
Respiratory Syncytial Virus Matrix Protein-Chromatin Association Is Key to Transcriptional Inhibition in Infected Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Constructs
2.2. Cell and Virus Culture
2.3. Transfection
2.4. CLSM Imaging and Image Analysis
2.5. In Situ RNA Synthesis
2.6. Bacterial Expression and Purification of RSV M Fusion Proteins
2.7. In Vitro Transcription Assay
2.8. In Vitro DNA Binding Assay
2.9. In Vitro RNA Binding Assay
2.10. Nuclear Association In Situ
2.11. Generation and Recovery of Mutant Recombinant RSV
2.12. Replication Kinetics of Recombinant RSV
2.13. Infection and Immunofluorescence
2.14. Real-Time RT-PCR to Quantify Viral RNA
2.15. Real-Time RT-PCR to Quantify Host Gene Expression
2.16. Animal Experiments
3. Results
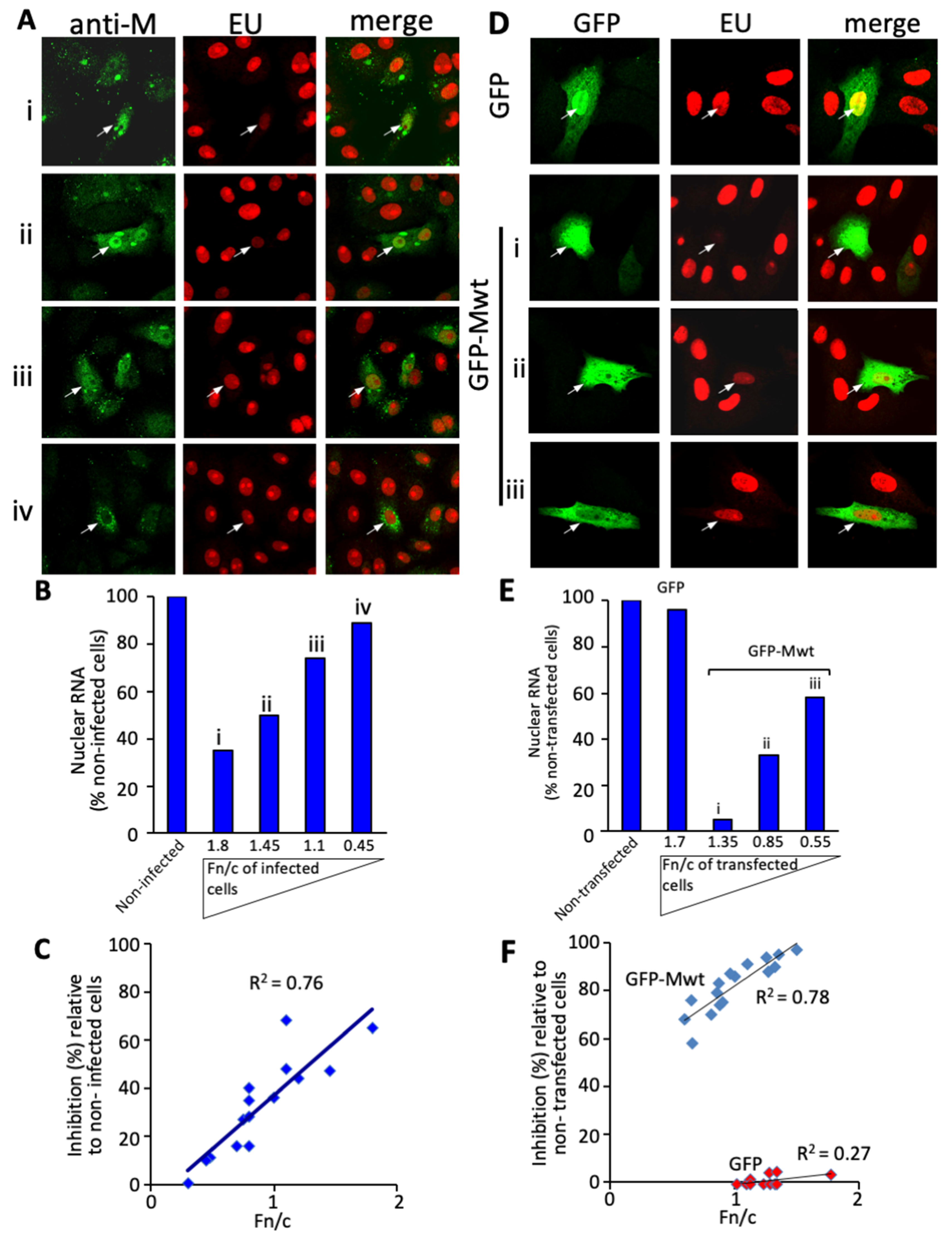
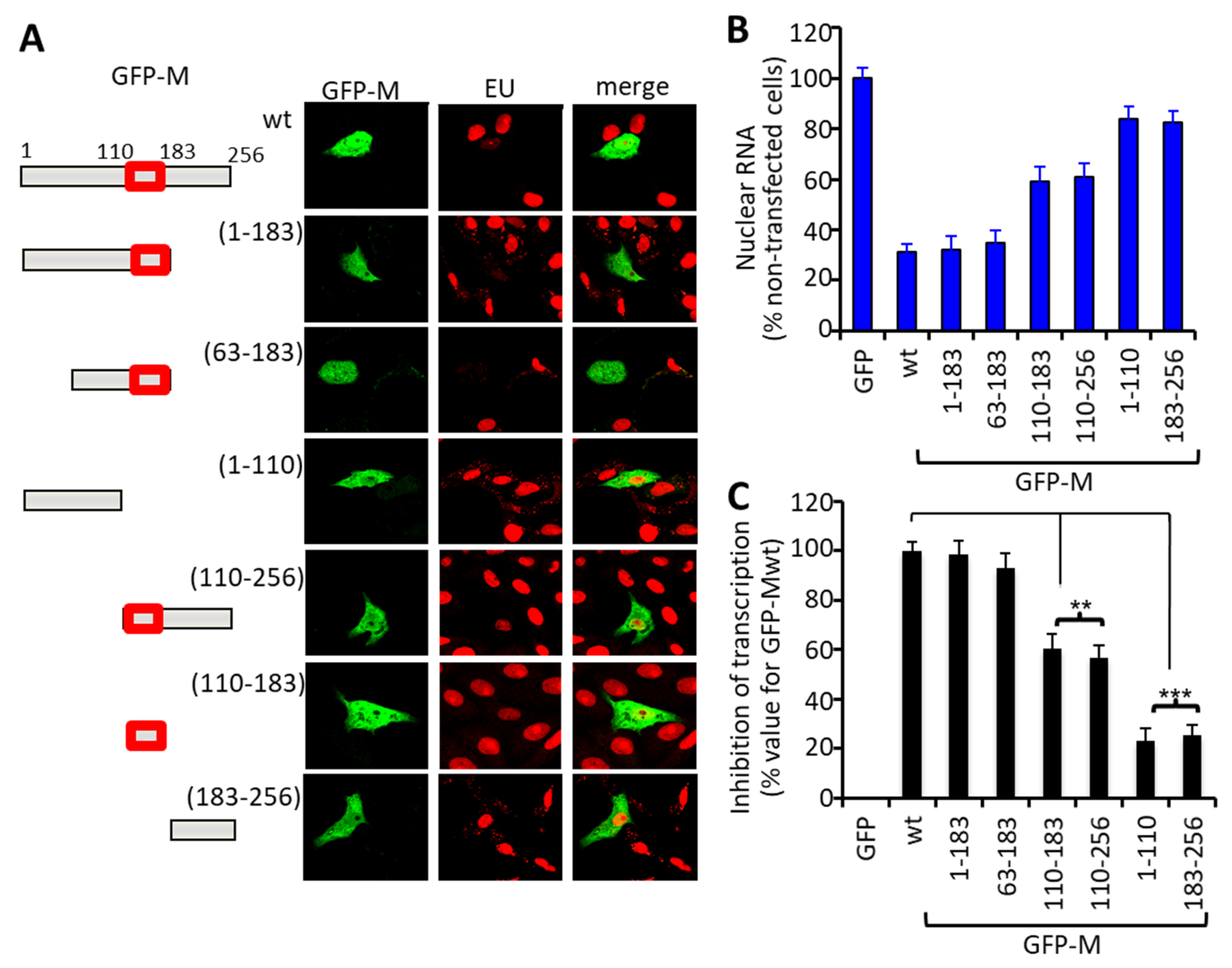
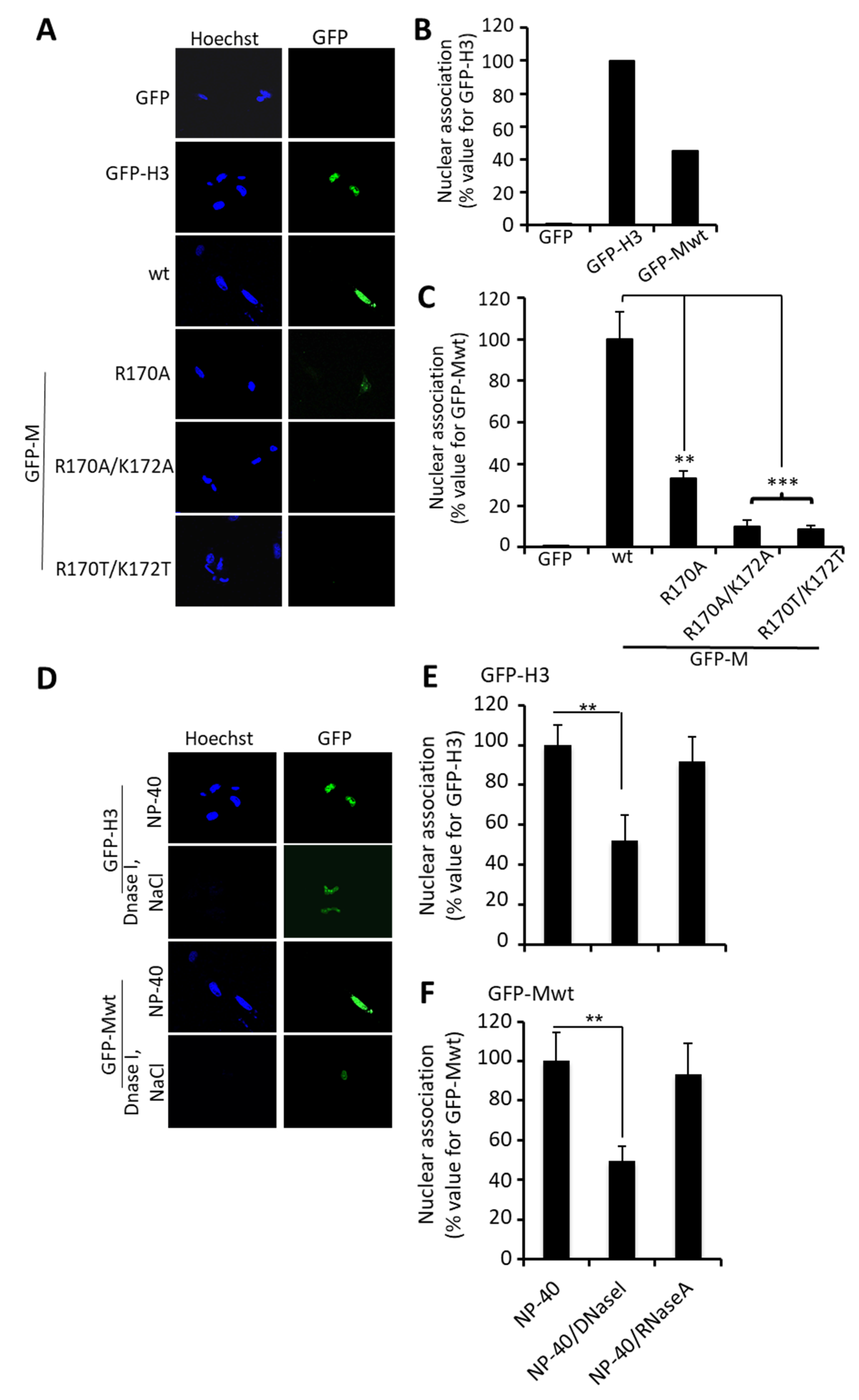
3.1. RSV M Inhibition of Host Cell Transcription Parallels the Extent of Nuclear Accumulation
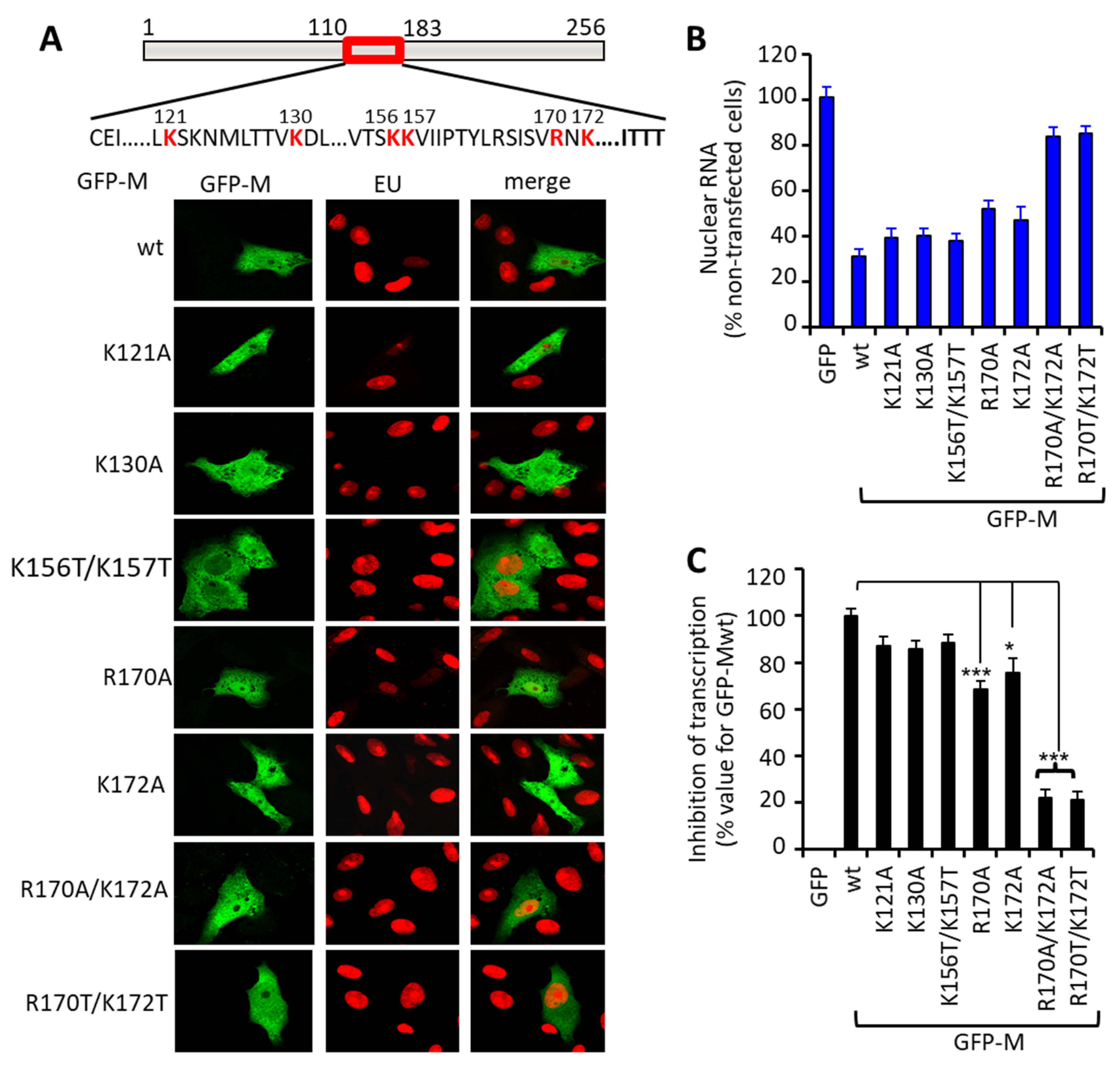
3.2. Arginine 170 and Lysine 172 within the Central Domain of M Are Critical for Transcriptional Inhibition
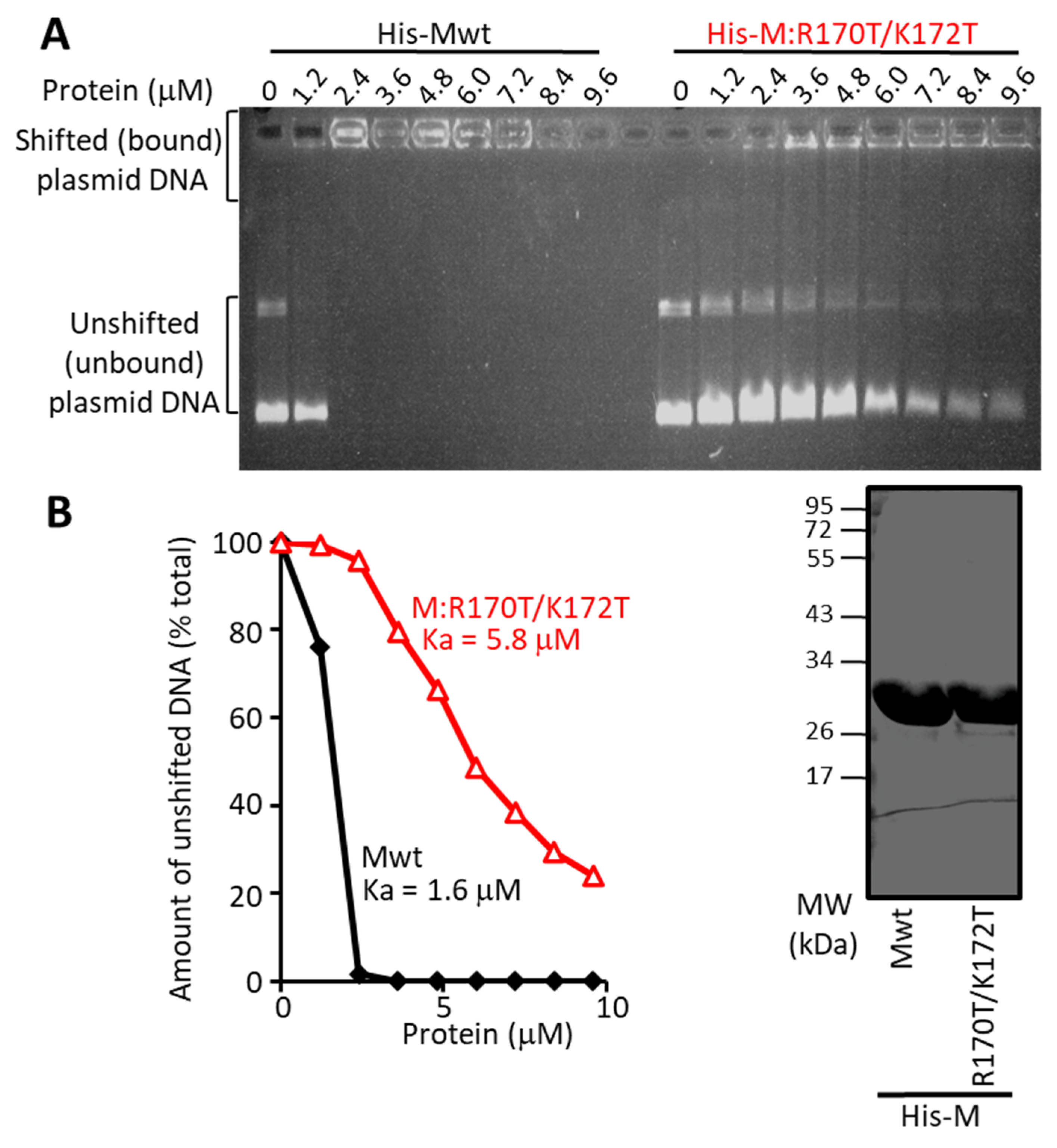
3.3. The R170T/K172T Mutant of M Binds RNA Normally, but Is Deficient in Transcriptional Inhibition and DNA Binding
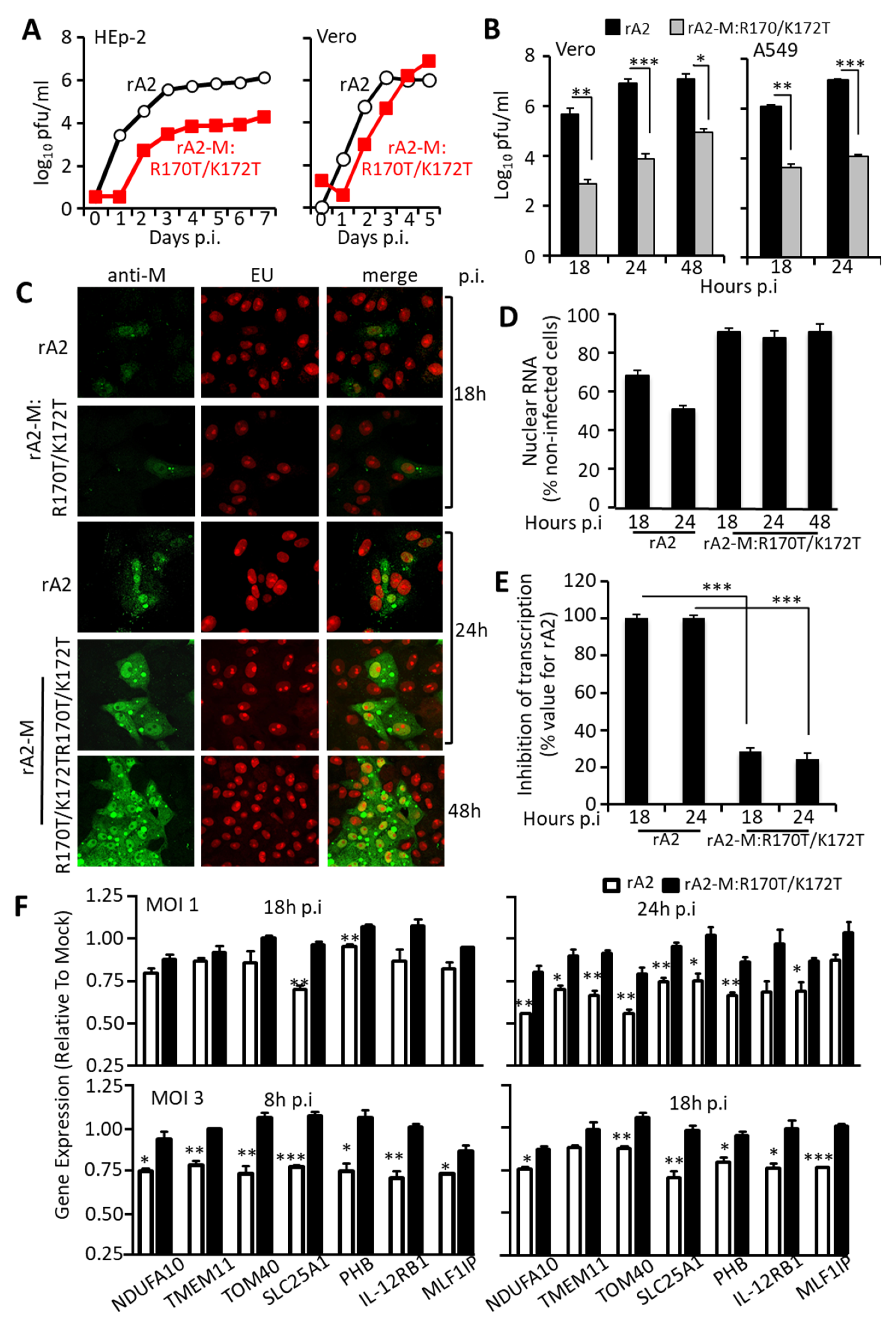
3.4. RSV Carrying Mutations in M Arginine 170/Lysine 172 Is Attenuated, Showing Concomitant Transcriptional Inhibition and Virus Production
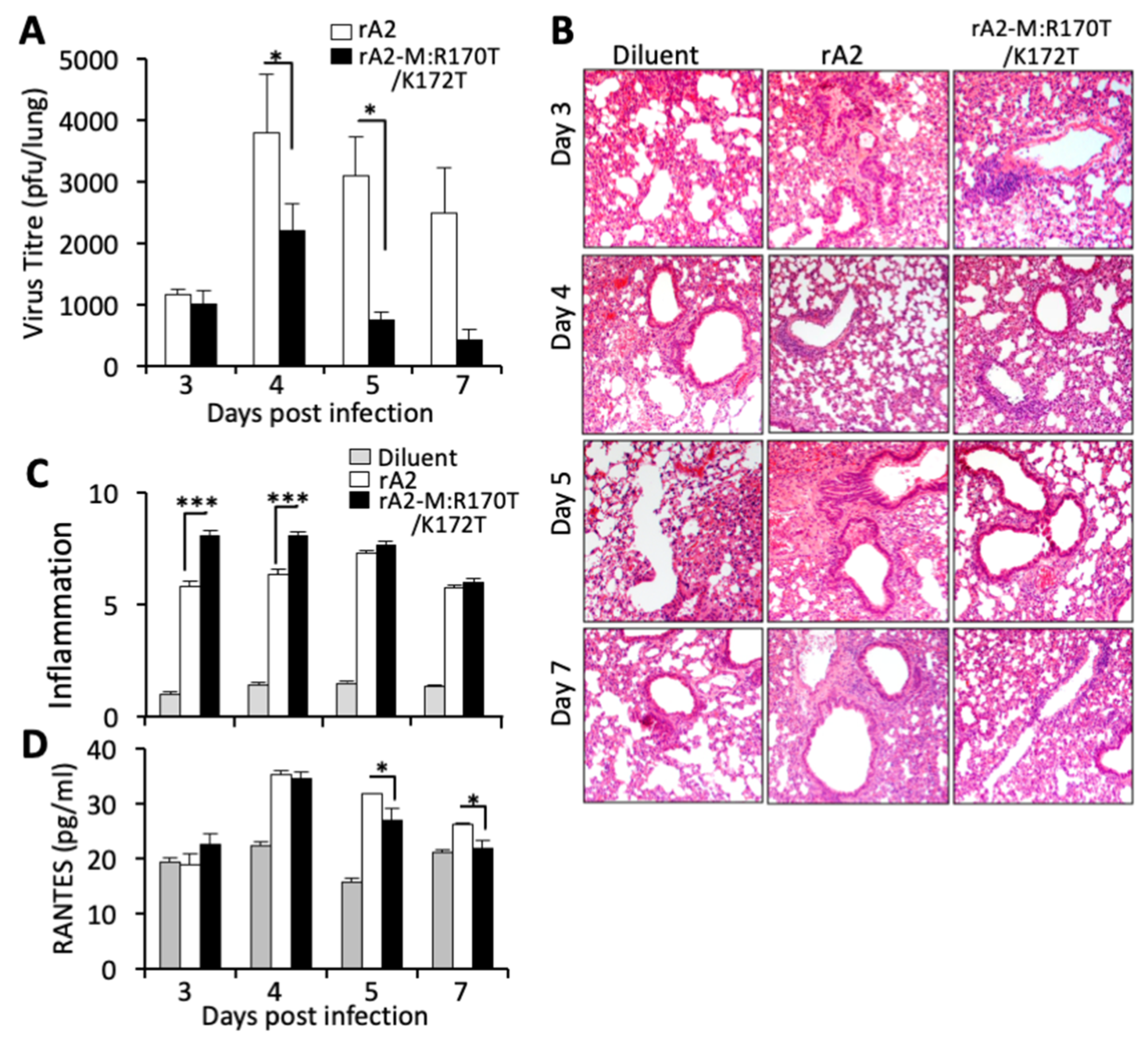
3.5. RSV Carrying Mutations in M Arginine 170/Lysine 172 Shows Reduced Viremia and Increased Anti-Viral Response in an Animal Model
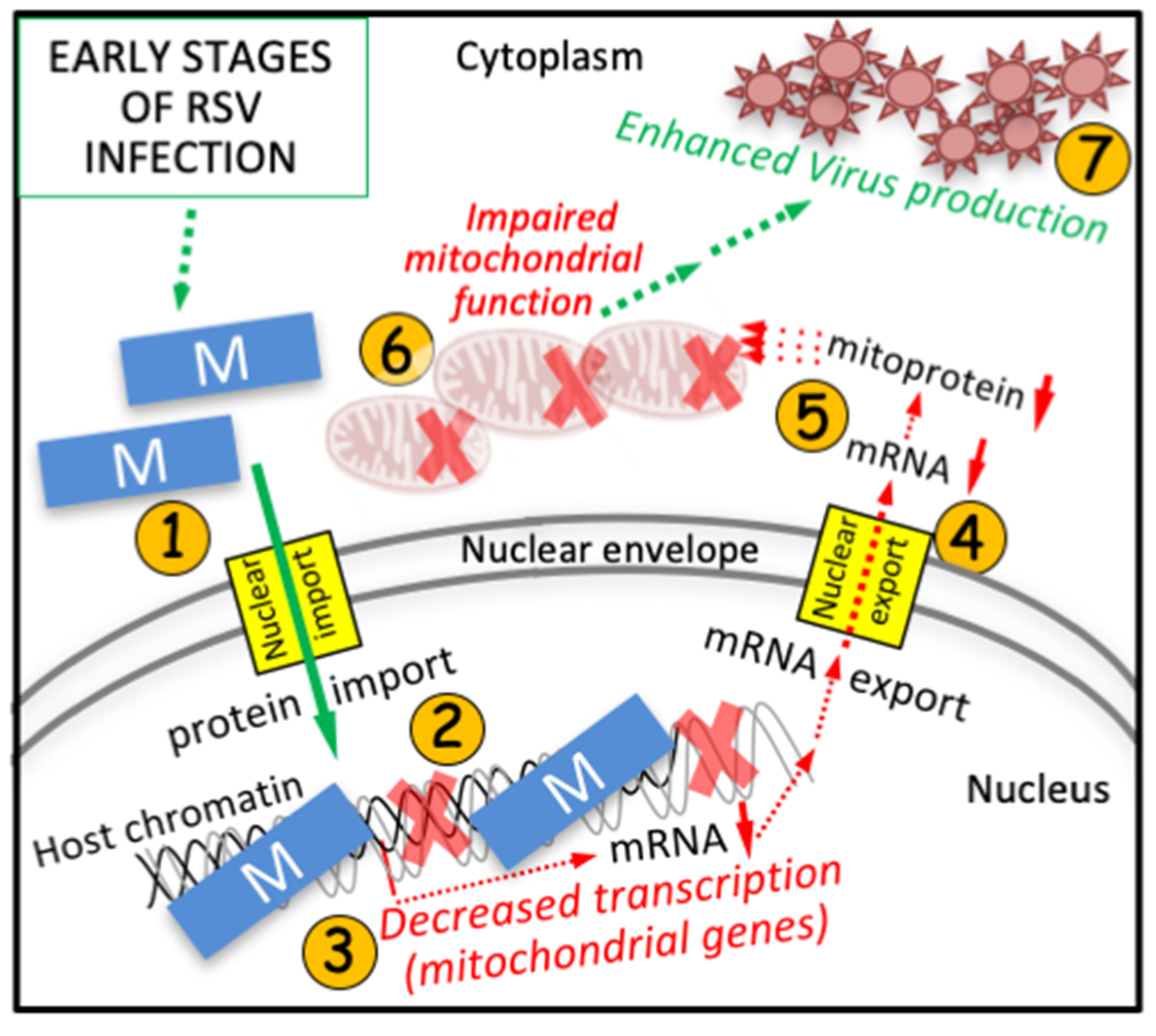
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the united states. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Collins, P.L.; Crowe, J. Respiratory syncytial virus and metapneumovirus. In Fields Virology, 5th ed.; Knipe, D., Howley, P., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2. [Google Scholar]
- Ghildyal, R.; Jans, D.A.; Bardin, P.G.; Mills, J. Protein-protein interactions in rsv assembly: Potential targets for attenuating rsv strains. Infect. Disord. Drug Targets 2012, 12, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghildyal, R.; Ho, A.; Jans, D.A. Central role of the respiratory syncytial virus matrix protein in infection. FEMS Microbiol. Rev. 2006, 30, 692–705. [Google Scholar] [CrossRef]
