
Synthesis and Characterization of Spirocyclic Mid-Block Containing Triblock Copolymer
 , and
, and
Abstract
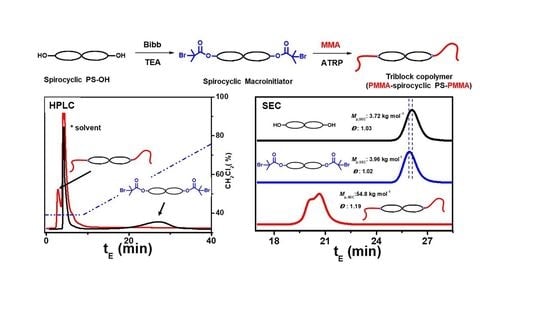
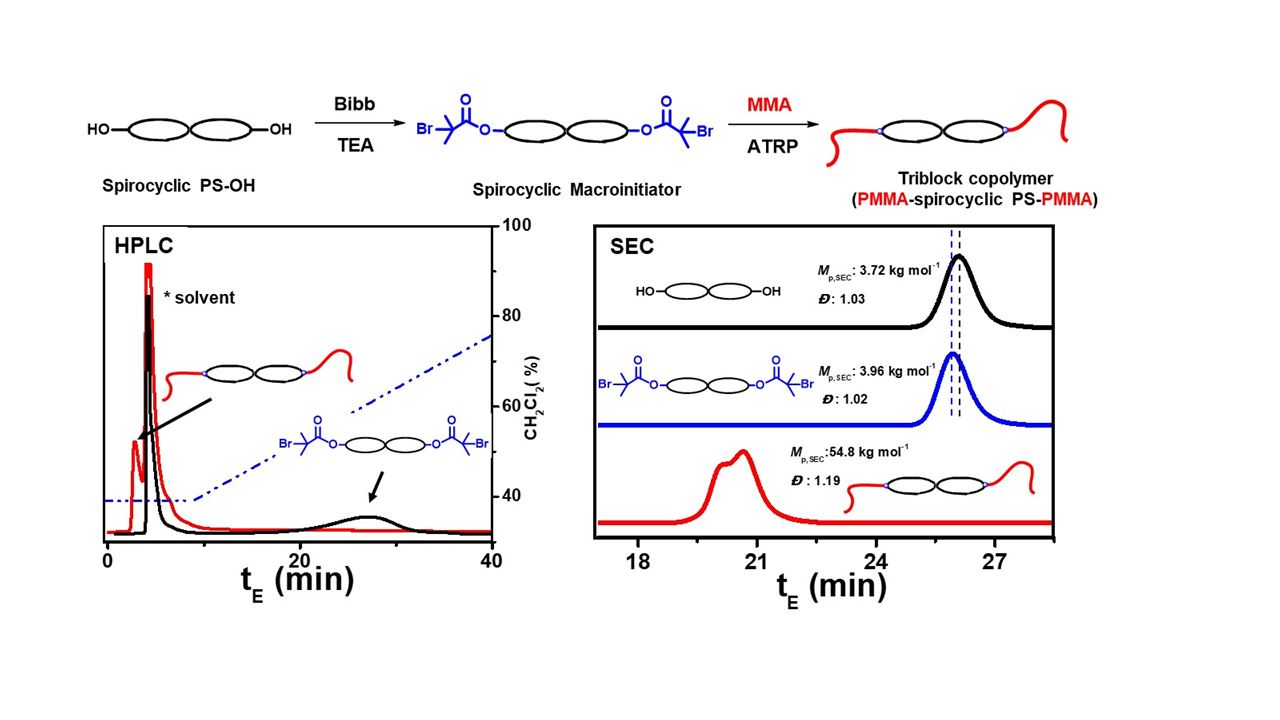
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Characterization
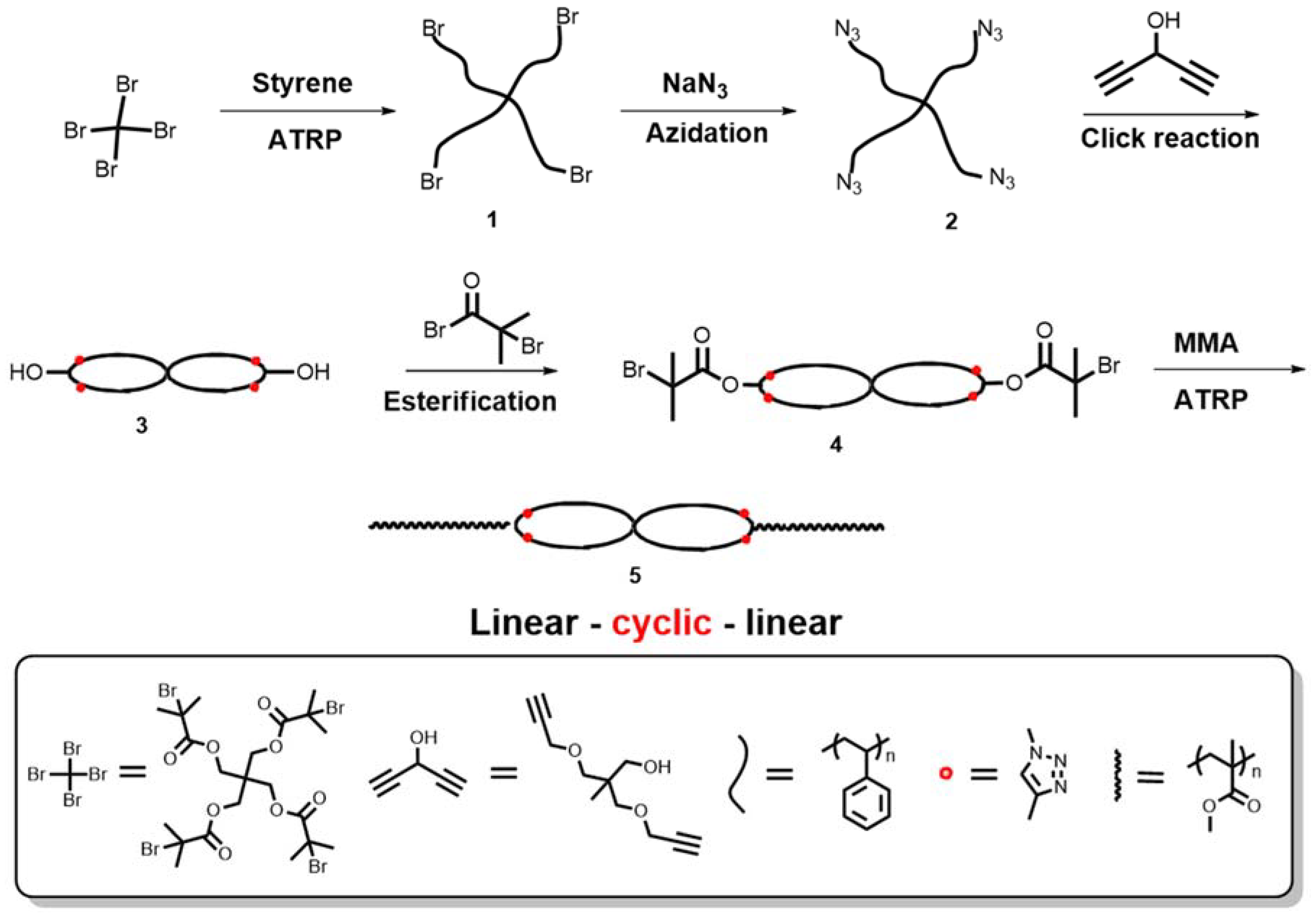
2.3. Syntheses
2.3.1. Tetra-Functional Initiator (Pentaerythritol Tetrakis (2-Bromoisobutyrate))
2.3.2. Tetra-Arm Polystyrene (1)
2.3.3. Azide Functionalized Tetra-Arm Polystyrene (2)
2.3.4. Coupling Agent (CA) of (2,2-Bis [(2-propyn-1-yloxy) methyl]-1-propanol)
2.3.5. cPS (3)
2.3.6. Spirocyclic Macroinitiator (4)
2.3.7. Triblock Copolymer (PMMA-b-cPS-b-PMMA) (5)
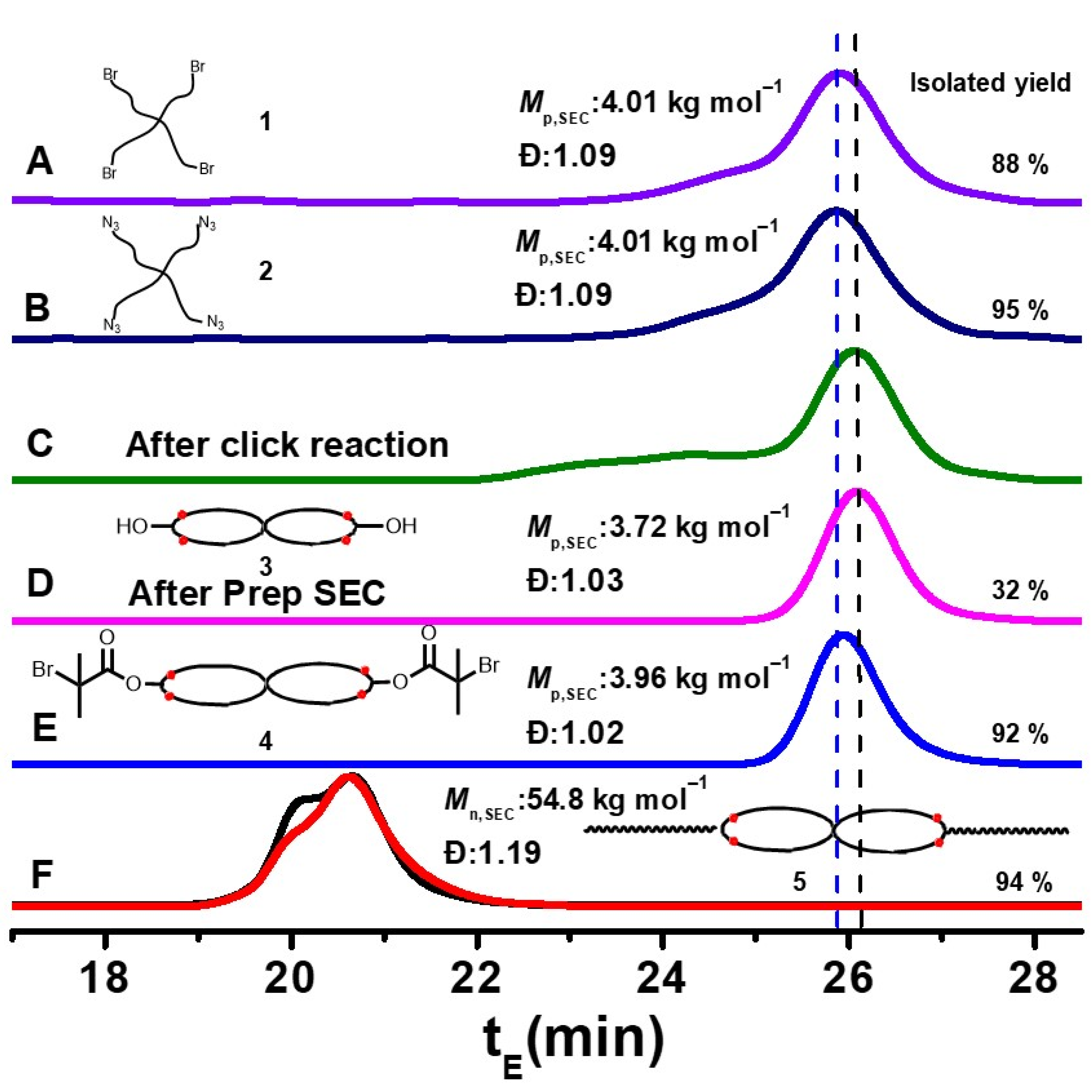
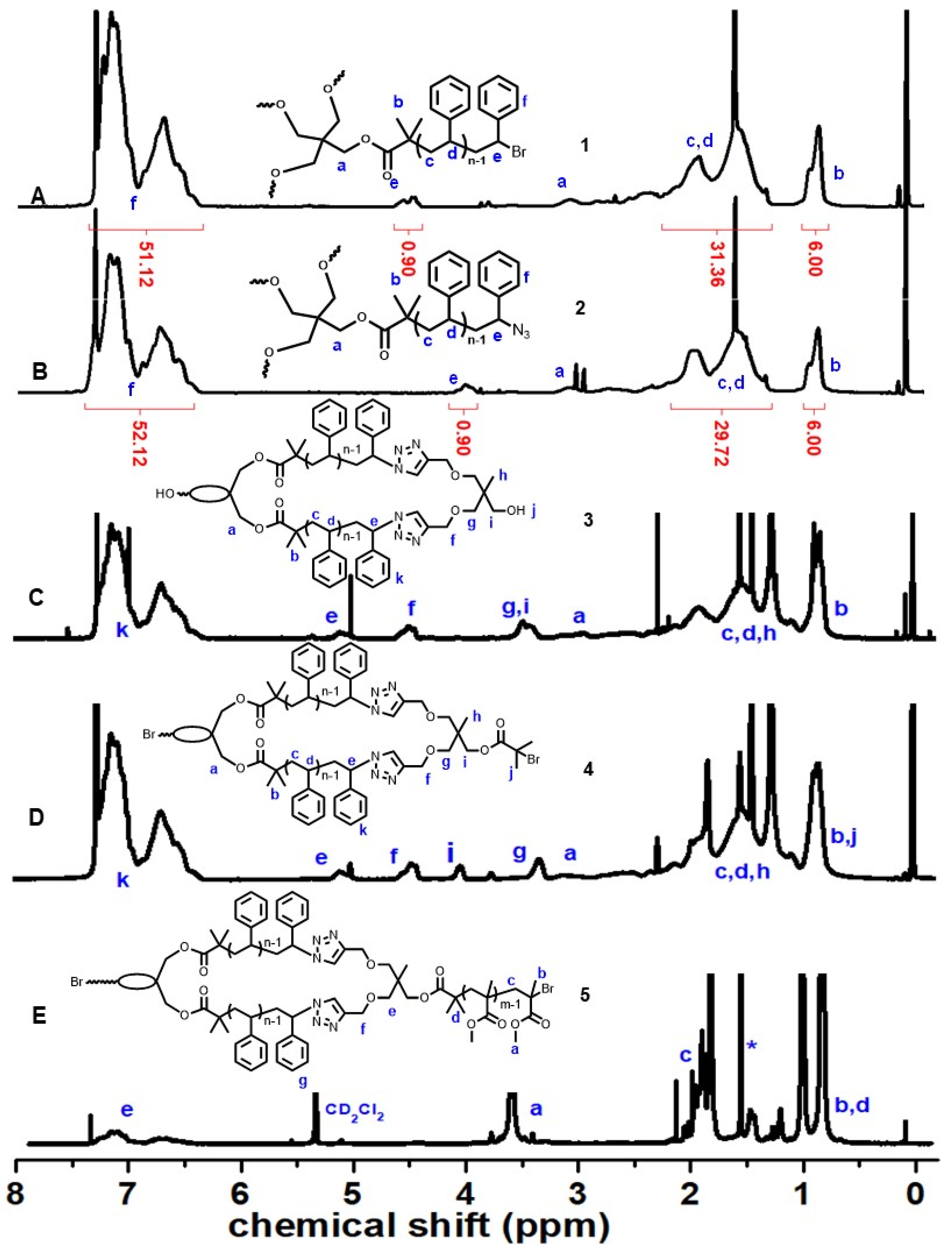
3. Results and Discussion
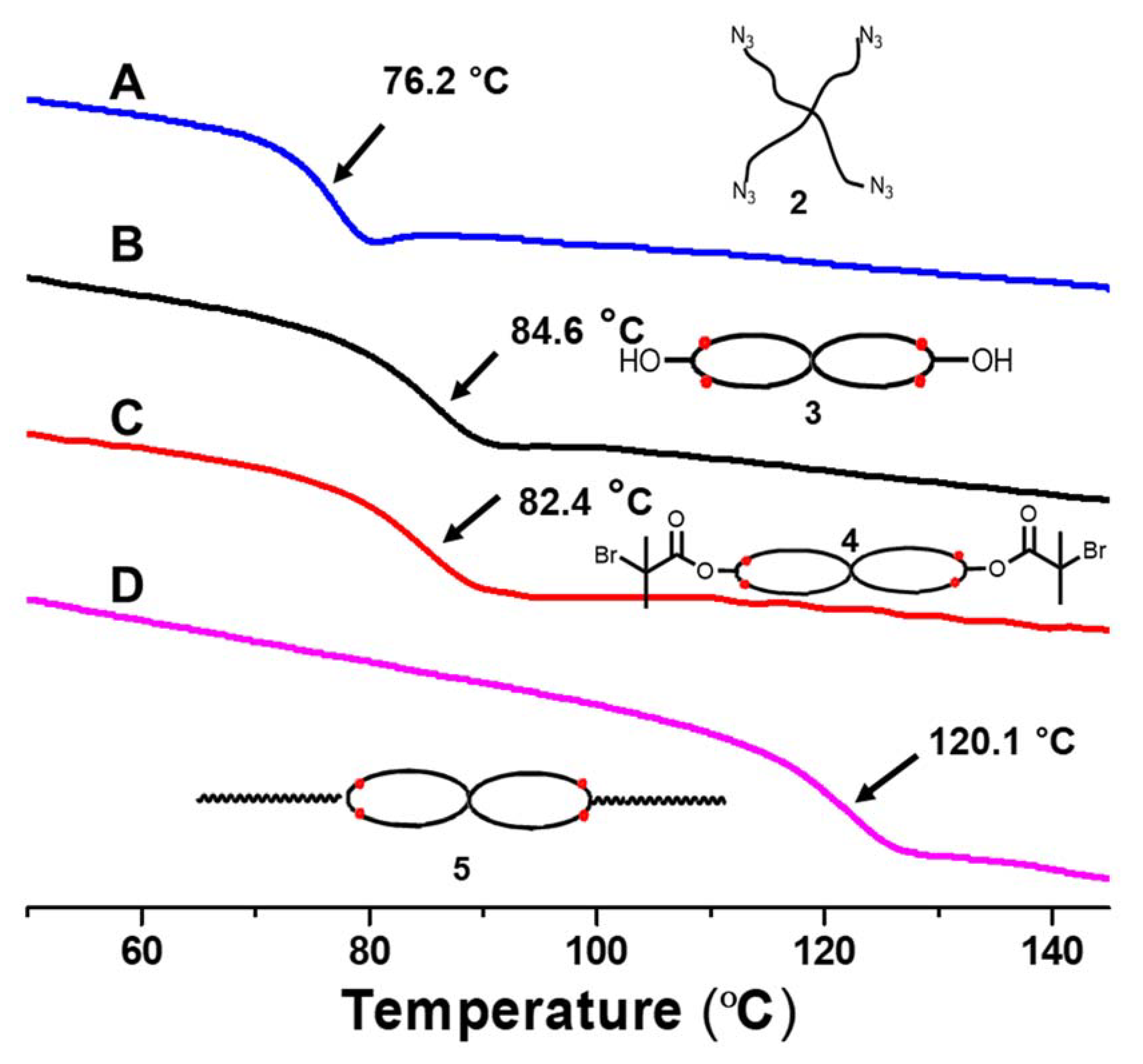
3.1. Tetra-Arm Polystyrene
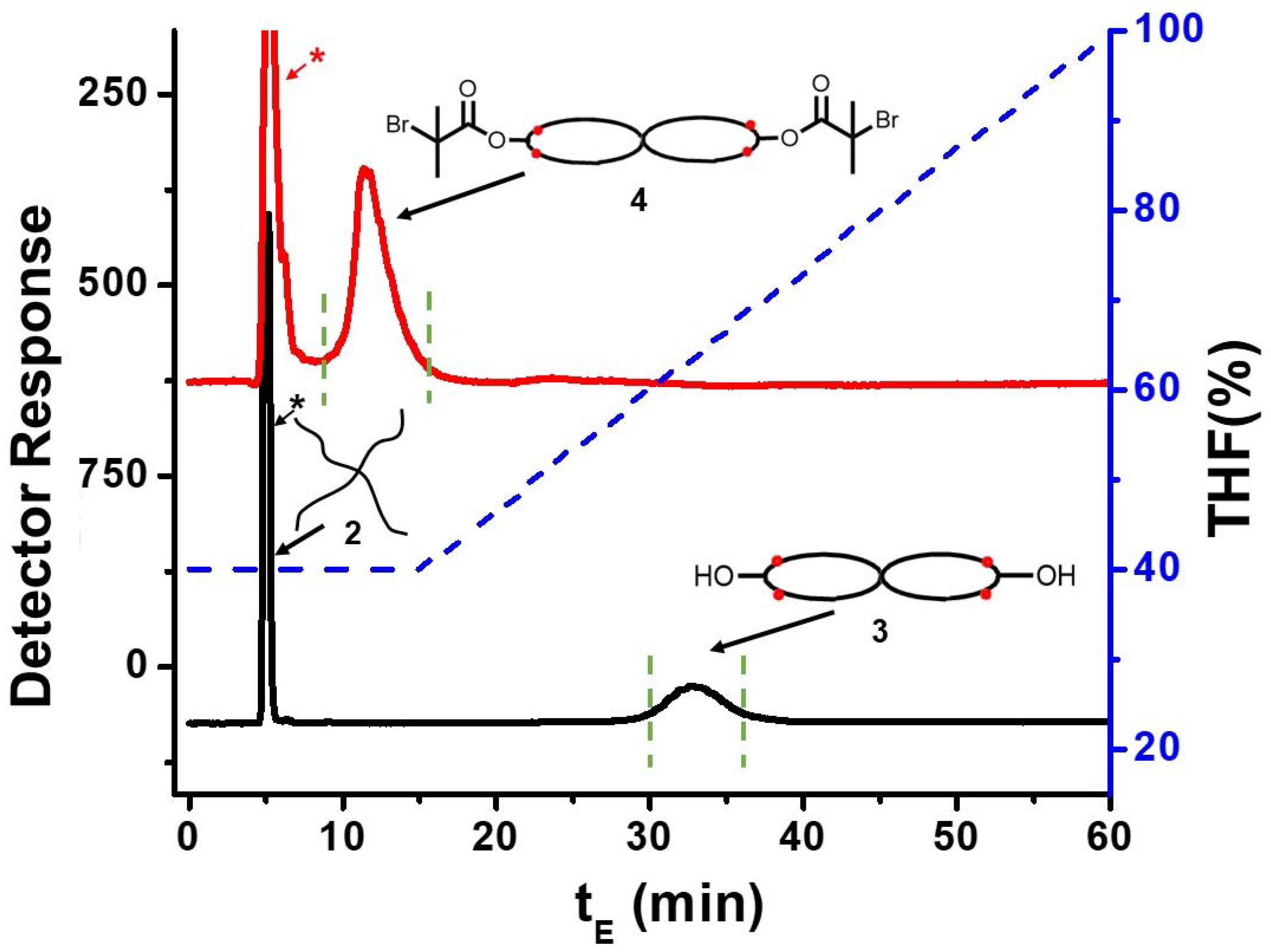
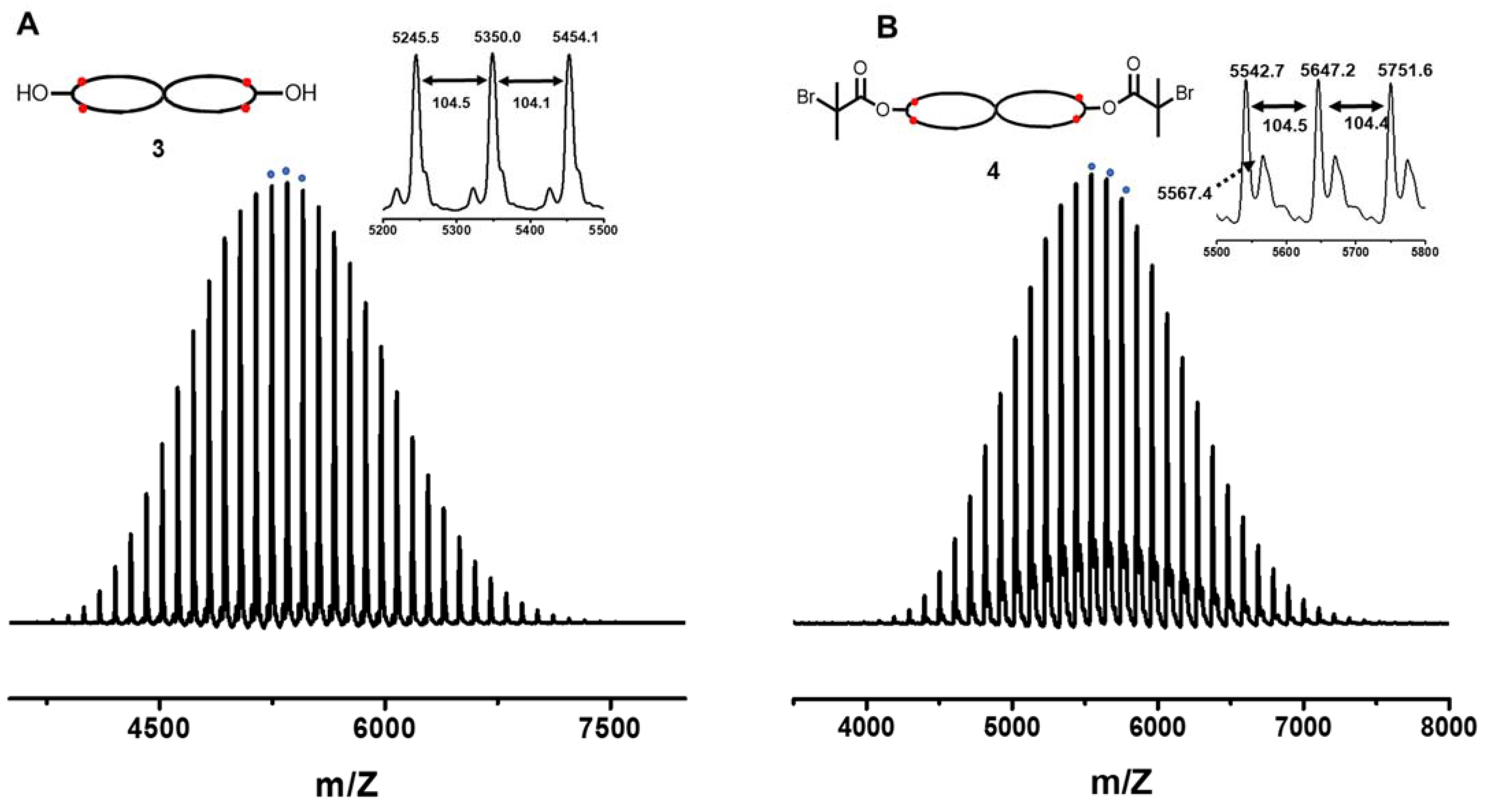
3.2. Spirocyclic PS
3.3. Spirocyclic Macroinitiator
3.4. Preparation of the Triblock Copolymer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, J.; Ye, J.; Liu, S. Synthesis of well-defined cyclic poly (n-isopropylacrylamide) via click chemistry and its unique thermal phase transition behavior. Macromolecules 2007, 40, 9103–9110. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Benitez, D.; Grubbs, R.H. An “endless” route to cyclic polymers. Science 2002, 297, 2041–2044. [Google Scholar] [CrossRef]
