
The Role of Polymer Structure in Formation of Various Nano- and Microstructural Materials: 30 Years of Research in the Laboratory of Nano- and Microstructural Materials at the Centre of Polymer and Carbon Materials PAS †

 , , , , and
, , , , and
Abstract
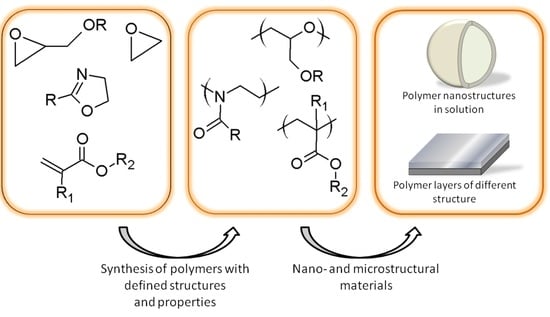
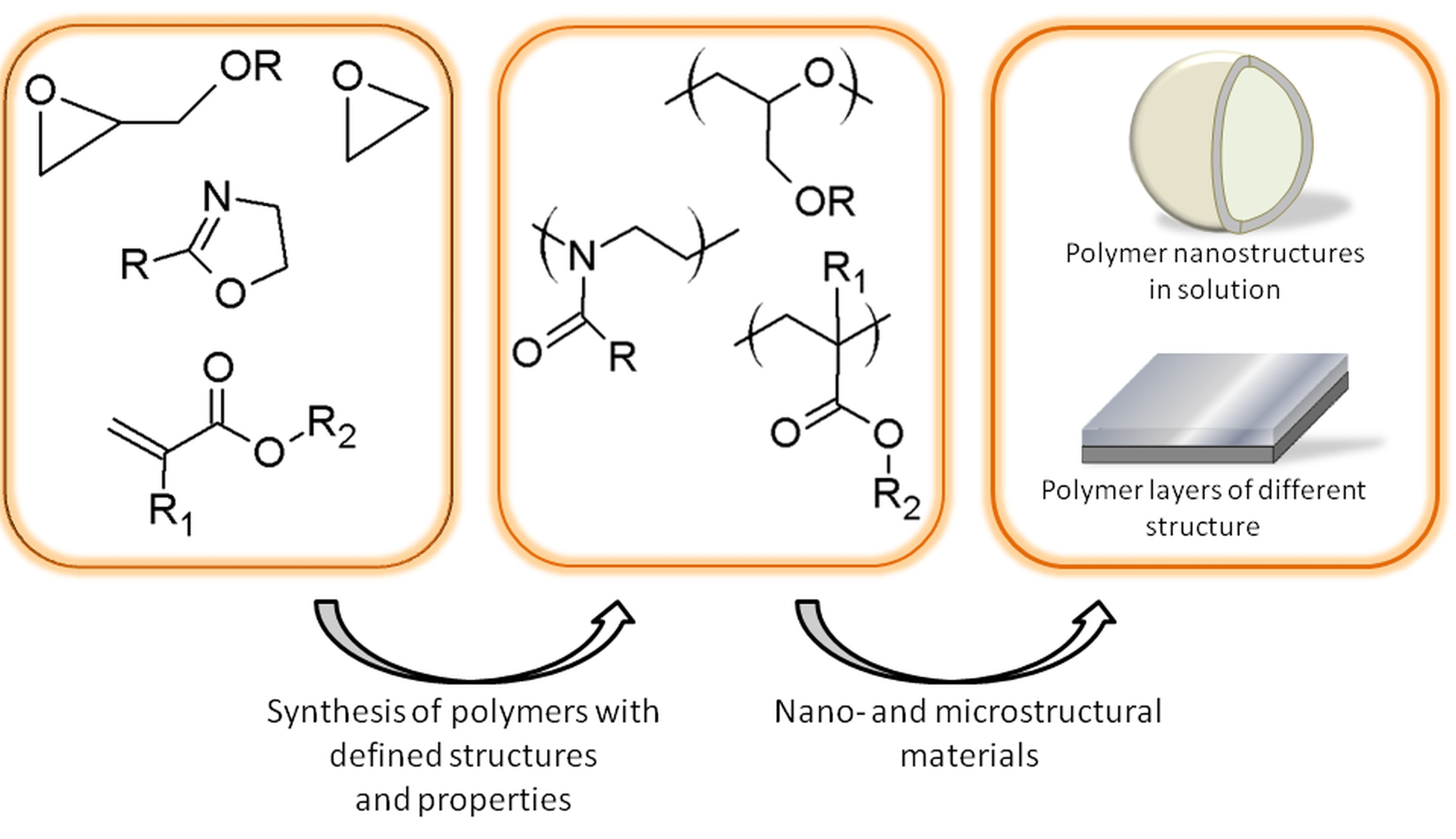
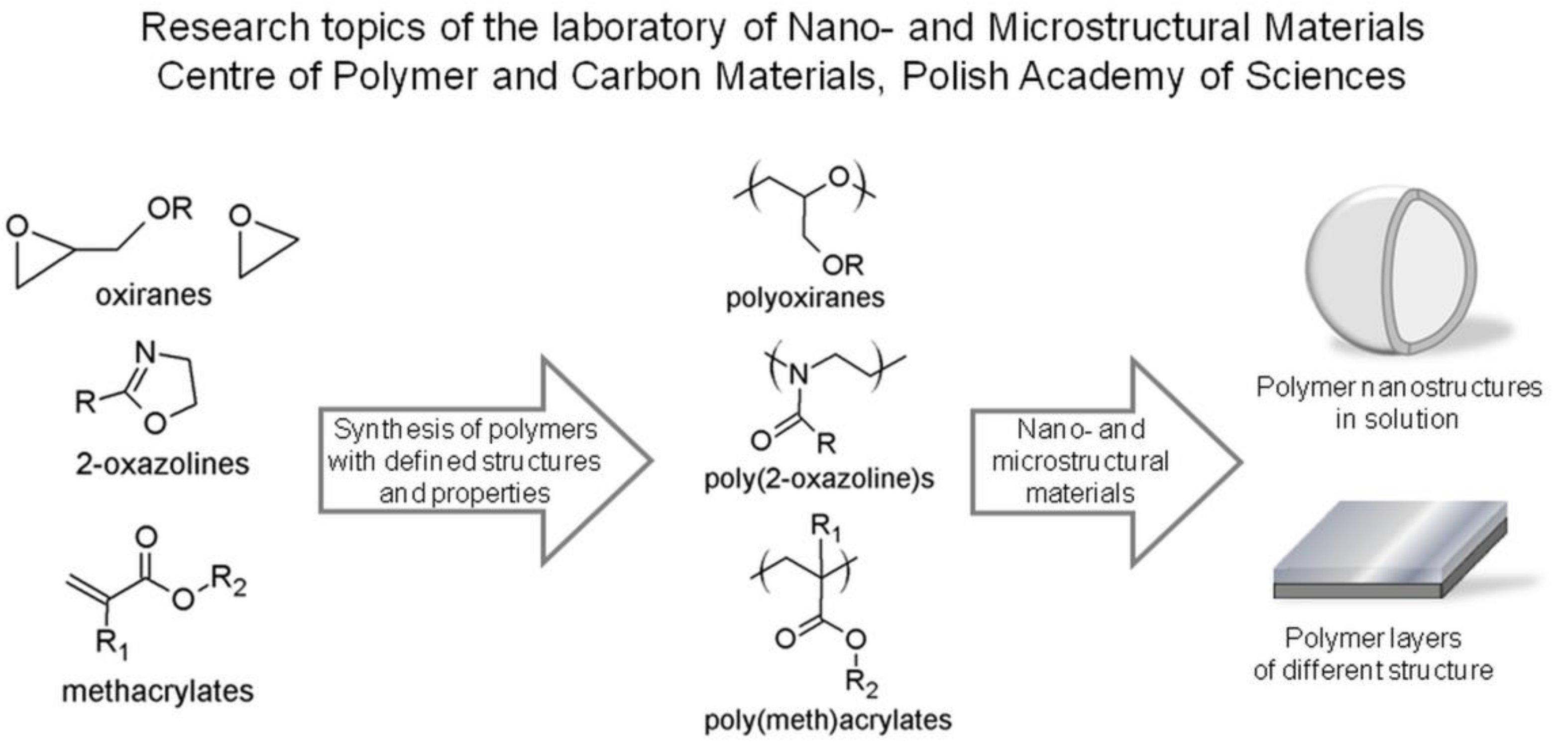
:
1. Introduction
2. Polymer Nano- and Microstructure
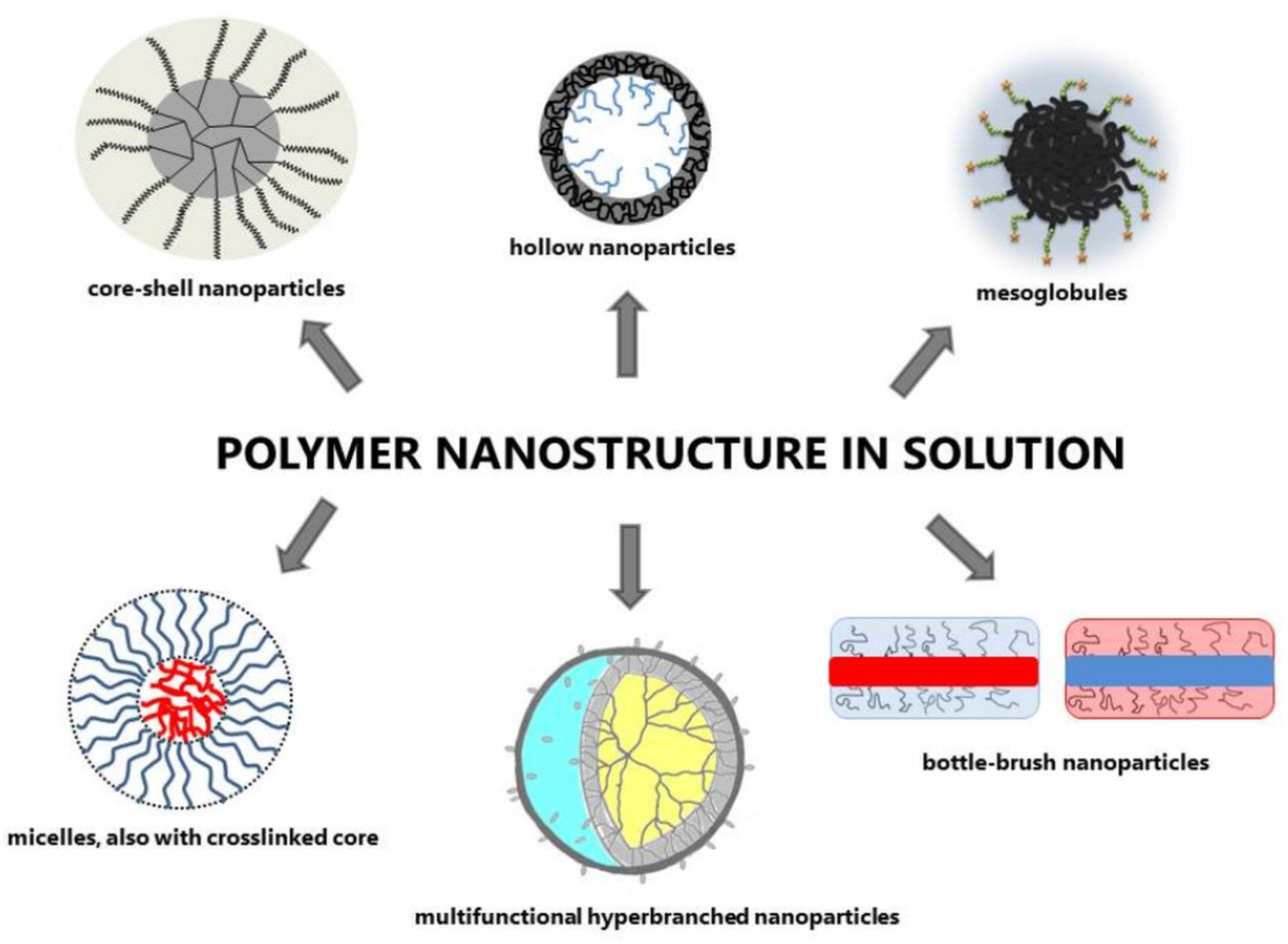
2.1. Polymer Nanostructure in Solution
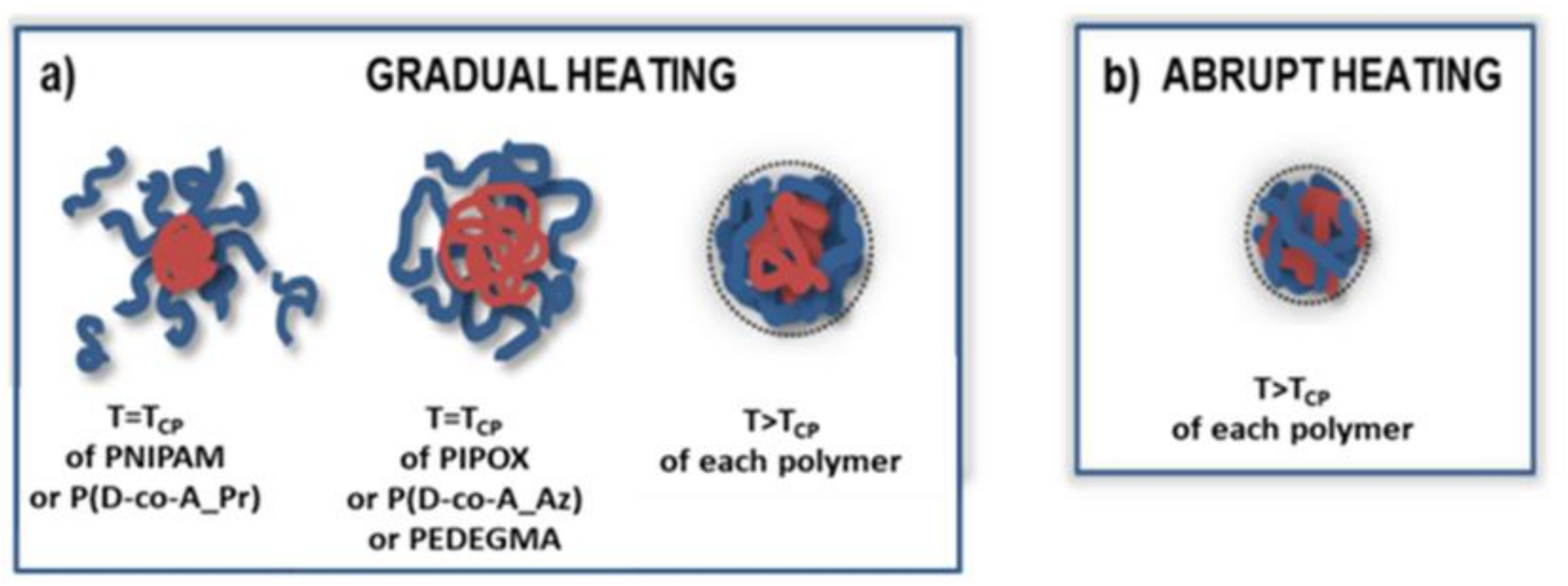
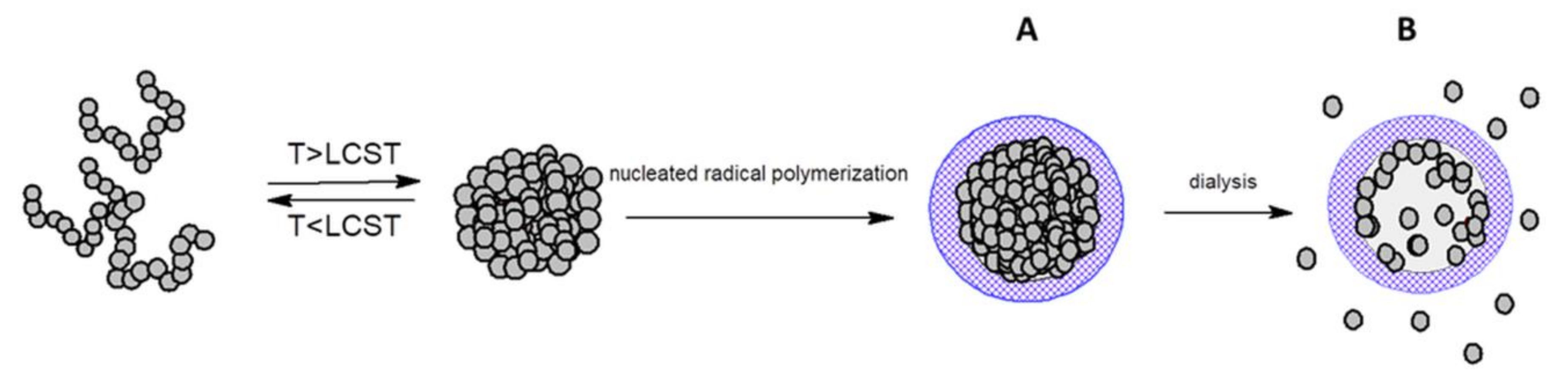