- Ghildyal, R.; Li, D.; Peroulis, I.; Shields, B.; Bardin, P.G.; Teng, M.N.; Collins, P.L.; Meanger, J.; Mills, J. Interaction between the respiratory syncytial virus g glycoprotein cytoplasmic domain and the matrix protein. J. Gen. Virol. 2005, 86, 1879–1884. [Google Scholar] [CrossRef]
- Ghildyal, R.; Mills, J.; Murray, M.; Vardaxis, N.; Meanger, J. Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J. Gen. Virol. 2002, 83, 753–757. [Google Scholar] [CrossRef]
- Li, D.; Jans, D.A.; Bardin, P.G.; Meanger, J.; Mills, J.; Ghildyal, R. Association of respiratory syncytial virus m protein with viral nucleocapsids is mediated by the m2-1 protein. J. Virol. 2008, 82, 8863–8870. [Google Scholar] [CrossRef] [Green Version]
- Marty, A.; Meanger, J.; Mills, J.; Shields, B.; Ghildyal, R. Association of matrix protein of respiratory syncytial virus with the host cell membrane of infected cells. Arch. Virol. 2004, 149, 199–210. [Google Scholar] [CrossRef]
- Teng, M.N.; Collins, P.L. Identification of the respiratory syncytial virus proteins required for formation and passage of helper-dependent infectious particles. J. Virol. 1998, 72, 5707–5716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghildyal, R.; Baulch-Brown, C.; Mills, J.; Meanger, J. The matrix protein of human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch. Virol. 2003, 148, 1419–1429. [Google Scholar] [CrossRef]
- Ghildyal, R.; Ho, A.; Wagstaff, K.M.; Dias, M.M.; Barton, C.L.; Jans, P.; Bardin, P.; Jans, D.A. Nuclear import of the respiratory syncytial virus matrix protein is mediated by importin beta1 independent of importin alpha. Biochemistry 2005, 44, 12887–12895. [Google Scholar] [CrossRef]
- Alvisi, G.; Rawlinson, S.M.; Ghildyal, R.; Ripalti, A.; Jans, D.A. Regulated nucleocytoplasmic trafficking of viral gene products: A therapeutic target? Biochim. Biophys. Acta 2008, 1784, 213–227. [Google Scholar] [CrossRef]