- Yan, W.; Divandari, M.; Rosenboom, J.-G.; Ramakrishna, S.N.; Trachsel, L.; Spencer, N.D.; Morgese, G.; Benetti, E.M. Design and characterization of ultrastable, biopassive and lubricious cyclic poly (2-alkyl-2-oxazoline) brushes. Polym. Chem. 2018, 9, 2580–2589. [Google Scholar] [CrossRef]
- Ishizu, K.; Akiyama, Y. Synthesis of cyclic polyethers. Polymer 1997, 38, 491–494. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, N.; Zhang, Z.; Sun, B.; Yang, Y.; Zhu, J.; Zhu, X. Cyclic polymers with pendent carbazole units: Enhanced fluorescence and redox behavior. Angew. Chem. Int. Ed. 2011, 50, 6615–6618. [Google Scholar] [CrossRef]
- Bannister, D.J.; Semlyen, J.A. Studies of cyclic and linear poly(dimethyl siloxanes): 6. Effect of heat. Polymer 1981, 22, 377. [Google Scholar] [CrossRef]
- Kimura, A.; Hasegawa, T.; Yamamoto, T.; Matsumoto, H.; Tezuka, Y. ESA-CF Synthesis of linear and cyclic polymers having densely appended perylene units and topology effects on their thin-film electron mobility. Macromolecules 2016, 49, 5831–5840. [Google Scholar] [CrossRef]
- Hossain, M.D.; Lu, D.; Jia, Z.; Monteiro, M.J. Glass transition temperature of cyclic stars. ACS Macro Lett. 2014, 3, 1254–1257. [Google Scholar] [CrossRef]
- Schappacher, M.; Deffieux, A. Imaging of catenated, figure-of-eight, and trefoil knot polymer rings. Angew. Chem. Int. Ed. 2009, 48, 5930–5933. [Google Scholar] [CrossRef]
