
Analysis of Elongational Viscosity of Entangled Poly (Propylene Carbonate) Melts by Primitive Chain Network Simulations


Abstract
:1. Introduction
2. Model and Simulations
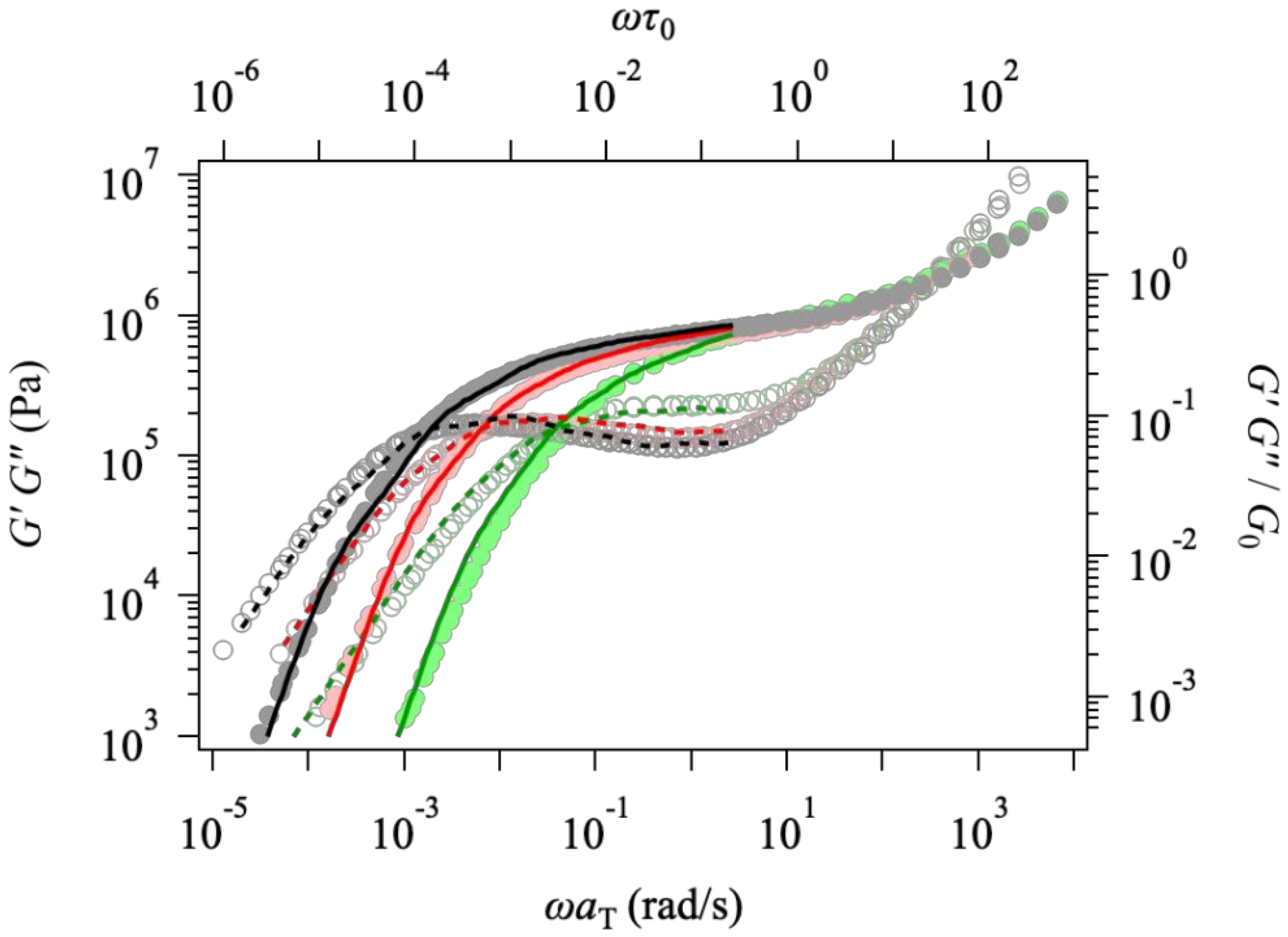
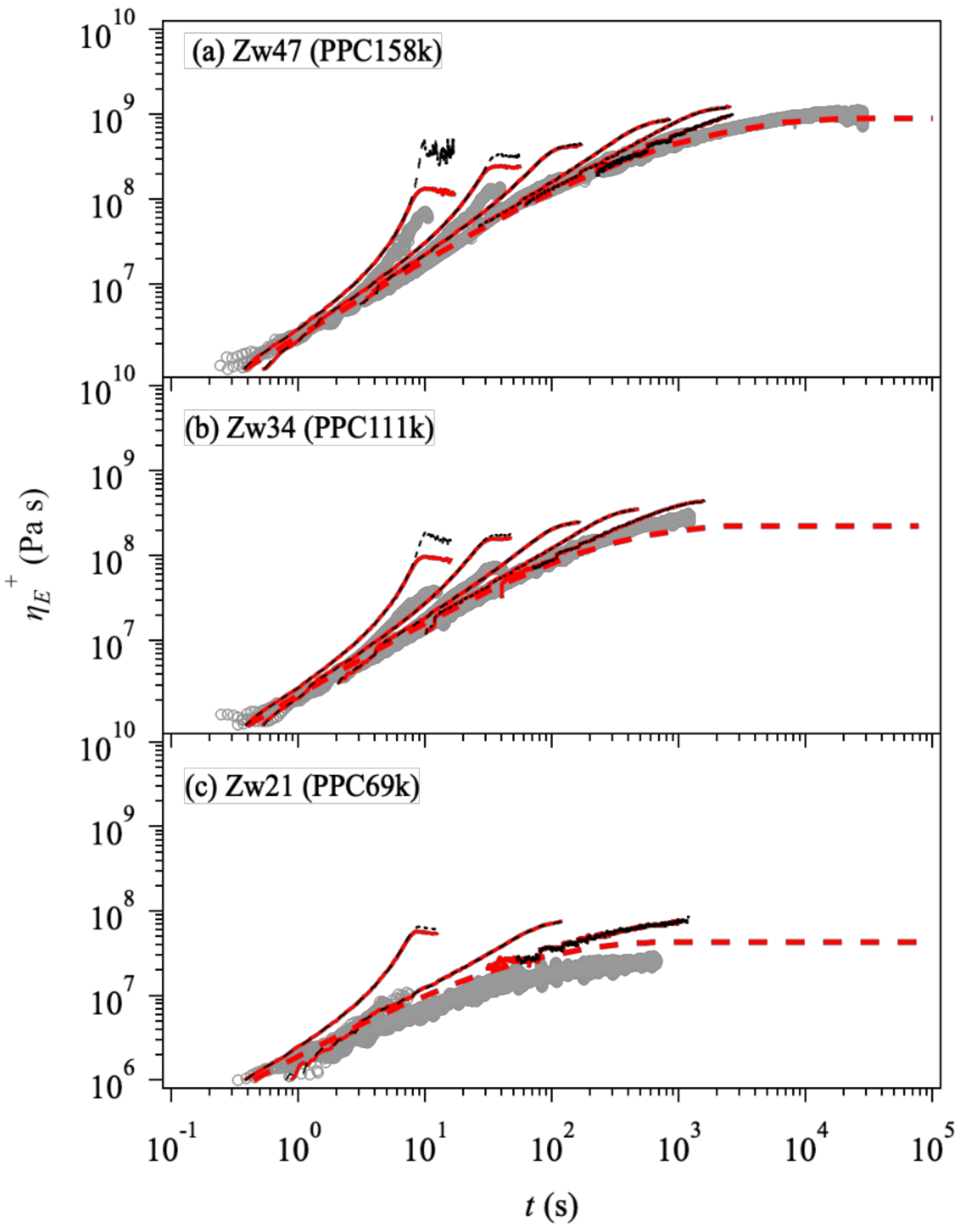
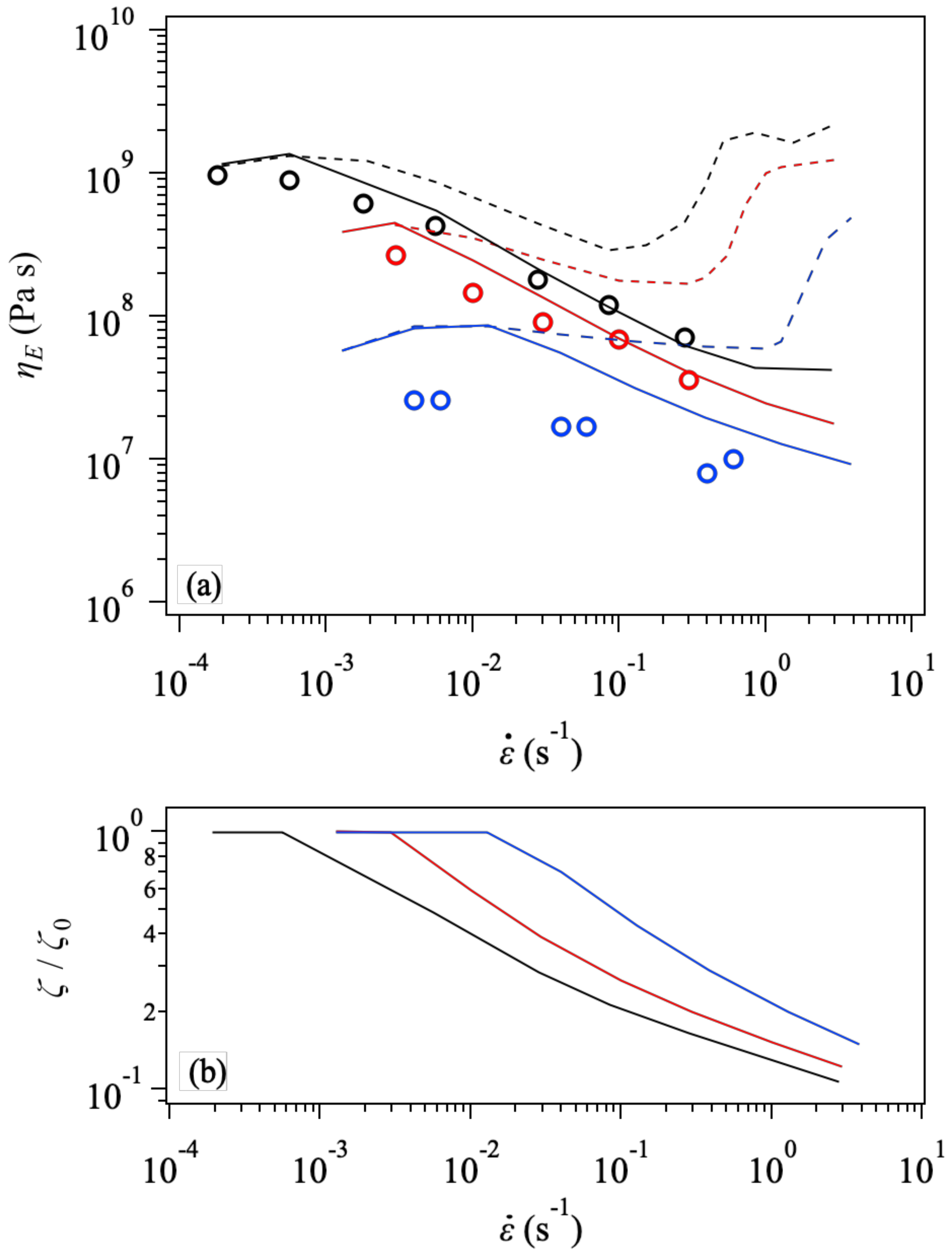
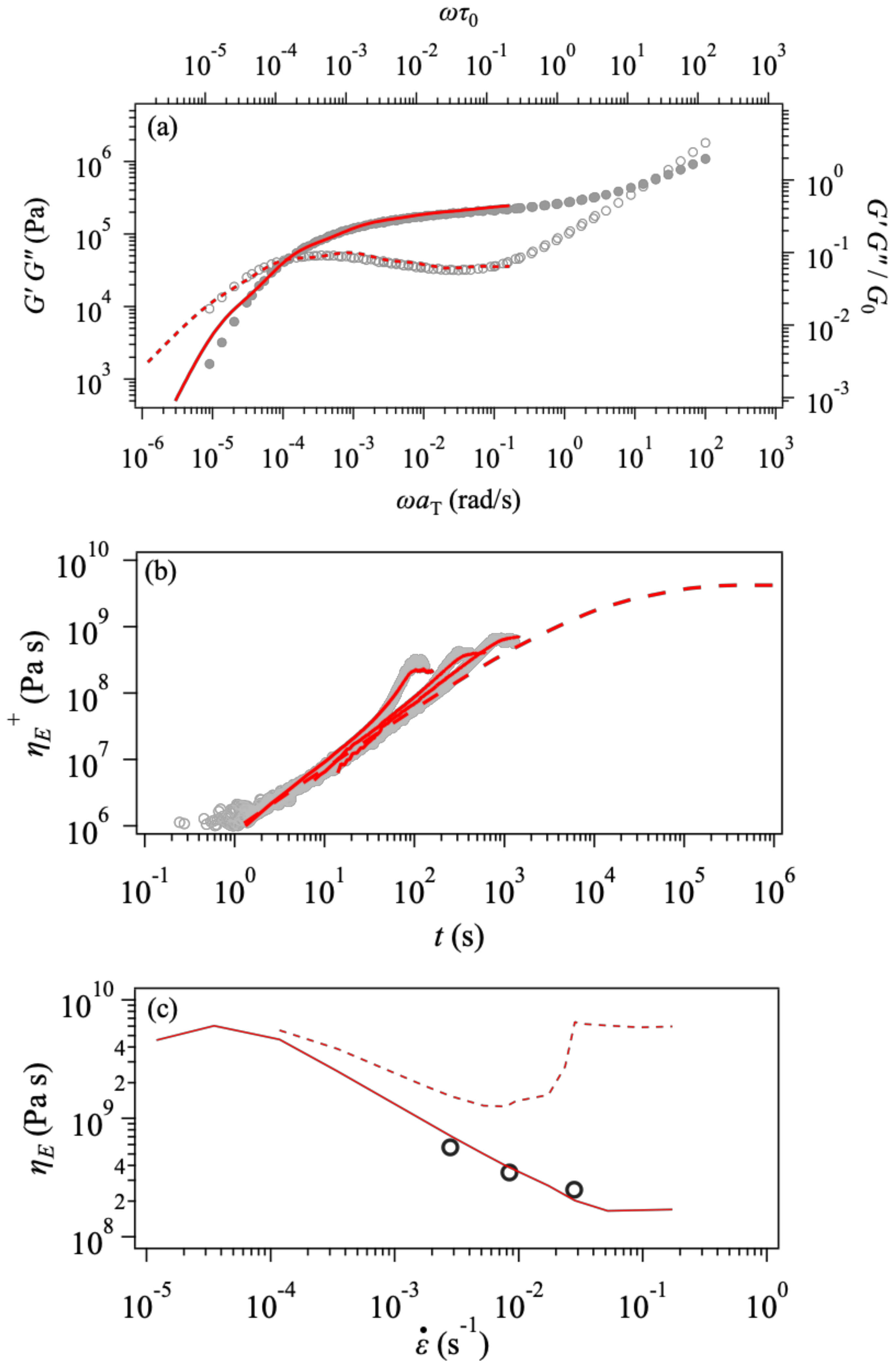
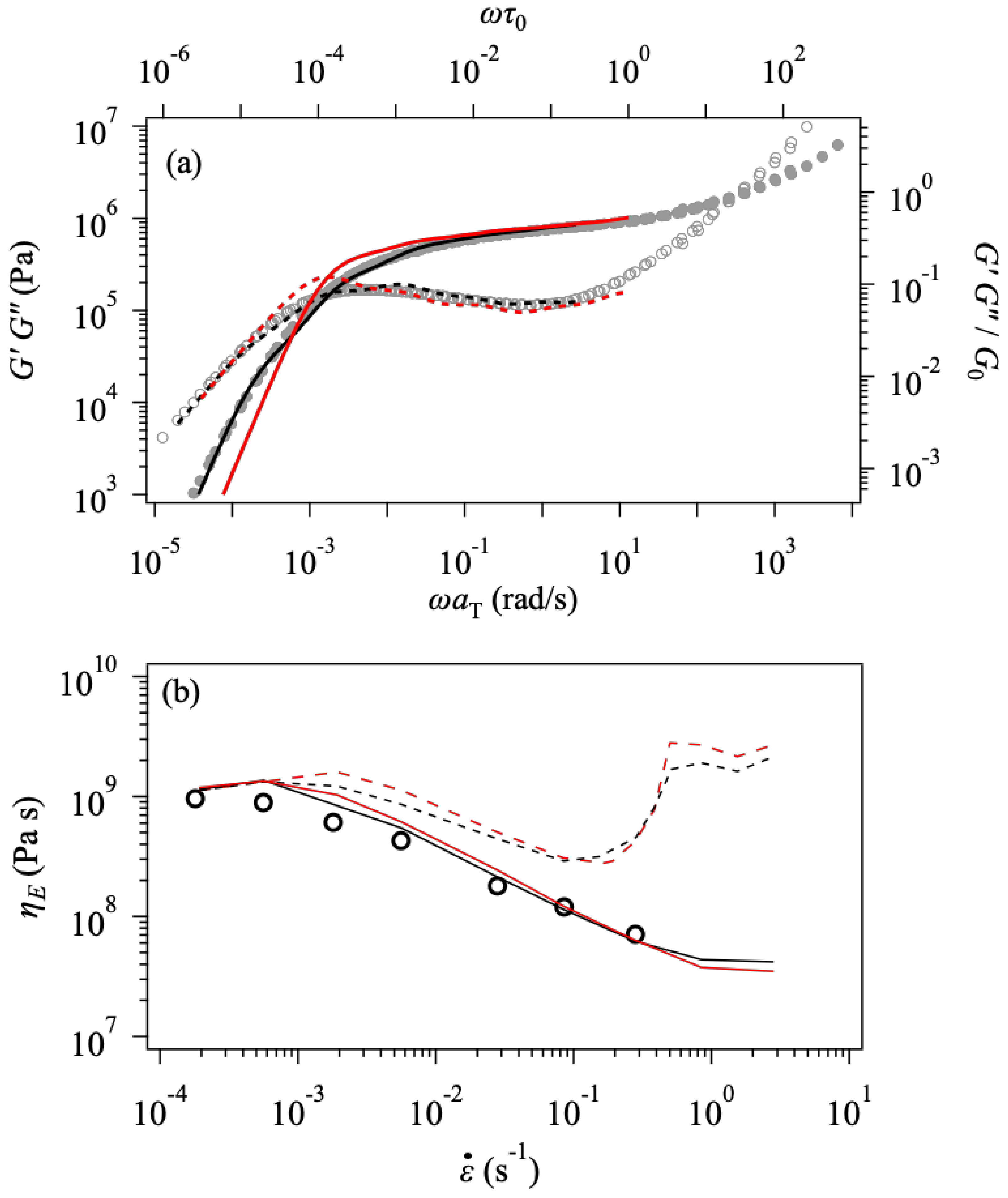
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ianniruberto, G.; Marrucci, G.; Masubuchi, Y. Melts of Linear Polymers in Fast Flows. Macromolecules 2020, 53, 5023–5033. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1986. [Google Scholar]
- Marrucci, G.; Grizzuti, N. Fast flows of concentrated polymers: Predictions of the tube model on chain stretching. Gazz. Chmica Ital. 1988, 118, 179–185. [Google Scholar]
- Bhattacharjee, P.K.; Nguyen, D.A.; McKinley, G.H.; Sridhar, T. Extensional stress growth and stress relaxation in entangled polymer solutions. J. Rheol. 2003, 47, 269. [Google Scholar] [CrossRef]
- Bhattacharjee, P.K.; Oberhauser, J.P.; McKinley, G.H.; Leal, L.G.; Sridhar, T. Extensional rheometry of entangled solutions. Macromolecules 2002, 35, 10131–10148. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Hengeller, L.; Alvarez, N.J.; Hassager, O. Bridging the Gap between Polymer Melts and Solutions in Extensional Rheology. Macromolecules 2015, 48, 4158–4163. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Mednova, O.; Rasmussen, H.K.; Alvarez, N.J.; Skov, A.L.; Almdal, K.; Hassager, O. Concentrated polymer solutions are different from melts: Role of entanglement molecular weight. Macromolecules 2013, 46, 5026–5035. [Google Scholar] [CrossRef]
- Sridhar, T.; Acharya, M.; Nguyen, D.A.; Bhattacharjee, P.K. On the Extensional Rheology of Polymer Melts and Concentrated Solutions. Macromolecules 2014, 47, 379–386. [Google Scholar] [CrossRef]
- Bach, A.; Almdal, K.; Rasmussen, H.K.; Hassager, O. Elongational viscosity of narrow molar mass distribution polystyrene. Macromolecules 2003, 36, 5174–5179. [Google Scholar] [CrossRef]