- Ghildyal, R.; Ho, A.; Dias, M.; Soegiyono, L.; Bardin, P.G.; Tran, K.C.; Teng, M.N.; Jans, D.A. The respiratory syncytial virus matrix protein possesses a crm1-mediated nuclear export mechanism. J. Virol. 2009, 83, 5353–5362. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, L.; Cuesta, I.; Asenjo, A.; Villanueva, N. Human respiratory syncytial virus matrix protein is an rna-binding protein: Binding properties, location and identity of the rna contact residues. J. Gen. Virol. 2004, 85, 709–719. [Google Scholar] [CrossRef]
- Henderson, G.; Murray, J.; Yeo, R.P. Sorting of the respiratory syncytial virus matrix protein into detergent-resistant structures is dependent on cell-surface expression of the glycoproteins. Virology 2002, 300, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Clarke, D.; Zhou, H.Z.; Cheng, X.; Coelingh, K.; Bryant, M.; Li, S. Recombinant human respiratory syncytial virus (rsv) from cdna and construction of subgroup a and b chimeric rsv. Virology 1998, 251, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Piechaczek, C.; Fetzer, C.; Baiker, A.; Bode, J.; Lipps, H.J. A vector based on the sv40 origin of replication and chromosomal s/mars replicates episomally in cho cells. Nucleic Acids Res. 1999, 27, 426–428. [Google Scholar] [CrossRef]
- Tannock, G.A.; Hierholzer, J.C.; Bryce, D.A.; Chee, C.F.; Paul, J.A. Freeze-drying of respiratory syncytial viruses for transportation and storage. J. Clin. Microbiol. 1987, 25, 1769–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, V.R.; Layfield, S.; Mitchell, C.L.; Forwood, J.K.; John, A.P.; Briggs, L.J.; McDowall, S.G.; Jans, D.A. Defective importin beta recognition and nuclear import of the sex-determining factor sry are associated with xy sex-reversing mutations. Proc. Natl. Acad. Sci. USA 2003, 100, 7045–7050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, M.H.; Thomas, R.J.; Loveland, K.L.; Schilders, S.; Gu, M.; Martin, T.J.; Gillespie, M.T.; Jans, D.A. Nuclear transport of parathyroid hormone (pth)-related protein is dependent on microtubules. Mol. Endocrinol. 2002, 16, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Fulcher, A.J.; Dias, M.M.; Jans, D.A. Binding of p110 retinoblastoma protein inhibits nuclear import of simian virus sv40 large tumor antigen. J. Biol. Chem. 2010, 285, 17744–17753. [Google Scholar] [CrossRef] [Green Version]