- Zhang, S.; Tezuka, Y.; Zhang, Z.; Li, N.; Zhang, W.; Zhu, X. Recent advances in the construction of cyclic grafted polymers and their potential applications. Polym. Chem. 2018, 9, 677–686. [Google Scholar] [CrossRef]
- Jiang, X.; Shi, Y.; Zhu, W.; Chen, Y.; Xi, F. Synthesis of mikto-topology star polymer containing one cyclic arm. J. Polym. Sci. Part A-1 Polym. Chem. 2012, 50, 4239–4245. [Google Scholar] [CrossRef]
- Kang, G.; Liu, Y.; Li, L.; Sun, L.; Ma, W.; Meng, C.; Ma, L.; Zheng, G.; Chang, C.; Wei, H. Modulation of cyclic topology toward enhanced drug delivery, from linear and tadpole-like to dumbbell-shaped copolymers. Chem. Commun. 2020, 56, 3003–3006. [Google Scholar] [CrossRef] [PubMed]
- Liénard, R.; De Winter, J.; Coulembier, O. Cyclic polymers: Advances in their synthesis, properties, and biomedical applications. J. Polym. Sci. 2020, 58, 1481–1502. [Google Scholar] [CrossRef]
- Chen, C.; Weil, T. Cyclic polymers: Synthesis, characteristics, and emerging applications. Nanoscale Horiz. 2022, 7, 1121–1135. [Google Scholar] [CrossRef]
- Golba, B.; Benetti, E.M.; De Geest, B.G. Biomaterials applications of cyclic polymers. Biomaterials 2021, 267, 120468. [Google Scholar] [CrossRef]
- Pang, X.; Wang, G.; Jia, Z.; Liu, C.; Huang, J. Preparation of the amphiphilic macro-rings of poly (ethylene oxide) with multi-polystyrene lateral chains and their extraction for dyes. J. Polym. Sci. Part A-1 Polym. Chem. 2007, 45, 5824–5837. [Google Scholar] [CrossRef]