2.1.1. Mesoglobules of Thermoresponsive Polymers
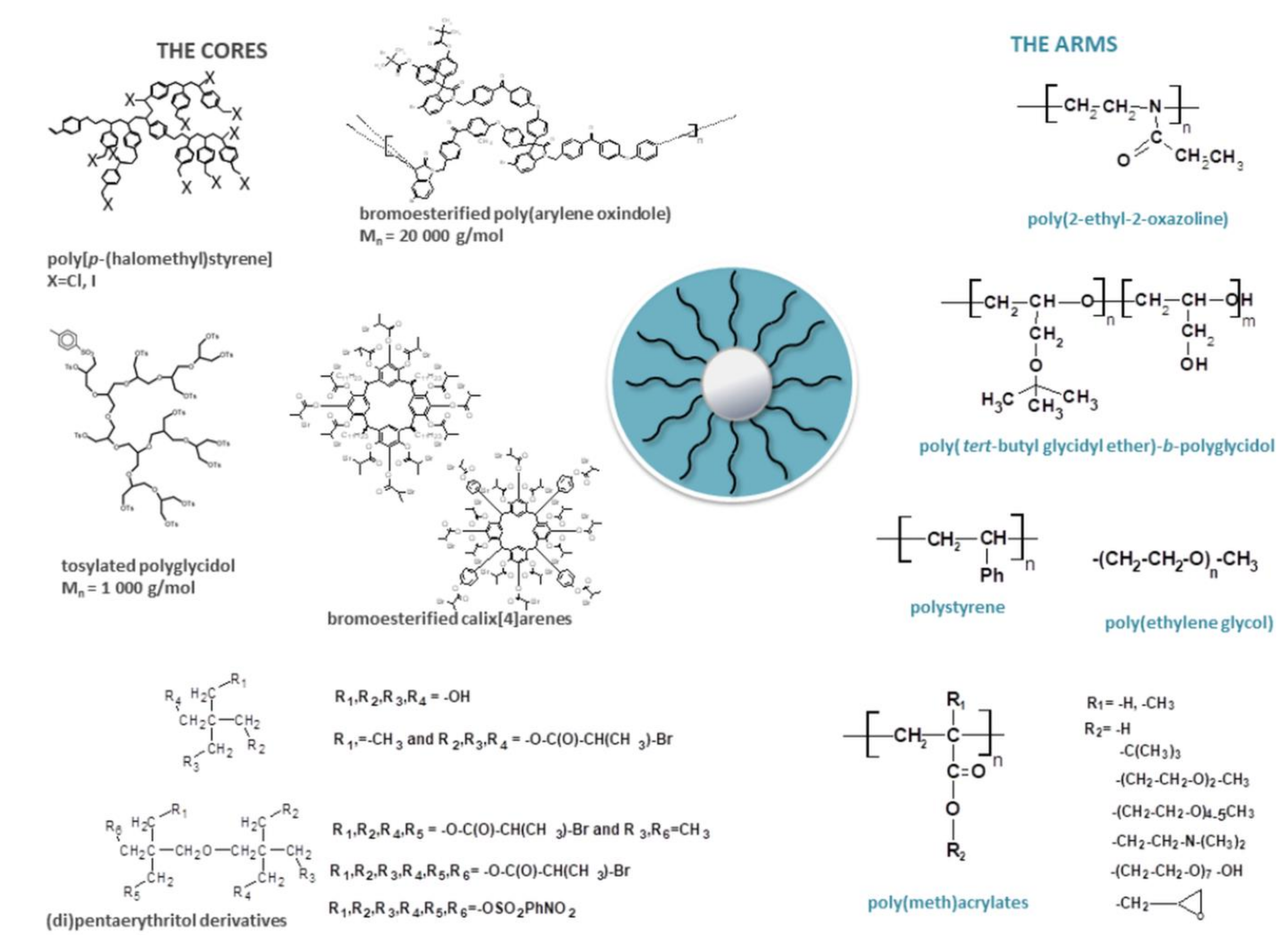
2.1.2. Branched Nanostructures
Hyperbranched Polymers
Bottle-Brush (Comb-Like) Macromolecules
Stars
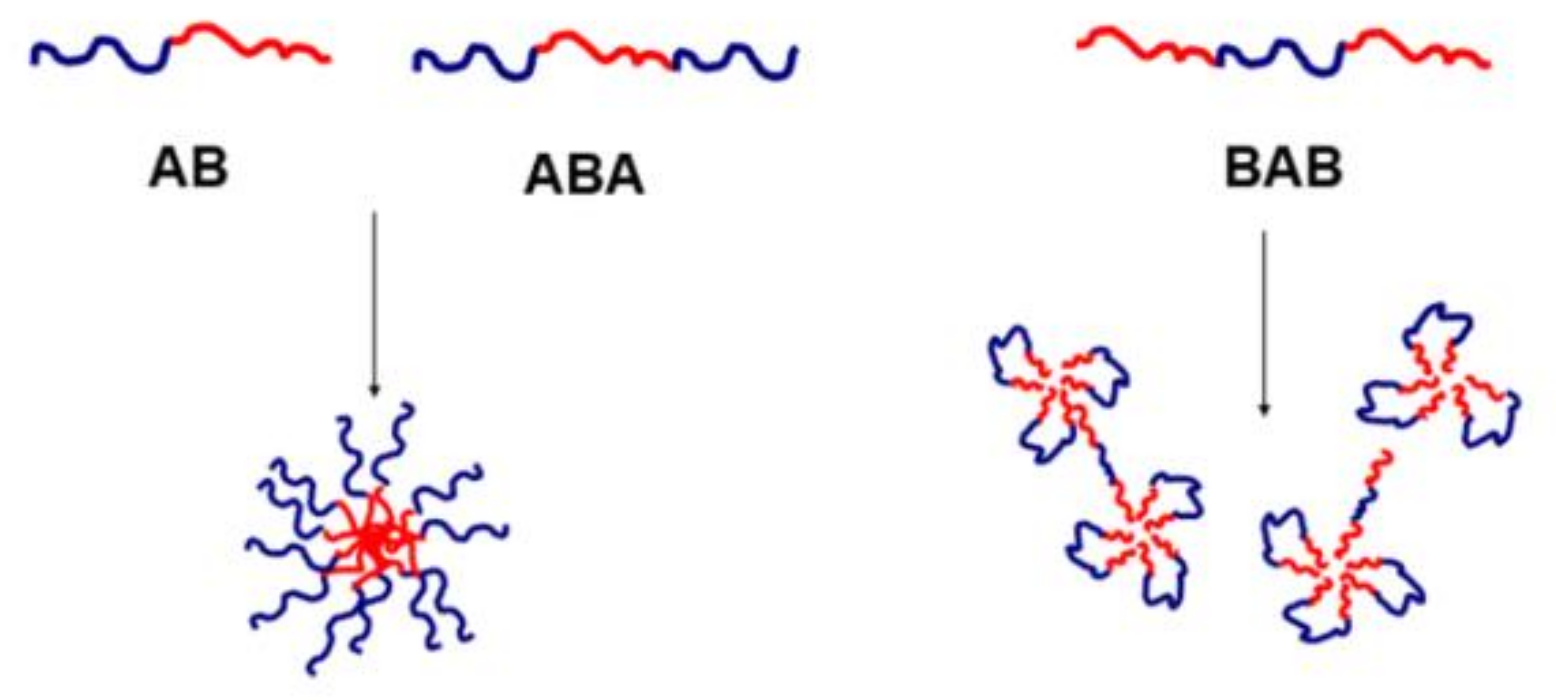
2.1.3. Nanostructure via Self-Assembling of Block Copolymers
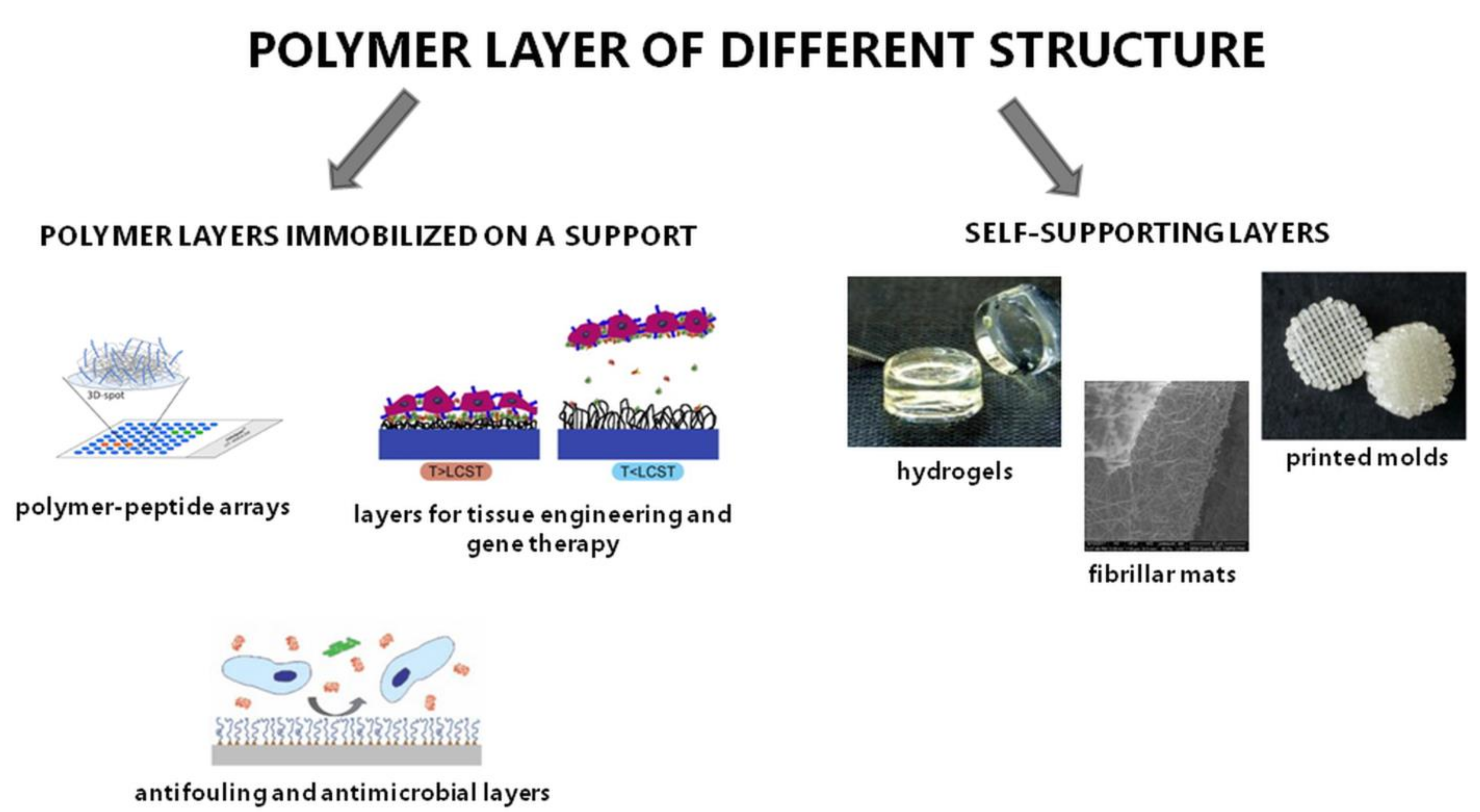
2.2. Polymer Layers of Different Structures
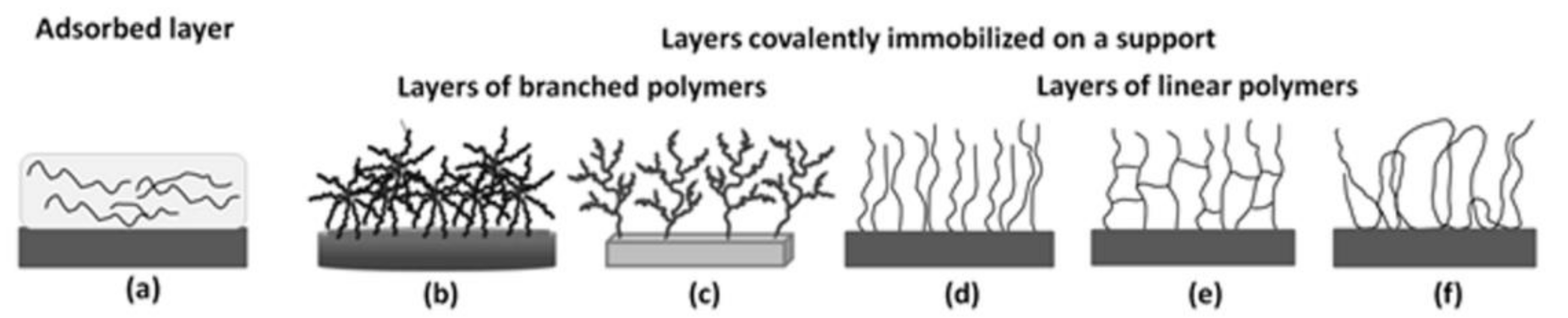
2.2.1. Polymer Layers Immobilized on a Support
Layers of Linear Polymers
The “Grafting from”—Layers of Brush Structures and Crosslinked Layers
Nanolayers of Branched Polymers
2.2.2. Self-Supporting Layers
Hydrogels
Fibrillar Mats
3. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kubisa, P.; Penczek, S. Cationic activated monomer polymerization of heterocyclic monomers. Prog. Polym. Sci. 1999, 24, 1409–1437. [Google Scholar] [CrossRef]
- Kubisa, P. Activated monomer mechanism in the cationic polymerization of cyclic ethers. Makromol. Chem. Macromol. Symp. 1988, 13–14, 203–210. [Google Scholar] [CrossRef]
- Dworak, A.; Schulz, R. Star polymers and block copolymers of 2-oxazolines using chloroformates as initiators. Die Makromol. Chem. 1991, 192, 437–445. [Google Scholar] [CrossRef]
- Schulz, R.C.; Schwarzenbach, E. Macromonomers on the basis of 2-phenyl-2-oxazoline. Makromol. Chem. Macromol. Symp. 1988, 13–14, 495–505. [Google Scholar] [CrossRef]
- Schulz, R.C.; Eifel, H.-W.; Perner, T.; Mühlbach, K. Some new examples of cationic polymerization. Makromol. Chem. Macromol. Symp. 1986, 3, 163–178. [Google Scholar] [CrossRef]
- Hoffman, A.S. Stimuli-responsive polymers: Biomedical applications and challenges for clinical translation. Adv. Drug Deliv. Rev. 2013, 65, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Priya James, H.; John, R.; Alex, A.; Anoop, K.R. Smart polymers for the controlled delivery of drugs—A concise overview. Acta