- Nielsen, J.K.; Rasmussen, H.K.; Hassager, O.; McKinley, G.H. Elongational viscosity of monodisperse and bidisperse polystyrene melts. J. Rheol. 2006, 50, 453–476. [Google Scholar] [CrossRef] [Green Version]
- Wingstrand, S.L.; Alvarez, N.J.; Huang, Q.; Hassager, O. Linear and Nonlinear Universality in the Rheology of Polymer Melts and Solutions. Phys. Rev. Lett. 2015, 115, 1–5. [Google Scholar] [CrossRef]
- Wagner, M.H.; Kheirandish, S.; Koyama, K.; Nishioka, A.; Minegishi, A.; Takahashi, T. Modeling strain hardening of polydisperse polystyrene melts by molecular stress function theory. Rheol. Acta 2005, 44, 235–243. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Kröger, M. From intermediate anisotropic to isotropic friction at large strain rates to account for viscosity thickening in polymer solutions. J. Chem. Phys. 2018, 148, 184903. [Google Scholar] [CrossRef]
- Ianniruberto, G.; Brasiello, A.; Marrucci, G. Simulations of fast shear flows of PS oligomers confirm monomeric friction reduction in fast elongational flows of monodisperse PS melts as indicated by rheooptical data. Macromolecules 2012, 45, 8058–8066. [Google Scholar] [CrossRef]
- Yaoita, T.; Isaki, T.; Masubuchi, Y.; Watanabe, H.; Ianniruberto, G.; Marrucci, G. Primitive chain network simulation of elongational flows of entangled linear chains: Stretch/orientation-induced reduction of monomeric friction. Macromolecules 2012, 45, 2773–2782. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Matsumiya, Y.; Watanabe, H. Test of Orientation/Stretch-Induced Reduction of Friction via Primitive Chain Network Simulations for Polystyrene, Polyisoprene, and Poly(n-butyl acrylate). Macromolecules 2014, 47, 6768–6775. [Google Scholar] [CrossRef]
- Takeda, K.; Sukumaran, S.K.; Sugimoto, M.; Koyama, K.; Masubuchi, Y. Primitive chain network simulations for elongational viscosity of bidisperse polystyrene melts. Adv. Modeling Simul. Eng. Sci. 2015, 2, 11. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Matsumiya, Y.; Watanabe, H.; Marrucci, G.; Ianniruberto, G. Primitive chain network simulations for Pom-Pom polymers in uniaxial elongational flows. Macromolecules 2014, 47, 3511–3519. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Ianniruberto, G.; Marrucci, G. Primitive Chain Network Simulations of Entangled Melts of Symmetric and Asymmetric Star Polymers in Uniaxial Elongational Flows. J. Soc. Rheol. Jpn. 2021, 3, 171–178. [Google Scholar] [CrossRef]
- Yaoita, T.; Masubuchi, Y.; Watanabe, H. Concept of Stretch/Orientation-Induced Friction Reduction Tested with a Simple Molecular Constitutive Equation. Nihon Reoroji Gakkaishi 2014, 42, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.S.; Larson, R.G. Constitutive model that shows extension thickening for entangled solutions and extension thinning for melts. J. Rheol. 2014, 58, 255–279. [Google Scholar] [CrossRef]
- Ianniruberto, G. Extensional Flows of Solutions of Entangled Polymers Confirm Reduction of Friction Coefficient. Macromolecules 2015, 48, 6306–6312. [Google Scholar] [CrossRef]
- Costanzo, S.; Huang, Q.; Ianniruberto, G.; Marrucci, G.; Hassager, O.; Vlassopoulos, D. Shear and Extensional Rheology of Polystyrene Melts and Solutions with the Same Number of Entanglements. Macromolecules 2016, 49, 3925–3935. [Google Scholar] [CrossRef] [Green Version]
- Park, G.W.; Ianniruberto, G. Flow-Induced Nematic Interaction and Friction Reduction Successfully Describe PS Melt and Solution Data in Extension Startup and Relaxation. Macromolecules 2017, 50, 4787–4796. [Google Scholar] [CrossRef]
- Matsumiya, Y.; Watanabe, H.; Masubuchi, Y.; Huang, Q.; Hassager, O. Nonlinear Elongational Rheology of Unentangled Polystyrene and Poly(p-tert-butylstyrene) Melts. Macromolecules 2018, 51, 9710–9729. [Google Scholar] [CrossRef] [Green Version]
- Morelly, S.L.; Palmese, L.; Watanabe, H.; Alvarez, N.J. Effect of Finite Extensibility on Nonlinear Extensional Rheology of Polymer Melts. Macromolecules 2019, 52, 915–922. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Yaoita, T.; Matsumiya, Y.; Watanabe, H.; Ianniruberto, G.; Marrucci, G. Stretch/orientation Induced Acceleration in Stress Relaxation in Coarse-grained Molecular Dynamics Simulations. Nihon Reoroji Gakkaishi 2013, 41, 35–37. [Google Scholar] [CrossRef] [Green Version]
- Ianniruberto, G.; Marrucci, G. Molecular Dynamics Reveals a Dramatic Drop of the Friction Coefficient in Fast Flows of Polymer Melts. Macromolecules 2020, 53, 2627–2633. [Google Scholar] [CrossRef]
- O’connor, T.C.; Hopkins, A.; Robbins, M.O. Stress Relaxation in Highly Oriented Melts of Entangled Polymers. Macromolecules 2019, 52, 8540–8550. [Google Scholar] [CrossRef]
- Kida, T.; Doi, Y.; Tanaka, R.; Uneyama, T.; Shiono, T.; Masubuchi, Y. Rheological properties of linear and short-chain branched polyethylene with nearly monodispersed molecular weight distribution. Rheol. Acta 2021, 60, 511–519. [Google Scholar] [CrossRef]
- Yang, L.; Uneyama, T.; Masubuchi, Y.; Doi, Y. Linear Rheological Properties of Poly (Propylene Carbonate) with Different Molecular Weights. Nihon Reoroji Gakkaishi 2021, 49, 267–274. [Google Scholar] [CrossRef]
- Yang, L.; Uneyama, T.; Masubuchi, Y.; Doi, Y. Nonlinear Shear and Elongational Rheology of Poly(Propylene Carbonate). Nihon Reoroji Gakkaishi 2022, 50, 127–135. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Takimoto, J.-I.; Koyama, K.; Ianniruberto, G.; Marrucci, G.; Greco, F. Brownian simulations of a network of reptating primitive chains. J. Chem. Phys. 2001, 115, 4387–4394. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Ianniruberto, G.; Greco, F.; Marrucci, G. Entanglement molecular weight and frequency response of sliplink networks. J. Chem. Phys. 2003, 119, 6925–6930. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Ianniruberto, G.; Greco, F.; Marrucci, G. Molecular simulations of the long-time behaviour of entangled polymeric liquids by the primitive chain network model. Model. Simul. Mater. Sci. Eng. 2004, 12, S91–S100. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Ianniruberto, G.; Greco, F.; Marrucci, G. Quantitative comparison of primitive chain network simulations with literature data of linear viscoelasticity for polymer melts. J. Non-Newton. Fluid Mech. 2008, 149, 87–92. [Google Scholar] [CrossRef]
