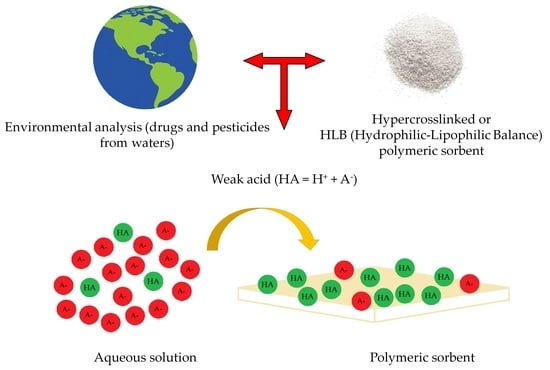
In the last decades, several new polymeric sorbents have been invented, which can bind strongly to almost any weakly hydrophobic compound. Two important groups of such polymers are the hypercrosslinked polymers [
7] and the hydrophilic–lipophilic balanced (HLB) polymers [
1]. These polymers share the strong binding property with activated carbon and other carbon materials, but in contrast, binding on the novel polymers is more reversible, and the bound substances may be eluted under mild conditions. This makes the novel polymer sorbents suitable for many practical applications. These include analytical sample preconcentration, even at extremely low analyte concentrations, and the passive sampling of environmental and other waters. With more recent and very expensive equipment, one may measure concentrations in this very low range without preconcentration [
8], but this does not make preconcentration generally superfluous, not even with MS detection [
9]. There are also many other possible applications beyond analytical chemistry. Some of these, including the preparation of nanocomposites, have been reported by the Davankov group [
10]. The possibilities may be further extended by the invention of a simple, versatile, fast, and green synthesis method involving hypercrosslinked polymers [
11].