- Alvisi, G.; Roth, D.M.; Camozzi, D.; Pari, G.S.; Loregian, A.; Ripalti, A.; Jans, D.A. The flexible loop of the human cytomegalovirus DNA polymerase processivity factor ppul44 is required for efficient DNA binding and replication in cells. J. Virol. 2009, 83, 9567–9576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, P.L.; Hill, M.G.; Camargo, E.; Grosfeld, H.; Chanock, R.M.; Murphy, B.R. Production of infectious human respiratory syncytial virus from cloned cdna confirms an essential role for the transcription elongation factor from the 5’ proximal open reading frame of the m2 mrna in gene expression and provides a capability for vaccine development. Proc. Natl. Acad. Sci. USA 1995, 92, 11563–11567. [Google Scholar]
- Teng, M.N.; Collins, P.L. Altered growth characteristics of recombinant respiratory syncytial viruses which do not produce ns2 protein. J. Virol. 1999, 73, 466–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, M.N.; Whitehead, S.S.; Collins, P.L. Contribution of the respiratory syncytial virus g glycoprotein and its secreted and membrane-bound forms to virus replication in vitro and in vivo. Virology 2001, 289, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Tran, K.C.; He, B.; Teng, M.N. Replacement of the respiratory syncytial virus nonstructural proteins ns1 and ns2 by the v protein of parainfluenza virus 5. Virology 2007, 368, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualano, R.C.; Pryor, M.J.; Cauchi, M.R.; Wright, P.J.; Davidson, A.D. Identification of a major determinant of mouse neurovirulence of dengue virus type 2 using stably cloned genomic-length cdna. J. Gen. Virol. 1998, 79 Pt 3, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Rawlinson, S.M.; Pryor, M.J.; Wright, P.J.; Jans, D.A. Crm1-mediated nuclear export of dengue virus rna polymerase ns5 modulates interleukin-8 induction and virus production. J. Biol. Chem. 2009, 284, 15589–15597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvell, C.; Norrby, E.; Mufson, M.A. Preparation and characterization of monoclonal antibodies directed against five structural components of human respiratory syncytial virus subgroup b. J. Gen. Virol. 1987, 68, 3125–3135. [Google Scholar] [CrossRef]