- Masubuchi, Y. PASTA and NAPLES: Rheology Simulator. In Computer Simulation of Polymeric Materials; Springer: Singapore, 2016; pp. 101–127. ISBN 9789811008153. [Google Scholar]
- Yaoita, T.; Isaki, T.; Masubuchi, Y.; Watanabe, H.; Ianniruberto, G.; Marrucci, G. Primitive chain network simulation of elongational flows of entangled linear chains: Role of finite chain extensibility. Macromolecules 2011, 44, 9675–9682. [Google Scholar] [CrossRef]
- Takeda, K.; Sukumaran, S.K.S.K.; Sugimoto, M.; Koyama, K.; Masubuchi, Y. Test of the Stretch/Orientation-Induced Reduction of Friction for Biaxial Elongational Flow via Primitive Chain Network Simulation. Nihon Reoroji Gakkaishi 2015, 43, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, P.K.; Nguyen, D.A.; Masubuchi, Y.; Sridhar, T. Extensional Step Strain Rate Experiments on an Entangled Polymer Solution. Macromolecules 2017, 50, 386–395. [Google Scholar] [CrossRef]
- Masubuchi, Y. Contraction of Entangled Polymers After Large Step Shear Deformations in Slip-Link Simulations. Polymers 2019, 11, 370. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Masubuchi, Y.; Sugimoto, M.; Koyama, K.; Sukumaran, S.K. Simulations of Startup Planar Elongation of an Entangled Polymer Melt. Nihon Reoroji Gakkaishi 2020, 48, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N. Atomistic molecular dynamics simulation of stress relaxation upon cessation of steady-state uniaxial elongational flow. Macromolecules 2000, 33, 8062–8076. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Mavrantzas, V.G. Quantitative predictions of the linear viscoelastic properties of entangled polyethylene and polybutadiene melts via modified versions of modern tube models on the basis of atomistic simulation data. J. Non-Newton. Fluid Mech. 2013, 200, 111–130. [Google Scholar] [CrossRef]