- Baba, E.; Yatsunami, T.; Yamamoto, T.; Tezuka, Y. A study on emulsion stabilization induced with linear and cyclized polystyrene-poly (ethylene oxide) block copolymer surfactants. Polym. J. 2015, 47, 408–412. [Google Scholar] [CrossRef]
- Poelma, J.E.; Ono, K.; Miyajima, D.; Aida, T.; Satoh, K.; Hawker, C.J. Cyclic block copolymers for controlling feature sizes in block copolymer lithography. ACS Nano 2012, 6, 10845–10854. [Google Scholar] [CrossRef]
- Lonsdale, D.E.; Monteiro, M.J. Synthesis and self-assembly of amphiphilic macrocyclic block copolymer topologies. J. Polym. Sci. Part A-1 Polym. Chem. 2011, 49, 4603–4612. [Google Scholar] [CrossRef]
- Xu, J.; Pu, L.; Ma, J.; Kumar, S.K.; Duan, H. Antibacterial properties of synthesized cyclic and linear cationic copolymers. Polym. Chem. 2020, 11, 6632–6639. [Google Scholar] [CrossRef]
- Haque, F.M.; Grayson, S.M. The synthesis, properties and potential applications of cyclic polymers. Nat. Chem. 2020, 12, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K. Advanced materials by atom transfer radical polymerization. Adv. Mater. Lett. 2018, 30, 1706441. [Google Scholar] [CrossRef]
- Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.J.; Matyjaszewski, K.; Boyer, C. Reversible-deactivation radical polymerization (Controlled/living radical polymerization): From discovery to materials design and applications. Prog. Polym. Sci. 2020, 111, 101311. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(i)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Chang, T. Fractionation of cyclic polystyrene from linear precursor by hplc at the chromatographic critical condition. Macromolecules 2000, 33, 8119–8121. [Google Scholar] [CrossRef]
- Cho, D.; Masuoka, K.; Koguchi, K.; Asari, T.; Kawaguchi, D.; Takano, A.; Matsushita, Y. Preparation and characterization of cyclic polystyrenes. Polym. J. 2005, 37, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Dreier, P.; Ahn, J.; Chang, T.; Frey, H. End group functionality of 95–99%: Epoxide functionalization of polystyryl-lithium evaluated via solvent gradient interaction chromatography. Macromol. Rapid Commun. 2022, 43, 2200560. [Google Scholar] [CrossRef]
- Oh, J.; Kuk, J.; Lee, T.; Ye, J.; Paik, H.J.; Lee, H.W.; Chang, T. Molecular weight distribution of living chains in polystyrene prepared by atom transfer radical polymerization. ACS Macro Lett. 2017, 6, 758–761. [Google Scholar] [CrossRef]
- Gao, L.; Oh, J.; Tu, Y.; Chang, T.; Li, C.Y. Glass transition temperature of cyclic polystyrene and the linear counterpart contamination effect. Polymer 2019, 170, 198–203. [Google Scholar] [CrossRef]