It is interesting to note that HLB polymeric sorbents were developed in the first place to make them directly water-wettable, without prewetting with an organic solvent [Waters™ Corporation, Milford, MA]. The OASIS® HLB sorbent, analyzed in the present work, is not a hypercrosslinked polymer, yet it is also highly adsorptive, well beyond the level that might be expected merely on the basis of its 800 sqm/g specific surface area. It is even more surprising that this styrene divinylbenzene (SDVB) copolymer, which contains a large proportion of hydrophilic N-vinylpyrrolidone groups, has similar binding properties to the purely SDVB hypercrosslinked sorbents.
5.1. Binding Isotherms
Before any practical application of a novel sorbent, it is useful to measure its adsorption isotherm. In case of weak acid or weak base adsorbates, the isotherms should be measured at different pH values.
The isotherms of modern liquid chromatography packing materials are usually linear over many concentration decades. It seems that the hypercrosslinked and HLB sorbents have been used very often without checking their isotherms, and therefore assuming isotherm linearity. Our experiments show that this assumption is not generally warranted. Overlooking isotherm nonlinearity may lead to serious analytical and technological errors.
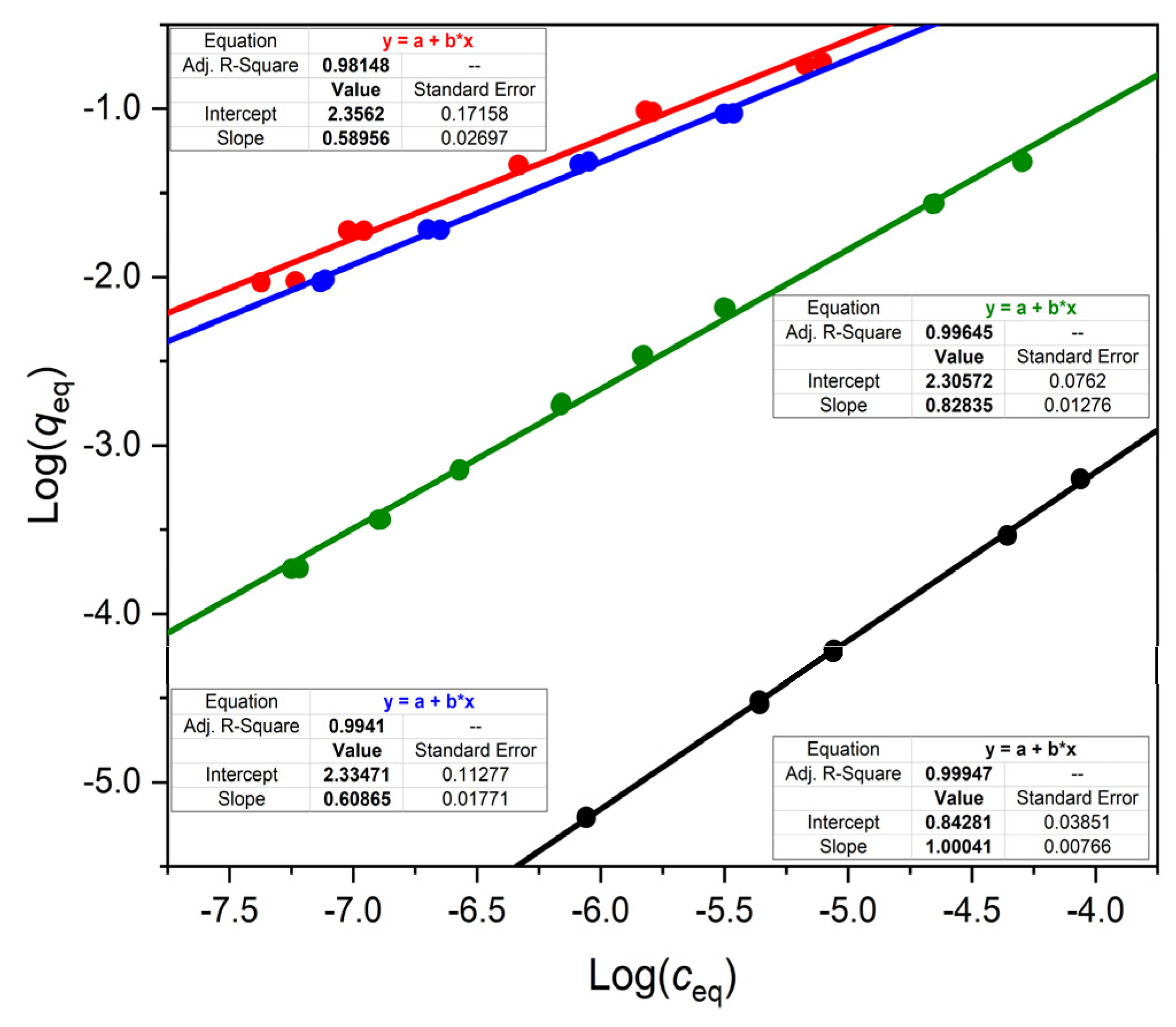
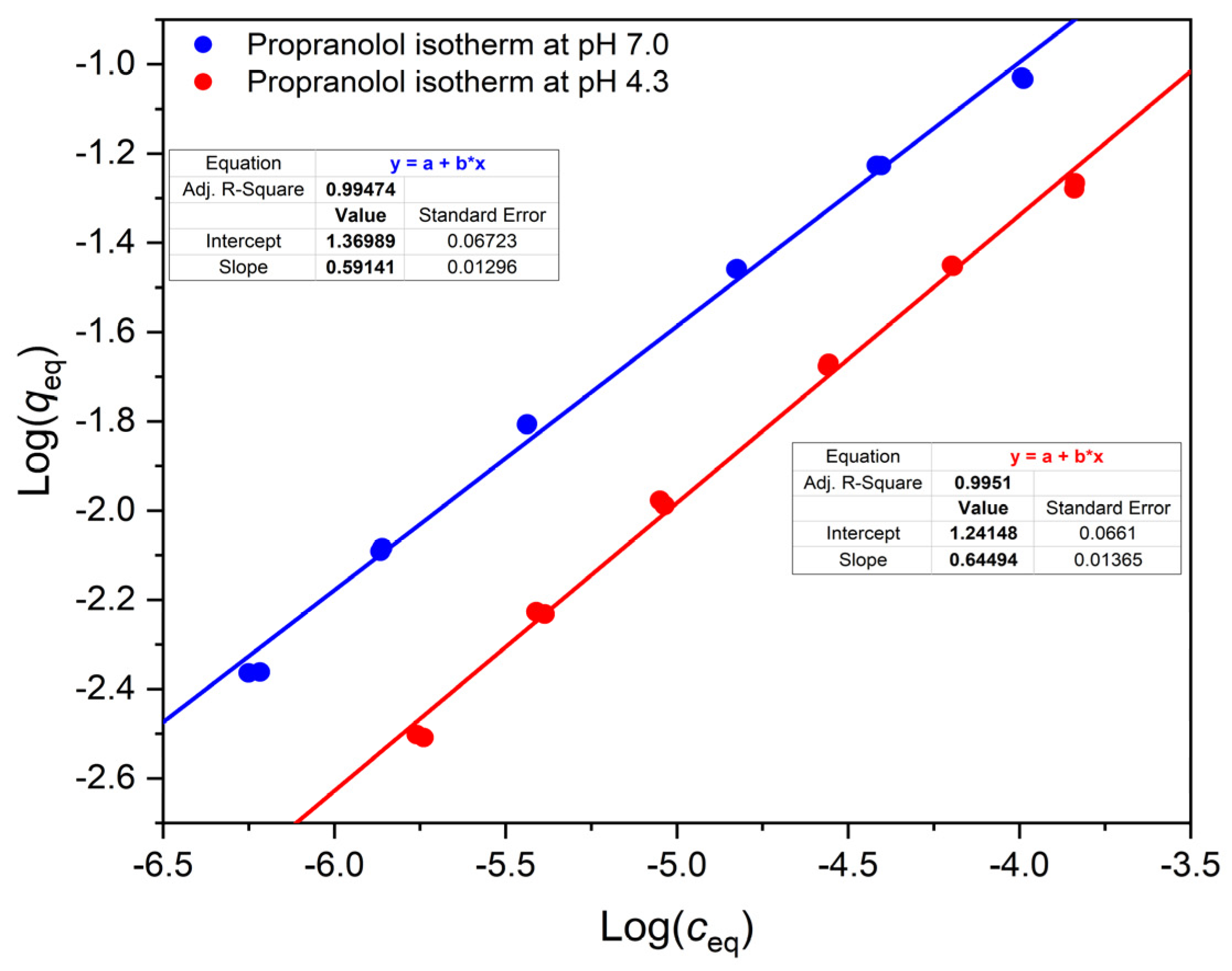
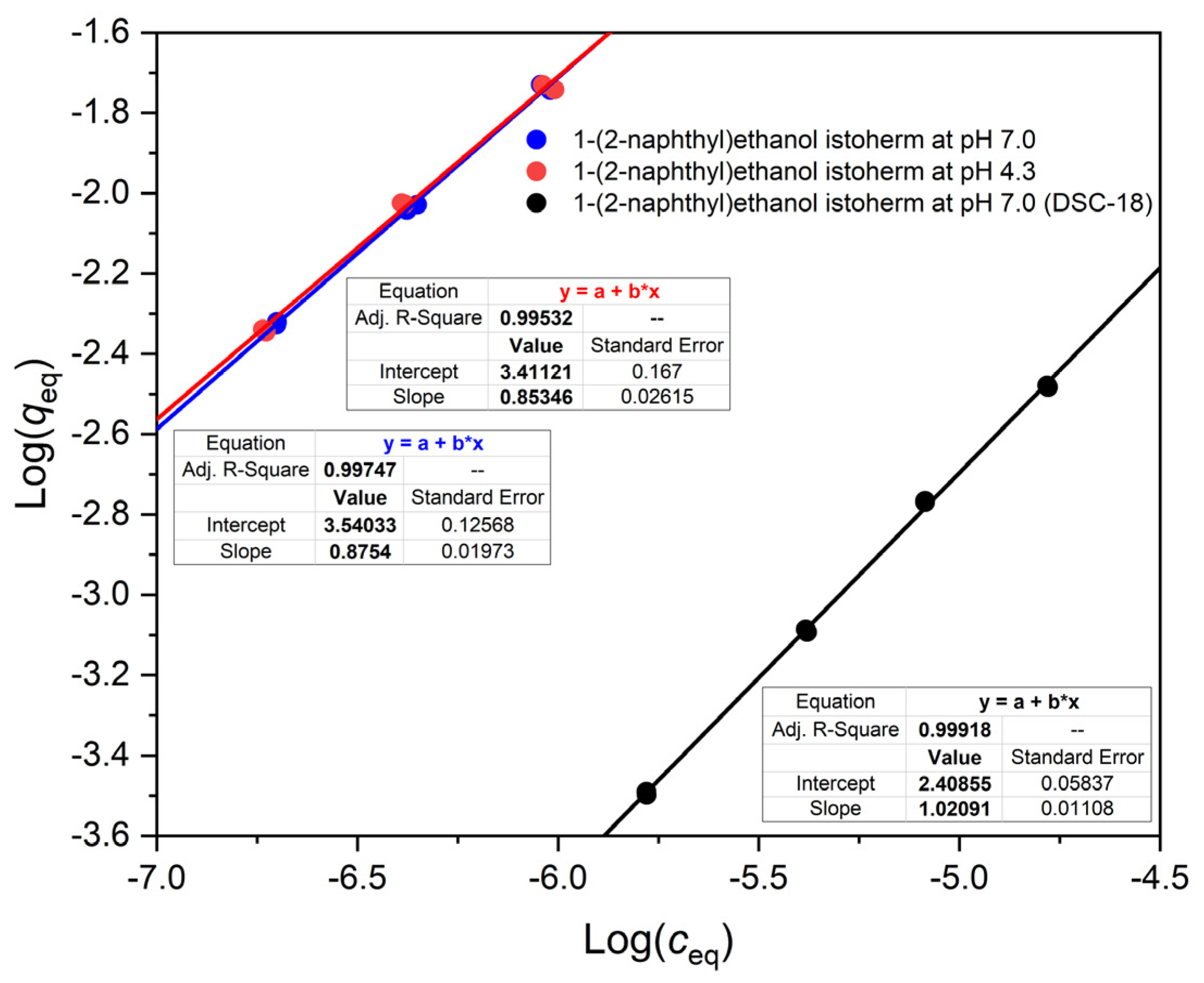
We have found that the isotherms of a weak acid, a weak base and a neutral substance are all nonlinear on OASIS® HLB. This is reflected by the slope m of their Freundlich lines (in log q-log c plots), which was less than 1.
If the slope in the log–log plot is significantly different from 1, then the
q vs.
c isotherm is a power function, and thus it is nonlinear. This has many consequences [
12]. If
m < 1, then the distribution coefficient of the substance is decreasing as the concentration of the substance increases. As a consequence of this, the shape of the breakthrough front in solid phase extraction will be sharper (more sudden) than with a linear isotherm. Moreover, if the analyte concentration is higher, the breakthrough will occur sooner. All this needs to be considered when optimizing an SPE method.
Jeong et al. [
2,
3] and Huysman et al. [
13] have also noted that the Freundlich exponents of many compounds deviated significantly from 1 on OASIS
® HLB. While we have only observed slope (
m) values less than 1 with OASIS
® HLB, these other investigators reported
m values both below and above 1. It is interesting that these authors observed
m > 1 (in their notation
n < 1, where
n = 1/
m) values. Isotherms with
m > 1 are usually a sign of multilayer adsorption [
12], but this appears to be unlikely at the very low
c values in the cited works. In any case, if the Freundlich exponent deviates from 1, the distribution coefficient varies with the analyte concentration, and this has important practical consequences, as pointed out above.
In the present work, we have particularly sought to study adsorption up to higher q values than others before us, i.e., even well above 0.01 mol/kg adsorbed concentration. Thus, we could show that OASIS® HLB also has unique properties (very strong adsorption, even of ions, and nonlinear isotherm) in this range. This is a concentration range where novel technical applications can be imagined, including interesting chemical reactions with adsorbed substances, on a preparative scale.
5.2. pH Dependence of Adsorption
An important feature of the novel sorbents which have been studied in this paper is their capability to adsorb ionizable organic compounds, such as many drugs and pesticides, at pH values where their ionic form dominates in solution, e.g., 2 pH units above their pKa values in case of weak acids. This property allows the simultaneous collection of neutral, weakly acidic and weakly basic compounds at near-neutral pH values. This is very important because multianalyte analyses, typically with chromatography, are in great demand these days.
In
Section 4.1 and
Section 5.1, it has been shown that the isotherms of a weak acid and a weak base are highly pH dependent on OASIS
® HLB. In the case of the acid, ketoprofen, binding at pH 7.0 was in the whole studied concentration range about two orders of magnitude less than at pH 3.2. Yet, the binding at pH 7.0 was still substantial, with the distribution coefficient around 1000. Since in the aqueous phase more than 99% of ketoprofen is in ionic form at pH 7.0 (see
Table 2), one might think that at this pH (and already from about pH 6) it would be almost exclusively the ionic form which is adsorbed. This view was indeed adopted when the novel adsorbents were first used for binding weak acids at neutral pH [
4].
We thought, however, that one cannot decide simply from the relative proportions of the ionic and the neutral forms (in the aqueous phase) the relative contributions of the ionic and neutral forms to the observed overall distribution coefficient. Indeed, according to Equation (6), the relative contributions of the two species to
D are:
As Equation (8) shows, at a given pH, the relative contributions of the neutral and the ionic forms to D depend on both the pH–pKa difference (which is equal to −log ([H]/Ka)), and on the ratio of the distribution constants KHA and KA. By way of an example: if KHA/KA = 200 (the neutral form is much better adsorbed than the ion), and if [H]/Ka = 0.01 (i.e., pH = pKa + 2), then the neutral to ionic ratio, [HA]/[A] is 0.01 (by virtue of Equation (1)), but DHA/DA is 2. This means that the neutral form, which accounts for only 1% of the compound in solution, has a two times higher contribution to D than the ionic form, which accounts for 99% in the solution. In other words, the concentration ratio of the neutral to the ionic form is 1:99 in the solution, but 2:1 in the adsorbed state.
Equation (6) also shows that even if the ionic form had zero distribution constant, the overall distribution coefficient might be high at pH values, where the ionic form is dominant in the solution. Indeed, if
KA = 0, Equation (6) gives:
If, for example, pH = pKa + 2, so that [H]/Ka = 0.01, then the denominator in Equation (9) is 1.01≈1, and D = 0.01 × KHA. On the novel sorbents, KHA values are very high, on the order of 1 × 105–1 × 106. Thus, even if the nonadsorbing ionic form dominates in the solution, D may be on the order of 1000–10,000, which are very high values.
Our experimental results obtained with the weak acid ketoprofen, and presented in
Table 2, confirm these conclusions. To prove this, we needed to calculate the values of
KHA,
KA and
pKa from the experimental data in
Table 2. Before showing how this was done, we shall point out the unique properties of OASIS
® HLB which follow from
Table 2.
The data in
Table 2 are indeed remarkable. At pH 3.2,
D is almost 200,000. This is a very high value. In HPLC a typical
D value would be only around 10. In solid phase extraction, the high
D value of ca. 200,000 would indicate a breakthrough volume of 200 L on a sorbent packing of only 1 g mass (according to the formula derived in
Appendix A). However, even at pH 7, i.e., for “neutral” water samples,
D of ketoprofen has been 974, which allows the percolation of 974 mL of sample through a 1 g extraction packing without full breakthrough. Thus our data support the observations made by others [
4] that sufficiently “hydrophobic” weak acids may be extracted with the novel sorbents even from neutral solutions with high recovery (little breakthrough).
The values of
KHA,
KA and
pKa may be calculated from the data in
Table 2 in the following way. Since at pH 10.0,
D is approximately equal to
KA, as follows from Equation (6) for a weak acid with pKa well below 7. Thus,
KA is about 468. One can also see from Equation (6), that at pH 3.2 and 4.3,
DA is less than
KA = 468, and thus negligible compared to the
D values measured at pH 3.2 and 4.3, respectively, which are both above 100,000. Thus, at pH 3.2 and 4.3
D is approximately equal to
DHA. This allows us to calculate
KHA and
Ka from the
D values measured at pH 3.2 and 4.3, respectively. The values obtained were
KHA = 211,173 and
Ka = 5.17 × 10
−5 (
pKa = 4.29). Having these two constants, one can calculate from the first part of Equation (5) (which is formally the same as Equation (6)) the pH dependence of the distribution coefficient
DHA for the neutral form of ketoprofen. The respective values, calculated for pH 6.0, 7.0 and 10.0 are shown in
Table 2. We see, that at pH 7.0 the calculated
DHA is still substantial, its value being 407, which is not negligible compared to the measured value, 974.
An estimation of the distribution constant of ketoprofen’s ionized form,
KA, from
Table 2 is difficult, but an approximate value can be obtained. This can be achieved if we compare the data in
Table 2 for pH 7.0 and 10.0. According to Equation (6), the difference between the measured
D and the calculated
DHA values at any pH should show the contribution of the anions to the distribution coefficient. This contribution should be almost constant in this pH range, because here
[H]/
Ka << 1, so that
DA≈
KA = constant. However, one can see that the apparent ionic contribution decreases substantially with increasing pH. If we assume that at pH 10.0 we are really measuring
KA, the distribution constant of the anion, then
KA is only 468, but it is certainly not higher than about 1000.
In any case, the estimated contribution of the neutral form of ketoprofen,
DHA, to the overall distribution coefficient,
D, is not negligible even at pH 7.0, and at pH 6.0 it is even higher, essentially equal to the overall
D of 3930 (
Table 2). Thus, at pH 6.0 and 7.0 the contribution of the neutral form to
D is substantial, and one cannot speak of pure ion-extraction. This experimental result is well explained by our theoretical calculations. The ratio of the experimental
KHA to
KA (211,173 to 468, i.e., 451) is indeed very high. Thus, on OASIS
® HLB the distribution constant of the neutral form of ketoprofen is 451 times higher than the distribution constant of the ketoprofen anion, and therefore even at pH values above
pKa + 2, the overall distribution is far from purely ionic. This situation may apply to many other environmentally relevant weak acids, which are fairly hydrophobic in their neutral form. For hydrophobic bases, an analogous conclusion is also warranted, as seen in
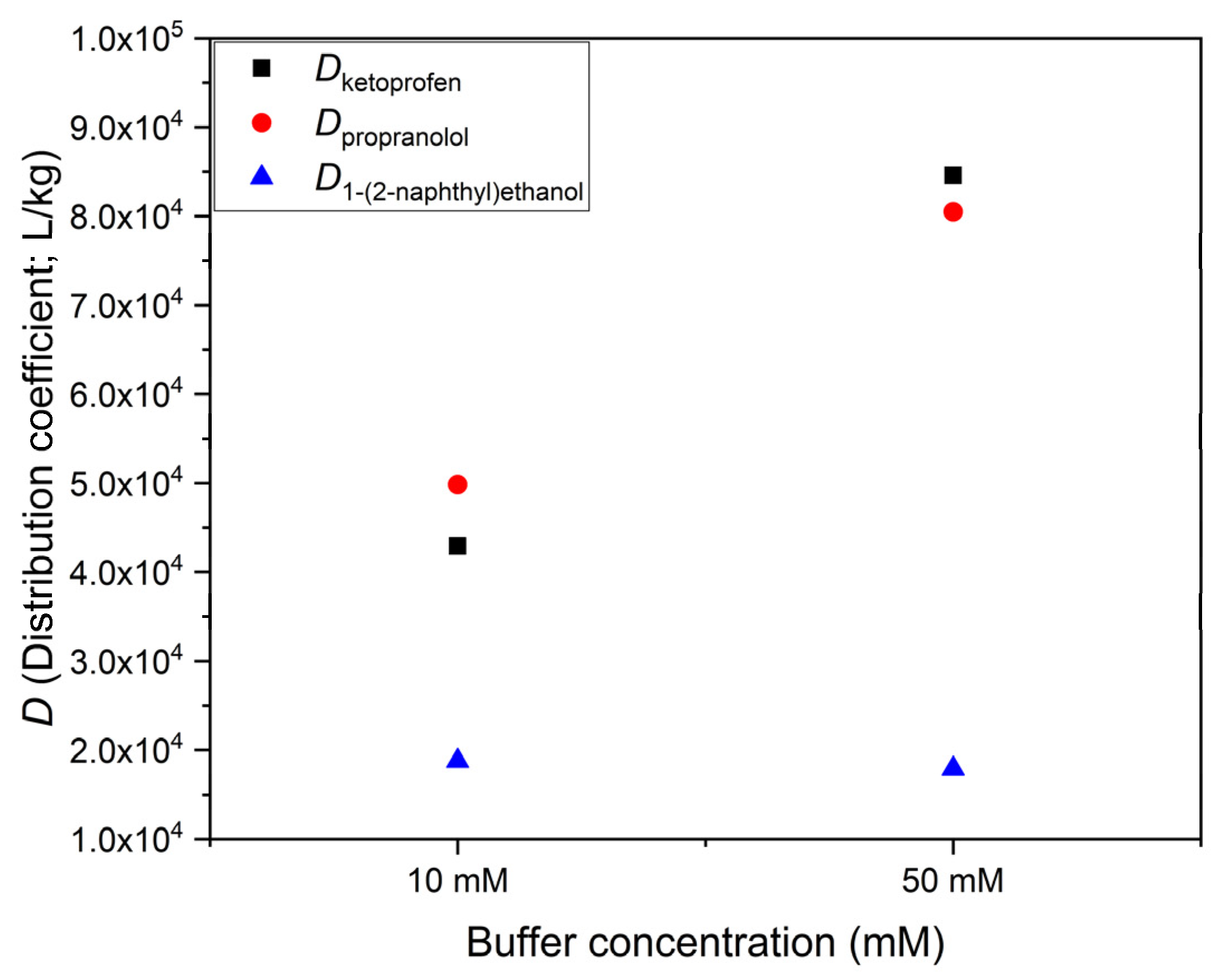
Table 1 for the case of propranolol (
pKa being about 9.5): the contribution of the neutral (basic) form to
D must be significant even if the pH is more than 2 units below the
pKa, since in our experiment at pH 7.0,
D is 4270, which is much higher than 1100, the
D at pH 4.3.
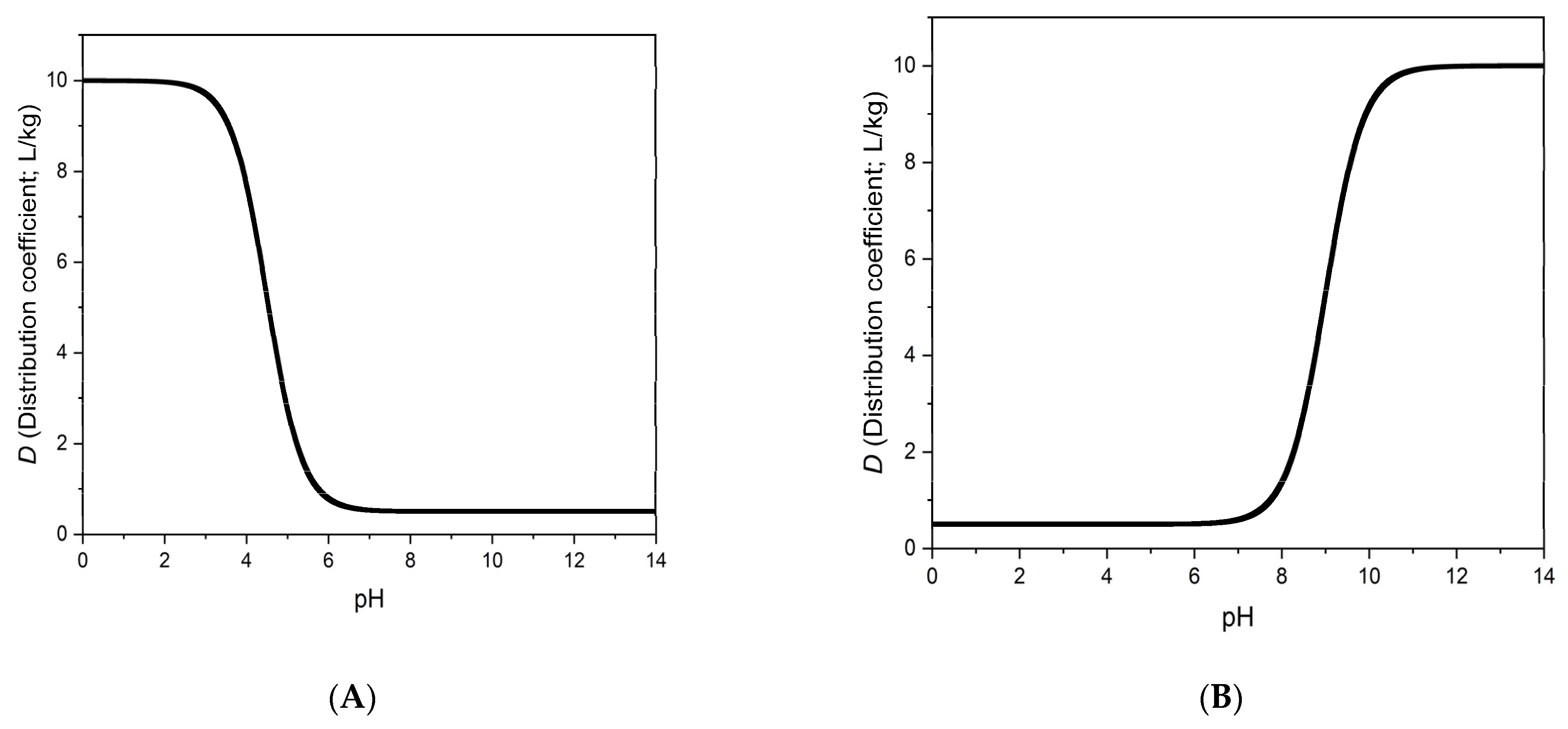
An important practical consequence of our theoretical results and of the experimental data in
Table 1 and
Table 2 is that the distribution coefficient of a weak acid (or a weak base) may vary substantially within the “neutral” pH range (between ca. pH 5.5 to 8.5). The
D values of ketoprofen at pH 6.0 and 7.0, which are ca. 4000 and 1000, respectively, show a fourfold change between pH 6.0 and 7.0. In real life applications, e.g., when working with natural water samples, which may have different pH values in this pH range, the breakthrough volumes may also vary by a factor of four (see
Appendix A), and therefore recoveries may vary from sample to sample. This problem is not limited to ketoprofen, it is a general problem, as shown by the following example. Let us assume, that
KHA/
KA = 100, which appears to be a typical order of magnitude for the novel sorbents. The
pKa of the acid should be about 5, which is a good approximation for many carboxylic acids. Then, according to Equation (5),
D is equal to 10 ×
KA at pH 6.0, 1.98 ×
KA at pH 7.0 and 1.1 ×
KA at pH 8.0. This is a nearly tenfold change of
D between pH 6.0 and 8.0.
5.4. Mechanism of Adsorption
Our experimental observations show that binding of ionizable compounds, particularly of their ionic form, by the novel sorbents must be related to hydrophobicity, but there must be some other factors, too. Hydrophobicity refers to the change of standard chemical potential when a species is transferred from one bulk phase into another bulk phase. In contrast to this, an adsorbed molecule or ion is generally not (fully) immersed into the solid sorbent. Rather, it is attached to the sorbent surface but remains partly in contact with the solvent. As is known for surfactants, hydrophilic “headgroups” may remain fully solvated by water.
Adsorption of (hydrophobic) ions on a neutral (i.e., not ion exchanger type) solid sorbent is not easy to treat theoretically. Gritti [
15] has recently summarized the different approaches for the quantitative prediction of retention, and thus of adsorption, in HPLC. He concluded that all known methods need improvement concerning the retention of ionizable solutes. One should add to this that the models for describing the adsorption of ionic surfactants at the water–air interface, which appears to be simpler than adsorption with solids, are still hotly debated [
16], and new approaches are appearing [
17]. For this reason, only a qualitative model will be used here. Nonetheless, the models used to describe surfactant adsorption on the water–air interface have relevance here, as the ionic form of many ionizable drugs have been shown to be reasonably good surfactants [
18,
19].
The ionized forms of the head groups of weak acids and bases are much more hydrophilic than in the neutral forms. This explains the more than one-hundredfold lower adsorption constant of the ionic form that was observed, compared to the neutral one. At near-neutral pH, both forms may comparably contribute to the overall adsorption: the ionic form because of its high relative abundance, while the neutral, much less abundant form by its much higher distribution constant.
The unique property of the novel (hypercrosslinked and HLB) sorbents, that they can strongly (with D between 500 and several thousand) bind weak acids and bases around pH 7, where their ionic forms dominate in solution, can also be explained by our results. The interaction of these novel polymers with the hydrophobic molecules, as expressed by their distribution constants, is 2–4 orders of magnitude stronger than that observed with comparable chromatographic sorbents, e.g., C18 modified silicas. Thus, the distribution coefficient of the neutral form is still quite high at around pH 7. On the other hand, due to the very strong adsorption of the hydrophobic part of the molecule, even the ionized form is significantly adsorbed (although its distribution constant is about two orders of magnitude lower than that of the neutral form). These effects are even more pronounced in purely aqueous solutions, than in the 20% methanolic solutions used in the present study. Approximate measurements showed that in the absence of methanol, the adsorption of the neutral form would have been unmeasurably high by UV-HPLC.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}