- Behbahani, A.F.; Schneider, L.; Rissanou, A.; Chazirakis, A.; Bačová, P.; Jana, P.K.; Li, W.; Doxastakis, M.; Polińska, P.; Burkhart, C.; et al. Dynamics and Rheology of Polymer Melts via Hierarchical Atomistic, Coarse-Grained, and Slip-Spring Simulations. Macromolecules 2021, 54, 2740–2762. [Google Scholar] [CrossRef]
- Spyriouni, T.; Tzoumanekas, C.; Theodorou, D.; Müller-Plathe, F.; Milano, G. Coarse-Grained and Reverse-Mapped United-Atom Simulations of Long-Chain Atactic Polystyrene Melts: Structure, Thermodynamic Properties, Chain Conformation, and Entanglements. Macromolecules 2007, 40, 3876–3885. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Uneyama, T.; Watanabe, H.; Ianniruberto, G.; Greco, F.; Marrucci, G. Structure of entangled polymer network from primitive chain network simulations. J. Chem. Phys. 2010, 132, 134902. [Google Scholar] [CrossRef] [Green Version]
- Masubuchi, Y.; Kida, T.; Doi, Y.; Uneyama, T. Radial Distribution Functions of Entanglements in Primitive Chain Network Simulations. Nihon Reoroji Gakkaishi 2021, 49, 337–345. [Google Scholar] [CrossRef]
- Masubuchi, Y. Simulating the Flow of Entangled Polymers. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 11–33. [Google Scholar] [CrossRef]
- Masubuchi, Y. Molecular modeling for polymer rheology. In Reference Module in Materials Science and Materials Engineering; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–7. ISBN 9780128035818. [Google Scholar]
- Masubuchi, Y.; Doi, Y.; Uneyama, T. Entanglement Molecular Weight. Nihon Reoroji Gakkaishi 2020, 48, 177–183. [Google Scholar] [CrossRef]
- Uneyama, T.; Masubuchi, Y. Detailed balance condition and effective free energy in the primitive chain network model. J. Chem. Phys. 2011, 135, 184904. [Google Scholar] [CrossRef]
- Uneyama, T.; Masubuchi, Y. Plateau Moduli of Several Single-Chain Slip-Link and Slip-Spring Models. Macromolecules 2021, 54, 1338–1353. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Uneyama, T. Comparison among multi-chain models for entangled polymer dynamics. Soft Matter 2018, 14, 5986–5994. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Watanabe, H.; Ianniruberto, G.; Greco, F.; Marrucci, G. Comparison among Slip-Link Simulations of Bidisperse Linear Polymer Melts. Macromolecules 2008, 41, 8275–8280. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Yaoita, T.; Matsumiya, Y.; Watanabe, H. Primitive chain network simulations for asymmetric star polymers. J. Chem. Phys. 2011, 134, 194905. [Google Scholar] [CrossRef] [Green Version]
- Masubuchi, Y.; Matsumiya, Y.; Watanabe, H.; Shiromoto, S.; Tsutsubuchi, M.; Togawa, Y. Primitive chain network simulations for comb-branched polymer under step shear deformations. Rheol. Acta 2012, 51, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Masubuchi, Y. Multichain Slip-Spring Simulations for Branch Polymers. Macromolecules 2018, 51, 10184–10193. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Ianniruberto, G.; Marrucci, G. Primitive chain network simulations for H-polymers under fast shear. Soft Matter 2020, 16, 1056–1065. [Google Scholar] [CrossRef]
- Furuichi, K.; Nonomura, C.; Masubuchi, Y.; Watanabe, H.; Ianniruberto, G.; Greco, F.; Marrucci, G. Entangled polymer orientation and stretch under large step shear deformations in primitive chain network simulations. Rheol. Acta 2008, 47, 591–599. [Google Scholar] [CrossRef]
- Furuichi, K.; Nonomura, C.; Masubuchi, Y.; Ianniruberto, G.; Greco, F.; Marrucci, G. Primitive Chain Network Simulations of Damping Functions for Shear, Uniaxial, Biaxial and Planar Deformations. Nihon Reoroji Gakkaishi 2007, 35, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Furuichi, K.; Nonomura, C.; Masubuchi, Y.; Watanabe, H. Chain contraction and nonlinear stress damping in primitive chain network simulations. J. Chem. Phys. 2010, 133, 174902. [Google Scholar] [CrossRef] [Green Version]
- Masubuchi, Y.; Watanabe, H. Origin of stress overshoot under start-up shear in primitive chain network simulation. ACS Macro Lett. 2014, 3, 1183–1186. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Ianniruberto, G.; Marrucci, G. Stress Undershoot of Entangled Polymers under Fast Startup Shear Flows in Primitive Chain Network Simulations. Nihon Reoroji Gakkaishi 2018, 46, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Peterlin, A. Gradient Dependence of Intrinsic Viscosity of Freely Flexible Linear Macromolecules. J. Chem. Phys. 1960, 33, 1799. [Google Scholar] [CrossRef]
- Boudara, V.A.H.; Read, D.J.; Ramírez, J. Reptate rheology software: Toolkit for the analysis of theories and experiments. J. Rheol. 2020, 64, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Yang, L. Linear and Nonlinear Rheological Properties of Poly(Propylene Carbonate); Nagoya University: Nagoya, Japan, 2022. [Google Scholar]
- Wagner, M.H.; Rolon-Garrido, V.H. Nonlinear rheology of linear polymer melts: Modeling chain stretch by interchain tube pressure and Rouse time. Korea-Aust. Rheol. J. 2009, 21, 203–211. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | ||||||
|---|---|---|---|---|---|---|
| Zw47 (PPC158k *) | 11 | 0.09 | 47.3 | 1.31 | 158 | 1.30 |
| 22 | 0.41 | |||||
| 44 | 0.41 | |||||
| 88 | 0.09 | |||||
| Zw34 (PPC111k *) | 8 | 0.09 | 33.7 | 1.30 | 111 | 1.30 |
| 16 | 0.41 | |||||
| 32 | 0.41 | |||||
| 62 | 0.09 | |||||
| Zw21 (PPC69k *) | 4 | 0.1 | 20.6 | 1.41 | 68.8 | 1.43 |
| 8 | 0.4 | |||||
| 18 | 0.4 | |||||
| 38 | 0.1 | |||||
| Z47 | 47 | 1 | 47 | 1 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masubuchi, Y.; Yang, L.; Uneyama, T.; Doi, Y. Analysis of Elongational Viscosity of Entangled Poly (Propylene Carbonate) Melts by Primitive Chain Network Simulations. Polymers 2022, 14, 741. https://doi.org/10.3390/polym14040741
Masubuchi Y, Yang L, Uneyama T, Doi Y. Analysis of Elongational Viscosity of Entangled Poly (Propylene Carbonate) Melts by Primitive Chain Network Simulations. Polymers. 2022; 14(4):741. https://doi.org/10.3390/polym14040741
Chicago/Turabian StyleMasubuchi, Yuichi, Lixin Yang, Takashi Uneyama, and Yuya Doi. 2022. "Analysis of Elongational Viscosity of Entangled Poly (Propylene Carbonate) Melts by Primitive Chain Network Simulations" Polymers 14, no. 4: 741. https://doi.org/10.3390/polym14040741