- Doi, Y.; Iwasa, Y.; Watanabe, K.; Nakamura, M.; Takano, A.; Takahashi, Y.; Matsushita, Y. Synthesis and characterization of comb-shaped ring polystyrenes. Macromolecules 2016, 49, 3109–3115. [Google Scholar] [CrossRef]
- Yokochi, H.; Ohira, M.; Oka, M.; Honda, S.; Li, X.; Aoki, D.; Otsuka, H. Topology transformation toward cyclic, figure-eight-shaped, and cross-linked polymers based on the dynamic behavior of a bis (hindered amino) disulfide linker. Macromolecules 2021, 54, 9992–10000. [Google Scholar] [CrossRef]
- Fan, X.; Huang, B.; Wang, G.; Huang, J. Synthesis of amphiphilic heteroeight-shaped polymer cyclic-[poly (ethylene oxide)-b-polystyrene]2 via “click” chemistry. Macromolecules 2012, 45, 3779–3786. [Google Scholar] [CrossRef]
- Lonsdale, D.E.; Bell, C.A.; Monteiro, M.J. Strategy for rapid and high-purity monocyclic polymers by CuAAC “click” reactions. Macromolecules 2010, 43, 3331–3339. [Google Scholar] [CrossRef]
- McKeown, G.R.; Fang, Y.; Obhi, N.K.; Manion, J.G.; Perepichka, D.F.; Seferos, D.S. Synthesis of macrocyclic poly (3-hexylthiophene) and poly (3-heptylselenophene) by alkyne homocoupling. ACS Macro Lett. 2016, 5, 1075–1079. [Google Scholar] [CrossRef] [Green Version]
- Chang, T. Structural characterization of ring polystyrene by liquid chromatography at the critical condition and MALDI-TOF mass spectrometry. Macromolecules 2001, 34, 7570–7572. [Google Scholar] [CrossRef]