Pharm. Sin. B 2014, 4, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Pal, K.; Banthia, A.K.; Majumdar, D.K. Polymeric hydrogels: Characterization and biomedical applications. Des. Monomers Polym. 2009, 12, 197–220. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Akiyama, Y.; Okano, T. Temperature-responsive polymer modified surface for cell sheet engineering. Polymers 2012, 4, 1478–1498. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.F. Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3459–3470. [Google Scholar] [CrossRef]
- Gosecki, M.; Gadzinowski, M.; Gosecka, M.; Basinska, T.; Slomkowski, S. Polyglycidol, its derivatives, and polyglycidol-containing copolymers-synthesis and medical applications. Polymers 2016, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Uchman, M. Poly(2-oxazoline)-based stimulus-responsive (Co)polymers: An overview of their design, solution properties, surface-chemistries and applications. Prog. Polym. Sci. 2020, 106, 101252. [Google Scholar] [CrossRef]
- Karayianni, M.; Pispas, S. Block copolymer solution self-assembly: Recent advances, emerging trends, and applications. J. Polym. Sci. 2021, 1–25. [Google Scholar] [CrossRef]
- Aseyev, V.; Hietala, S.; Laukkanen, A.; Nuopponen, M.; Confortini, O.; Du Prez, F.E.; Tenhu, H. Mesoglobules of thermoresponsive polymers in dilute aqueous solutions above the LCST. Polymer 2005, 46, 7118–7131. [Google Scholar] [CrossRef]
- Rangelov, S.; Simon, P.; Toncheva-Moncheva, N.; Dimitrov, P.; Gajewska, B.; Tsvetanov, C.B. Nanosized colloidal particles from thermosensitive poly(methoxydiethyleneglycol methacrylate)s in aqueous media. Polym. Bull. 2012, 68, 2175–2185. [Google Scholar] [CrossRef]
- Trzebicka, B.; Szweda, D.; Rangelov, S.; Kowalczuk, A.; Mendrek, B.; Utrata-Wesołek, A.; Dworak, A. (Co)polymers of oligo(ethylene glycol) methacrylates—Temperature-induced aggregation in aqueous solution. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 614–623. [Google Scholar] [CrossRef]
- Trzebicka, B.; Weda, P.; Utrata-Wesołek, A.; Dworak, A.; Tsvetanov, C. Mesoglobules of random copolyethers as templates for nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 4074–4083. [Google Scholar] [CrossRef]
- Wu, C.; Li, W.; Zhu, X.X. Viscoelastic Effect on the Formation of Mesoglobular Phase in Dilute Solutions. Macromolecules 2004, 37, 4989–4992. [Google Scholar] [CrossRef]
- Junk, M.J.N.; Li, W.; Schlüter, A.D.; Wegner, G.; Spiess, H.W.; Zhang, A.; Hinderberger, D. