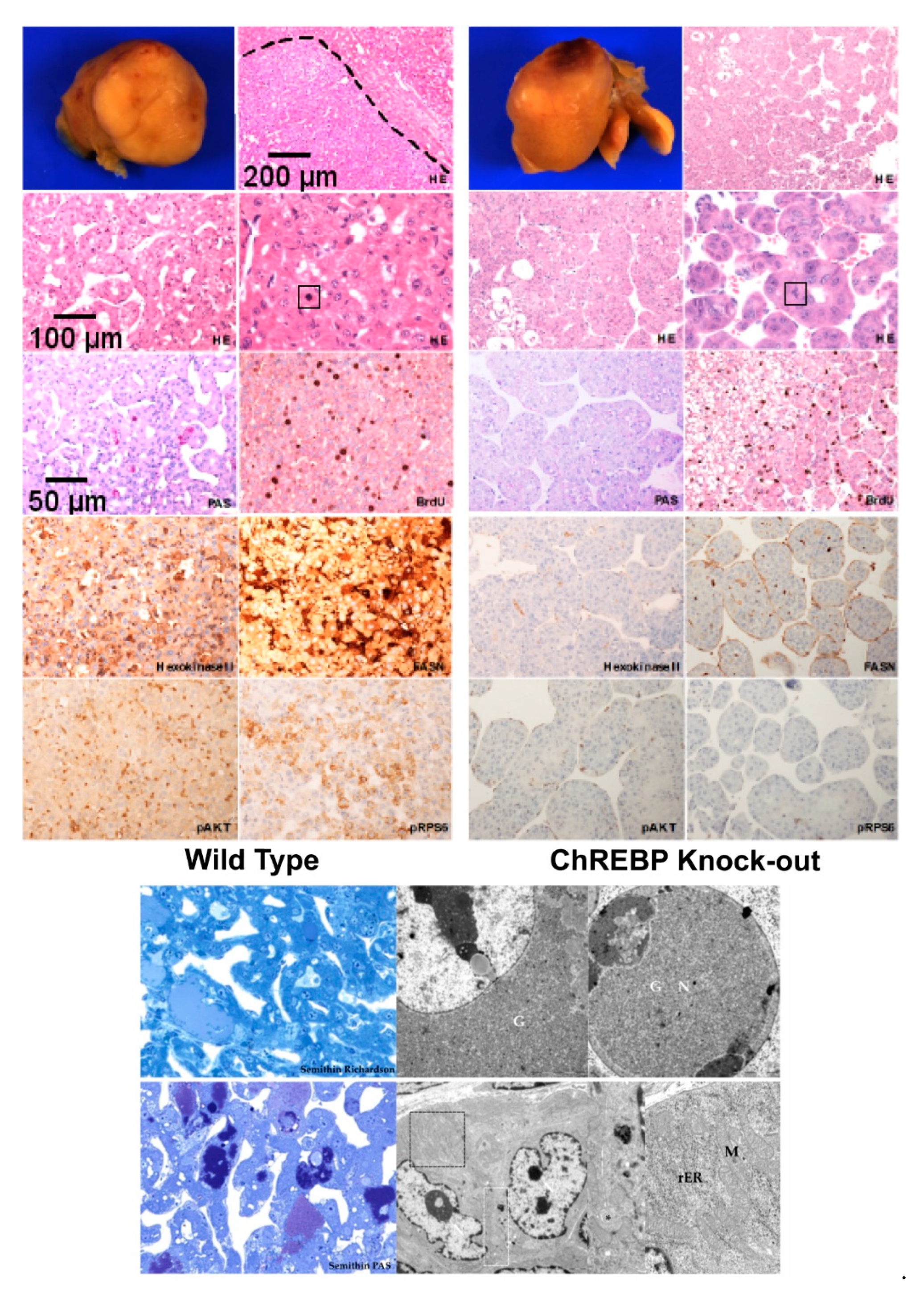
3.1. Hormonally Induced Hepatocarcinogenesis Leads to CCF of Altered Hepatocytes
3.1.1. ChREBP Is Associated with Distinct Morphological Alterations
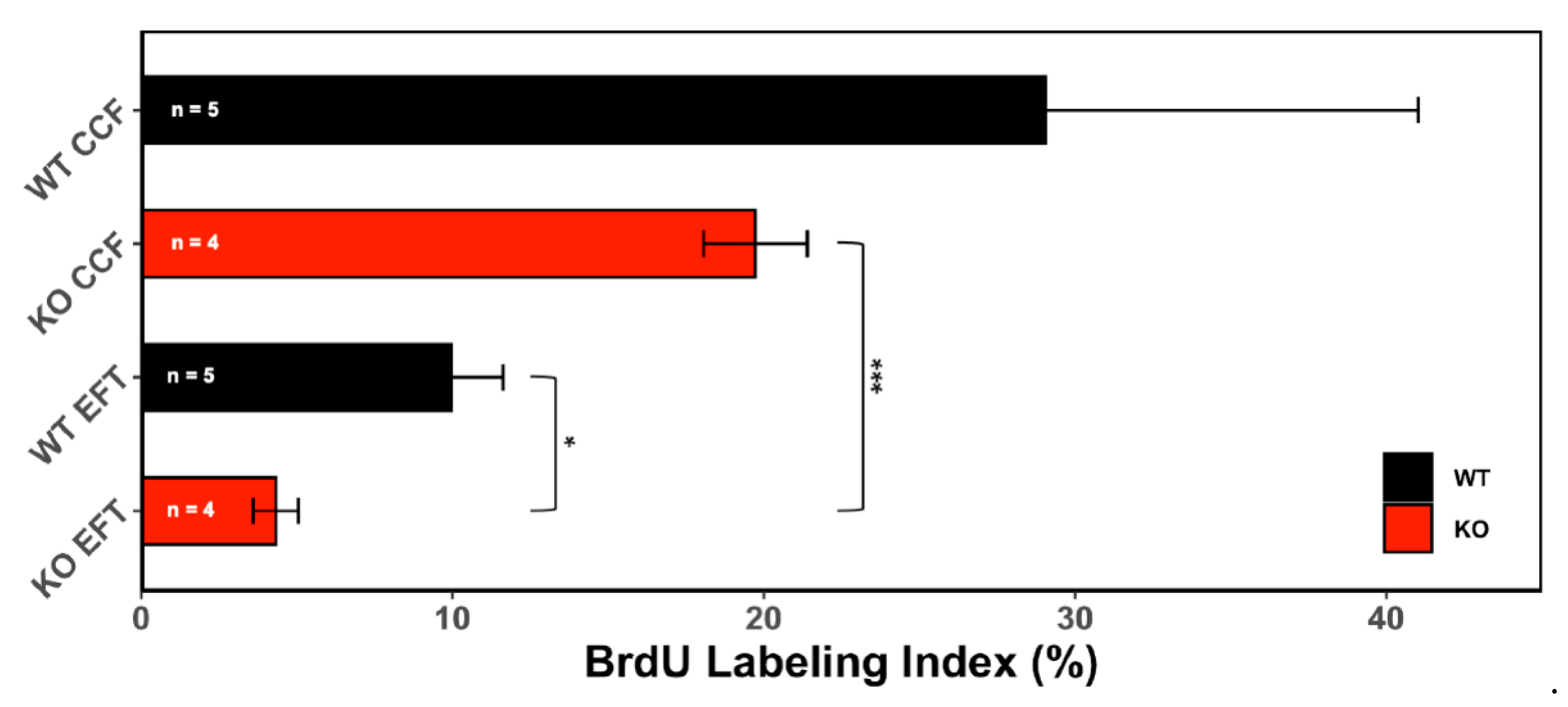
3.1.2. Proliferative Activity
3.2. CCF Signature Leads to Hepatocellular Adenomas (HCAs) and Carcinomas (HCCs)
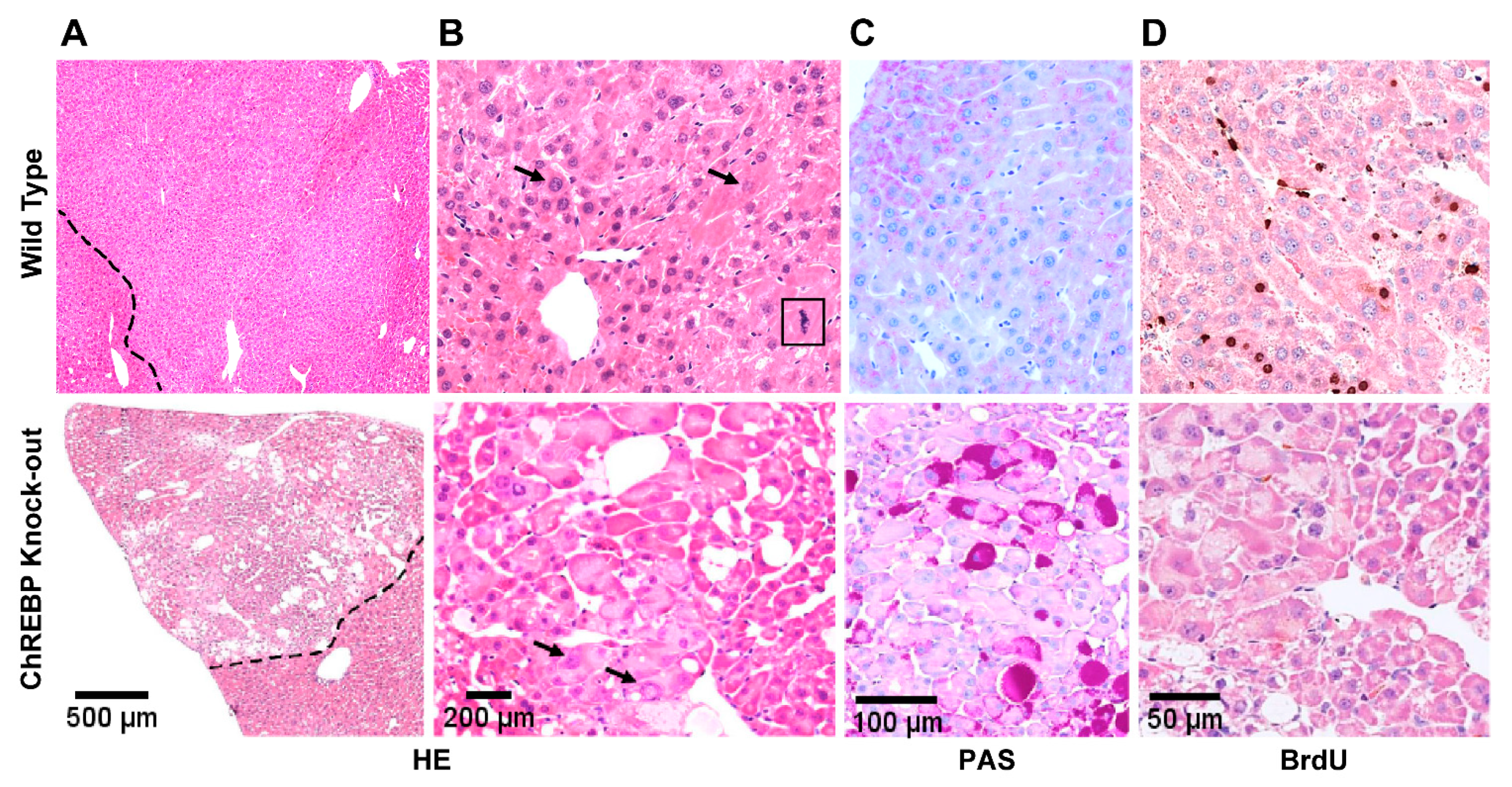
3.2.1. HCAs and HCCs Are Associated with Distinct Morphological Alterations
3.2.2. Immunohistochemical Expression Patterns of Glycolytic, Lipogenic and Molecular Pathways
3.3. Glycogen Storage in Unaltered Liver Tissue
3.4. Proliferative Activity of Unaltered Liver Tissue
3.5. Blood Glucose Level, Body Weight, and Serum ALT and AST Levels
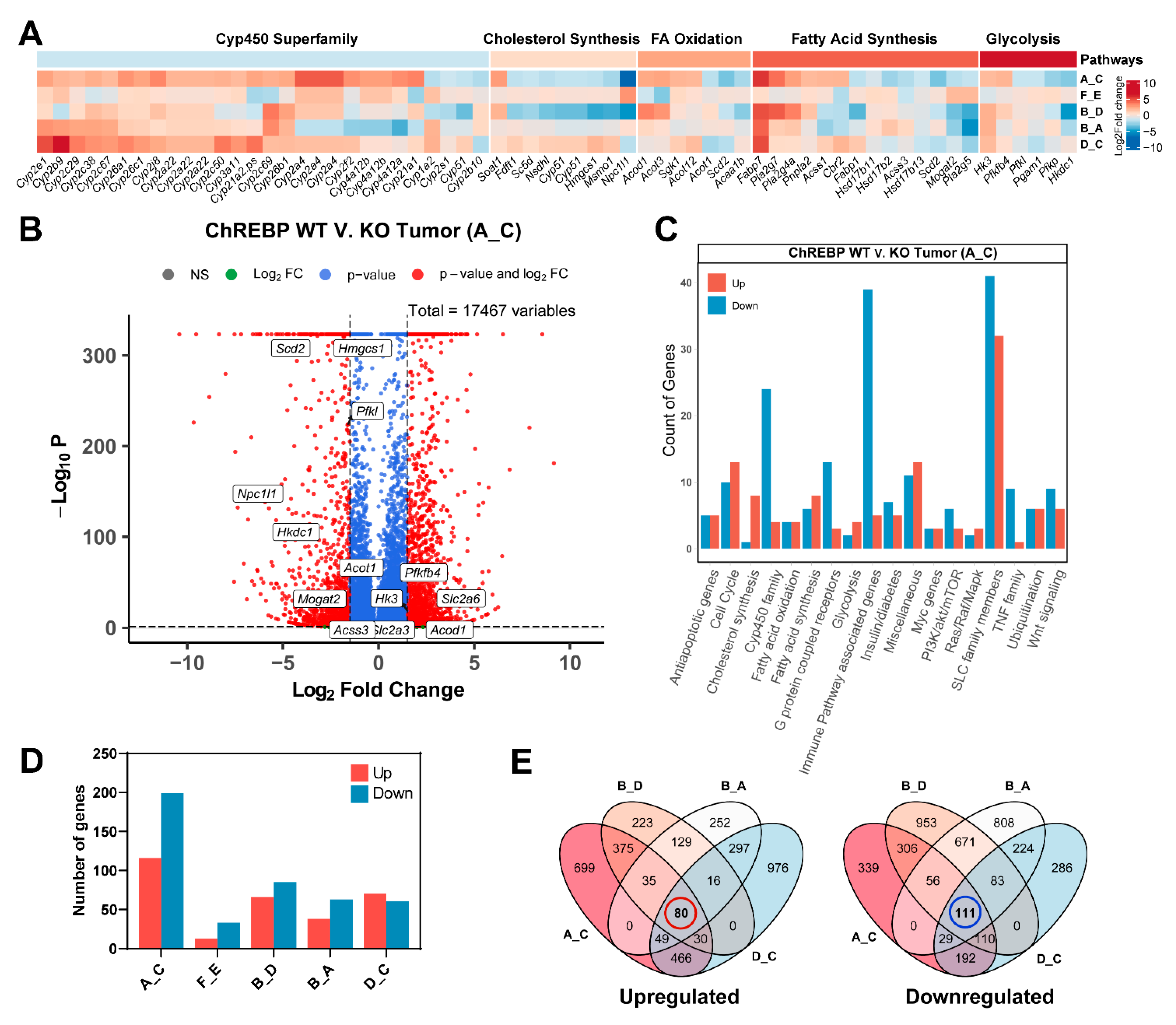
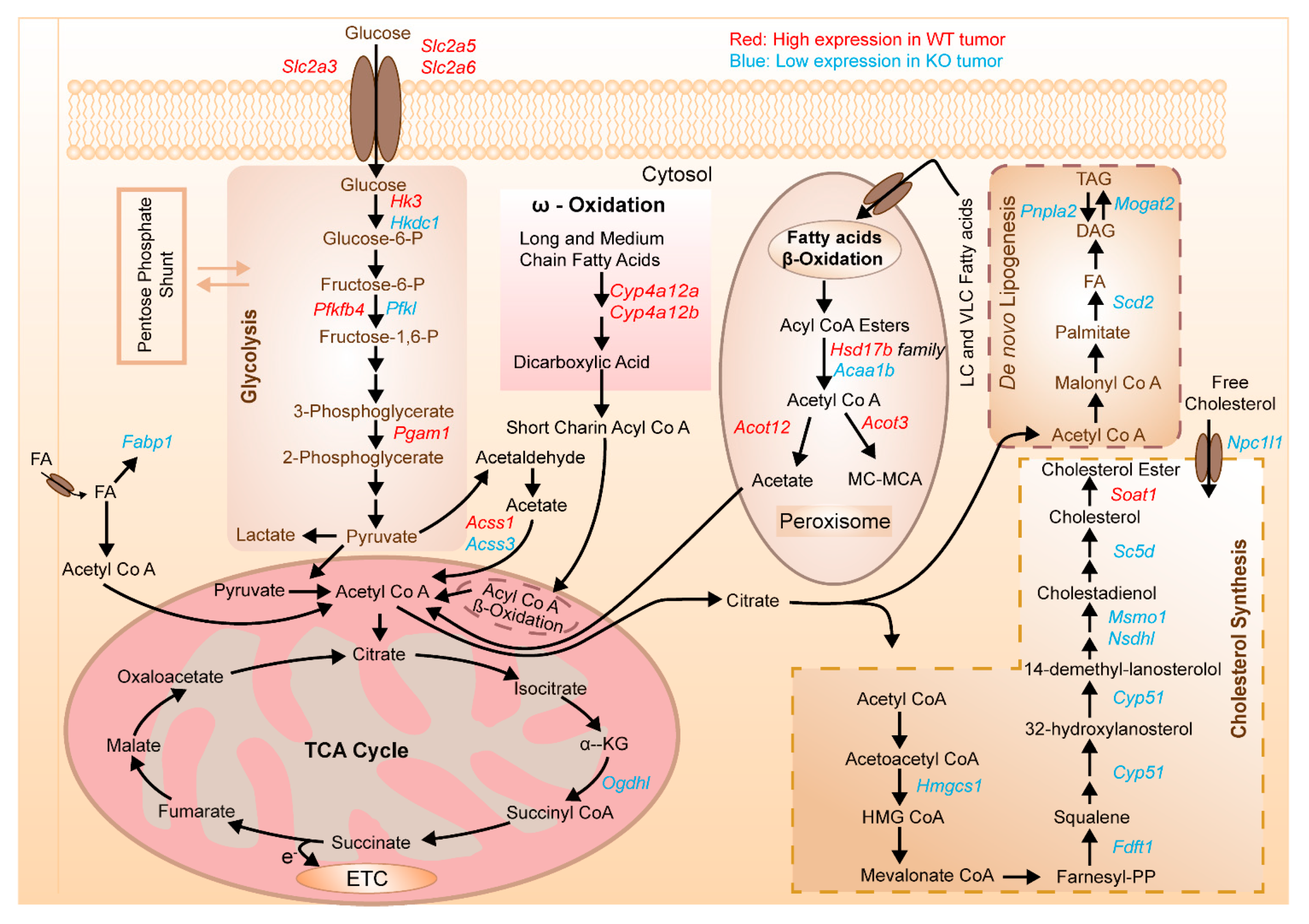
3.6. Transcriptional Profiling of Liver Tissues (Tumor and Extrafocal Liver Tissue) to Characterize Prominent Dysregulated Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Kasmari, A.J.; Welch, A.; Liu, G.; Leslie, D.; McGarrity, T.; Riley, T. Independent of Cirrhosis, Hepatocellular Carcinoma Risk Is Increased with Diabetes and Metabolic Syndrome. Am. J. Med. 2017, 130, 746.e741–746.e747. [Google Scholar] [CrossRef] [Green Version]
- Evert, M.; Dombrowski, F. Hepatocellular carcinoma in the non-cirrhotic liver. Pathologe 2008, 29, 47–52. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e121. [Google Scholar] [CrossRef] [Green Version]
- Bannasch, P. Pathogenesis of hepatocellular carcinoma: Sequential cellular, molecular, and metabolic changes. Prog. Liver Dis. 1996, 14, 161–197. [Google Scholar]
- Dombrowski, F.; Filsinger, E.; Bannasch, P.; Pfeifer, U. Altered liver acini induced in diabetic rats by portal vein islet isografts resemble preneoplastic hepatic foci in their enzymic pattern. Am. J. Pathol. 1996, 148, 1249–1256. [Google Scholar] [PubMed]
- Dombrowski, F.; Lehringer-Polzin, M.; Pfeifer, U. Hyperproliferative liver acini after intraportal islet transplantation in streptozotocin-induced diabetic rats. Lab. Investig. 1994, 71, 688–699. [Google Scholar] [PubMed]
- Evert, M.; Calvisi, D.F.; Evert, K.; De Murtas, V.; Gasparetti, G.; Mattu, S.; Destefanis, G.; Ladu, S.; Zimmermann, A.; Delogu, S.; et al. V-AKT murine thymoma viral oncogene homolog/mammalian target of rapamycin activation induces a module of metabolic changes contributing to growth in insulin-induced hepatocarcinogenesis. Hepatology 2012, 55, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, F.; Mathieu, C.; Evert, M. Hepatocellular neoplasms induced by low-number pancreatic islet transplants in autoimmune diabetic BB/Pfd rats. Cancer Res. 2006, 66, 1833–1843. [Google Scholar] [CrossRef] [Green Version]
- Dombrowski, F.; Bannasch, P.; Pfeifer, U. Hepatocellular neoplasms induced by low-number pancreatic islet transplants in streptozotocin diabetic rats. Am. J. Pathol. 1997, 150, 1071–1087. [Google Scholar]
- Ribback, S.; Calvisi, D.F.; Cigliano, A.; Sailer, V.; Peters, M.; Rausch, J.; Heidecke, C.D.; Birth, M.; Dombrowski, F. Molecular and metabolic changes in human liver clear cell foci resemble the alterations occurring in rat hepatocarcinogenesis. J. Hepatol. 2013, 58, 1147–1156. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Su, Q.; Benner, A.; Hofmann, W.J.; Otto, G.; Pichlmayr, R.; Bannasch, P. Human hepatic preneoplasia: Phenotypes and proliferation kinetics of foci and nodules of altered hepatocytes and their relationship to liver cell dysplasia. Virchows Arch. 1997, 431, 391–406. [Google Scholar] [CrossRef]
- Ribback, S.; Sonke, J.; Lohr, A.; Frohme, J.; Peters, K.; Holm, J.; Peters, M.; Cigliano, A.; Calvisi, D.F.; Dombrowski, F. Hepatocellular glycogenotic foci after combined intraportal pancreatic islet transplantation and knockout of the carbohydrate responsive element binding protein in diabetic mice. Oncotarget 2017, 8, 104315–104329. [Google Scholar] [CrossRef]
- Wang, H.; Dolezal, J.M.; Kulkarni, S.; Lu, J.; Mandel, J.; Jackson, L.E.; Alencastro, F.; Duncan, A.W.; Prochownik, E.V. Myc and ChREBP transcription factors cooperatively regulate normal and neoplastic hepatocyte proliferation in mice. J. Biol. Chem. 2018, 293, 14740–14757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizuka, K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 474–485. [Google Scholar] [CrossRef]
- Ishii, S.; Iizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef] [Green Version]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ido-Kitamura, Y.; Sasaki, T.; Kobayashi, M.; Kim, H.J.; Lee, Y.S.; Kikuchi, O.; Yokota-Hashimoto, H.; Iizuka, K.; Accili, D.; Kitamura, T. Hepatic FoxO1 integrates glucose utilization and lipid synthesis through regulation of Chrebp O-glycosylation. PLoS ONE 2012, 7, e47231. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Girard, J.; Postic, C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005, 87, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Martin, J.; Wang, Z. Statistical methods for identifying differentially expressed genes in RNA-Seq experiments. Cell Biosci. 2012, 2, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, A.; Kato, M.; Fukasawa, M.; Ishiguro, M.; Furuya, E.; Sakakibara, R. Cloning of cDNA encoding for a novel isozyme of fructose 6-phosphate, 2-kinase/fructose 2,6-bisphosphatase from human placenta. J. Biochem. 1996, 119, 506–511. [Google Scholar] [CrossRef]
- Mahlknecht, U.; Chesney, J.; Hoelzer, D.; Bucala, R. Cloning and chromosomal characterization of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 gene (PFKFB3, iPFK2). Int. J. Oncol. 2003, 23, 883–891. [Google Scholar] [CrossRef]
- Yalcin, A.; Telang, S.; Clem, B.; Chesney, J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp. Mol. Pathol. 2009, 86, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Pusec, C.M.; De Jesus, A.; Khan, M.W.; Terry, A.R.; Ludvik, A.E.; Xu, K.; Giancola, N.; Pervaiz, H.; Daviau Smith, E.; Ding, X.; et al. Hepatic HKDC1 Expression Contributes to Liver Metabolism. Endocrinology 2019, 160, 313–330. [Google Scholar] [CrossRef] [Green Version]
- Ribback, S.; Che, L.; Pilo, M.G.; Cigliano, A.; Latte, G.; Pes, G.M.; Porcu, A.; Pascale, R.M.; Li, L.; Qiao, Y.; et al. Oncogene-dependent addiction to carbohydrate-responsive element binding protein in hepatocellular carcinoma. Cell Cycle 2018, 17, 1496–1512. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.H.; Savas, U.; Griffin, K.J.; Johnson, E.F. Human cytochrome p450 family 4 enzymes: Function, genetic variation and regulation. Drug Metab. Rev. 2007, 39, 515–538. [Google Scholar] [CrossRef]
- Yamashita, T.; Honda, M.; Takatori, H.; Nishino, R.; Minato, H.; Takamura, H.; Ohta, T.; Kaneko, S. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J. Hepatol. 2009, 50, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6 Months | 12 Months | |||||||
|---|---|---|---|---|---|---|---|---|
| Transplantation | Control | Transplantation | Control | |||||
| Diabetic | Nondiabetic | Diabetic | Nondiabetic | Diabetic | Nondiabetic | Diabetic | Nondiabetic | |
| Wildtype C57Bl/6J | n = 36 | n = 20 | n = 20 | n = 19 | n = 33 | n = 19 | n = 13 | n = 20 |
| ChREBP-knockout | n = 18 | n = 19 | n = 18 | n = 20 | n = 12 | n = 19 | n = 13 | n = 21 |
| Experimental Groups | 6 Months (Mean ± S.E.M. (n)) | 12 Months (Mean ± S.E.M. (n)) |
|---|---|---|
| WT diabetic transplanted | 8.76 ± 0.90% (20) 1 | 10.63 ± 1.77% (20) 2 |
| KO diabetic transplanted | 5.16 ± 1.04% (9) 1 | 2.24 ± 0.61% (9) 2 |
| WT transplanted | 1.35 ± 0.27% (10) | 1.49 ± 0.43% (10) |
| KO transplanted | 6.10 ± 2.51% (10) | 3.89 ± 2.33% (10) |
| WT diabetic | 6.11 ± 2.18% (6) | 2.57 ± 0.93% (9) |
| KO diabetic | 2.11 ± 0.60% (9) | 4.20 ± 1.31% (8) |
| WT non-diabetic | 1.54 ± 0.33% (10) | 1.03 ± 0.21% (10) |
| KO non-diabetic | 2.21 ± 0.87% (10) | 1.69 ± 0.47% (15) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuernberger, V.; Mortoga, S.; Metzendorf, C.; Burkert, C.; Ehricke, K.; Knuth, E.; Zimmer, J.; Singer, S.; Nath, N.; Karim, M.; et al. Hormonally Induced Hepatocellular Carcinoma in Diabetic Wild Type and Carbohydrate Responsive Element Binding Protein Knockout Mice. Cells 2021, 10, 2787. https://doi.org/10.3390/cells10102787
Nuernberger V, Mortoga S, Metzendorf C, Burkert C, Ehricke K, Knuth E, Zimmer J, Singer S, Nath N, Karim M, et al. Hormonally Induced Hepatocellular Carcinoma in Diabetic Wild Type and Carbohydrate Responsive Element Binding Protein Knockout Mice. Cells. 2021; 10(10):2787. https://doi.org/10.3390/cells10102787
Chicago/Turabian StyleNuernberger, Vincent, Sharif Mortoga, Christoph Metzendorf, Christian Burkert, Katrina Ehricke, Elisa Knuth, Jenny Zimmer, Stephan Singer, Neetika Nath, Majedul Karim, and et al. 2021. "Hormonally Induced Hepatocellular Carcinoma in Diabetic Wild Type and Carbohydrate Responsive Element Binding Protein Knockout Mice" Cells 10, no. 10: 2787. https://doi.org/10.3390/cells10102787