- Mentel, R.; Wegner, U.; Bruns, R.; Gurtler, L. Real-time pcr to improve the diagnosis of respiratory syncytial virus infection. J. Med. Microbiol. 2003, 52, 893–896. [Google Scholar] [CrossRef]
- Murphy, N.; Ring, M.; Heffron, C.C.; King, B.; Killalea, A.G.; Hughes, C.; Martin, C.M.; McGuinness, E.; Sheils, O.; O’Leary, J.J. P16ink4a, cdc6, and mcm5: Predictive biomarkers in cervical preinvasive neoplasia and cervical cancer. J. Clin. Pathol. 2005, 58, 525–534. [Google Scholar] [CrossRef]
- Desfarges, S.; Abderrahmani, A.; Hernandez-Novoa, B.; Munoz, M.; Ciuffi, A. Ledgf/p75 tata-less promoter is driven by the transcription factor sp1. J. Mol. Biol. 2011, 414, 177–193. [Google Scholar] [CrossRef] [Green Version]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and stable knockdown of the integrase cofactor ledgf/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussey, D.J.; Moore, S.; Nicola, M.; Dobrovic, A. Fusion of the nup98 gene with the ledgf/p52 gene defines a recurrent acute myeloid leukemia translocation. BMC Genet. 2001, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein drp1 in neurons from patients with alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Hwang, S.; Mahadevan, S.; Qadir, F.; Hutchison, I.L.; Costea, D.E.; Neppelberg, E.; Liavaag, P.G.; Waseem, A.; Teh, M.T. Identification of foxm1-induced epigenetic markers for head and neck squamous cell carcinomas. Cancer 2013, 119, 4249–4258. [Google Scholar] [CrossRef]
- Shiina, M.; Kobayashi, K.; Satoh, H.; Niitsuma, H.; Ueno, Y.; Shimosegawa, T. Ribavirin upregulates interleukin-12 receptor and induces t cell differentiation towards type 1 in chronic hepatitis c. J. Gastroenterol. Hepatol. 2004, 19, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.F.; Cheong, W.F.; Shu, M.H.; Teh, C.H.; Chan, K.L.; AbuBakar, S. Eurycomanone suppresses expression of lung cancer cell tumor markers, prohibitin, annexin 1 and endoplasmic reticulum protein 28. Phytomedicine 2012, 19, 138–144. [Google Scholar] [CrossRef]
- Kolukula, V.K.; Sahu, G.; Wellstein, A.; Rodriguez, O.C.; Preet, A.; Iacobazzi, V.; D’Orazi, G.; Albanese, C.; Palmieri, F.; Avantaggiati, M.L. Slc25a1, or cic, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Tran, K.C.; Collins, P.L.; Teng, M.N. Effects of altering the transcription termination signals of respiratory syncytial virus on viral gene expression and growth in vitro and in vivo. J. Virol. 2004, 78, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Ford, J.G.; Rennick, D.; Donaldson, D.D.; Venkayya, R.; McArthur, C.; Hansell, E.; Kurup, V.P.; Warnock, M.; Grunig, G. Il-13 and ifn-gamma: Interactions in lung inflammation. J. Immunol. 2001, 167, 1769–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foronjy, R.F.; Dabo, A.J.; Taggart, C.C.; Weldon, S.; Geraghty, P. Respiratory syncytial virus infections enhance cigarette smoke induced copd in mice. PLoS ONE 2014, 9, e90567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemeijer, M.C.; Vonk, A.M.; Monastyrska, I.; Rottier, P.J.; de Haan, C.A. Visualizing coronavirus rna synthesis in time by using click chemistry. J. Virol. 2012, 86, 5808–5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, I.K.; Oro, C.; Dias, M.M.; Zhang, J.; Jans, D.A. Apoptin nuclear accumulation is modulated by a crm1-recognized nuclear export signal that is active in normal but not in tumor cells. Cancer Res. 2005, 65, 7059–7064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preiss, S.; Argentaro, A.; Clayton, A.; John, A.; Jans, D.A.; Ogata, T.; Nagai, T.; Barroso, I.; Schafer, A.J.; Harley, V.R. Compound effects of point mutations causing campomelic dysplasia/autosomal sex reversal upon SOX9 structure, nuclear transport, DNA binding, and transcriptional activation. J. Biol. Chem. 2001, 276, 27864–27872. [Google Scholar] [CrossRef] [Green Version]
- Orrego, M.; Ponte, I.; Roque, A.; Buschati, N.; Mora, X.; Suau, P. Differential affinity of mammalian histone H1 somatic subtypes for DNA and chromatin. BMC Biol. 2007, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Ling, Z.; Tran, K.C.; Arnold, J.J.; Teng, M.N. Purification and characterization of recombinant human respiratory syncytial virus nonstructural protein NS1. Protein Expr. Purif. 2008, 57, 261–270. [Google Scholar] [CrossRef]
- Zhang, Y.; Jamaluddin, M.; Wang, S.; Tian, B.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Ribavirin treatment up-regulates antiviral gene expression via the interferon-stimulated response element in respiratory syncytial virus-infected epithelial cells. J. Virol. 2003, 77, 5933–5947. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Schulze, K.E.; Ghildyal, R.; Henstridge, D.C.; Kolanowski, J.L.; New, E.J.; Hong, Y.; Hsu, A.C.; Hansbro, P.M.; Wark, P.A.; et al. Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. eLife 2019, 8, e42448. [Google Scholar] [CrossRef]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Impact of respiratory syncytial virus infection on host functions: Implications for antiviral strategies. Physiol. Rev. 2020, 100, 1527–1594. [Google Scholar] [CrossRef]
- Hu, M.; Li, H.M.; Bogoyevitch, M.A.; Jans, D.A. Mitochondrial protein p32/hapb1/gc1qr/c1qbp is required for efficient respiratory syncytial virus production. Biochem. Biophys. Res. Commun. 2017, 489, 460–465. [Google Scholar] [CrossRef]
- Mehra, D.; Geraghty, P.M.; Hardigan, A.A.; Foronjy, R. A comparison of the inflammatory and proteolytic effects of dung biomass and cigarette smoke exposure in the lung. PLoS ONE 2012, 7, e52889. [Google Scholar] [CrossRef]
- Munday, D.C.; Howell, G.; Barr, J.N.; Hiscox, J.A. Proteomic analysis of mitochondria in respiratory epithelial cells infected with human respiratory syncytial virus and functional implications for virus and cell biology. J. Pharm. Pharmacol. 2015, 67, 300–318. [Google Scholar] [CrossRef] [PubMed]
- Van Diepen, A.; Brand, H.K.; Sama, I.; Lambooy, L.H.; van den Heuvel, L.P.; van der Well, L.; Huynen, M.; Osterhaus, A.D.; Andeweg, A.C.; Hermans, P.W. Quantitative proteome profiling of respiratory virus-infected lung epithelial cells. J. Proteom. 2010, 73, 1680–1693. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Subversion of host cell mitochondria by RSV to favor virus production is dependent on inhibition of mitochondrial complex I and ROS generation. Cells 2019, 8, 1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broussard, G.W.; Hatfull, G.F. Evolution of genetic switch complexity. Bacteriophage 2013, 3, e24186. [Google Scholar] [CrossRef] [Green Version]
- Lou, C.; Yang, X.; Liu, X.; He, B.; Ouyang, Q. A quantitative study of lambda-phage switch and its components. Biophys. J. 2007, 92, 2685–2693. [Google Scholar] [CrossRef] [Green Version]
- Rokney, A.; Kobiler, O.; Amir, A.; Court, D.L.; Stavans, J.; Adhya, S.; Oppenheim, A.B. Host responses influence on the induction of lambda prophage. Mol. Microbiol. 2008, 68, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Schubert, R.A.; Dodd, I.B.; Egan, J.B.; Shearwin, K.E. Cro’s role in the CI Cro bistable switch is critical for l’s transition from lysogeny to lytic development. Genes Dev. 2007, 21, 2461–2472. [Google Scholar] [CrossRef] [Green Version]
- Peeples, M.E. Differential detergent treatment allows immunofluorescent localization of the newcastle disease virus matrix protein within the nucleus of infected cells. Virology 1988, 162, 255–259. [Google Scholar] [CrossRef]
- Peeples, M.E.; Wang, C.; Gupta, K.C.; Coleman, N. Nuclear entry and nucleolar localization of the newcastle disease virus (NDV) matrix protein occur early in infection and do not require other NDV proteins. J. Virol. 1992, 66, 3263–3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentecost, M.; Vashisht, A.A.; Lester, T.; Voros, T.; Beaty, S.M.; Park, A.; Wang, Y.E.; Yun, T.E.; Freiberg, A.N.; Wohlschlegel, J.A.; et al. Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among paramyxovirinae matrix proteins. PLoS Pathog. 2015, 11, e1004739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Nagai, Y.; Yoshii, S.; Maeno, K.; Matsumoto, T.; Hoshino, M. Membrane (M) protein of HVJ (sendai virus): Its role in virus assembly. Virology 1976, 71, 143–161. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.-M.; Ghildyal, R.; Hu, M.; Tran, K.C.; Starrs, L.M.; Mills, J.; Teng, M.N.; Jans, D.A. Respiratory Syncytial Virus Matrix Protein-Chromatin Association Is Key to Transcriptional Inhibition in Infected Cells. Cells 2021, 10, 2786. https://doi.org/10.3390/cells10102786
Li H-M, Ghildyal R, Hu M, Tran KC, Starrs LM, Mills J, Teng MN, Jans DA. Respiratory Syncytial Virus Matrix Protein-Chromatin Association Is Key to Transcriptional Inhibition in Infected Cells. Cells. 2021; 10(10):2786. https://doi.org/10.3390/cells10102786
Chicago/Turabian StyleLi, Hong-Mei, Reena Ghildyal, Mengjie Hu, Kim C. Tran, Lora M. Starrs, John Mills, Michael N. Teng, and David A. Jans. 2021. "Respiratory Syncytial Virus Matrix Protein-Chromatin Association Is Key to Transcriptional Inhibition in Infected Cells" Cells 10, no. 10: 2786. https://doi.org/10.3390/cells10102786