- Braun, D. Functionality type analysis of carboxy-terminated oligostyrenes by gradient HPLC. Macromol. Chem. Phys. 1997, 198, 3365–3367. [Google Scholar] [CrossRef]
- Gao, H.; Siegwart, D.J.; Jahed, N.; Sarbu, T.; Matyjaszewski, K. Characterization of α,ω-dihydroxypolystyrene by gradient polymer elution chromatography and two-dimensional liquid chromatography. Des. Monomers Polym. 2012, 8, 533–546. [Google Scholar] [CrossRef]
- Challal, S.; Queiroz, E.F.; Debrus, B.; Kloeti, W.; Guillarme, D.; Gupta, M.P.; Wolfender, J.L. Rational and efficient preparative isolation of natural products by MPLC-UV-elsd based on HPLC to MPLC gradient transfer. Planta Med. 2015, 81, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.D.; Jia, Z.; Monteiro, M.J. Complex polymer topologies built from tailored multifunctional cyclic polymers. Macromolecules 2014, 47, 4955–4970. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Y.; Muhammad, S.; Jia, Q.; Ding, Y.; Chen, Y. A facile end-functionalization of polystyrene by ATRP and click chemistry: Chain end effect on the glass transition temperature. React. Funct. Polym. 2020, 151, 104566. [Google Scholar] [CrossRef]
- Lee, J.; Lee, K.; Park, J.; Kim, J.K.; Chang, T. Characterization and fractionation of PS-b-PMMA diblock copolymer synthesized via click chemistry. Polymer 2015, 80, 46–51. [Google Scholar] [CrossRef]
- Park, S.; Park, I.; Chang, T.; Ryu, C.Y. Interaction-controlled HPLC for block copolymer analysis and separation. J. Am. Chem. Soc. 2004, 126, 8906–8907. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.R.; Kirkland, J.J.; Glajch, J.L. Practical HPLC Method Development, 2nd ed.; A Wiley-Interscience: Toronto, Canada, 1997; pp. 233–236. [Google Scholar]
- Johnson, R.M.; Fraser, C.L. Iron tris (bipyridine)-centered star block copolymers: Chelation of triblock macroligands generated by ROP and ATRP. Macromolecules 2004, 37, 2718–2727. [Google Scholar] [CrossRef]
- Qin, S.-H.; Qiu, K.-Y. A facile method for synthesis of ABA triblock copolymers with macro-iniferter technique. Polymer 2001, 42, 3033–3042. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aswale, S.; Kim, M.; Kim, D.; Mohanty, A.K.; Jeon, H.B.; Cho, H.Y.; Paik, H.-j. Synthesis and Characterization of Spirocyclic Mid-Block Containing Triblock Copolymer. Polymers 2023, 15, 1677. https://doi.org/10.3390/polym15071677
Aswale S, Kim M, Kim D, Mohanty AK, Jeon HB, Cho HY, Paik H-j. Synthesis and Characterization of Spirocyclic Mid-Block Containing Triblock Copolymer. Polymers. 2023; 15(7):1677. https://doi.org/10.3390/polym15071677
Chicago/Turabian StyleAswale, Suraj, Minji Kim, Dongwoo Kim, Aruna Kumar Mohanty, Heung Bae Jeon, Hong Y. Cho, and Hyun-jong Paik. 2023. "Synthesis and Characterization of Spirocyclic Mid-Block Containing Triblock Copolymer" Polymers 15, no. 7: 1677. https://doi.org/10.3390/polym15071677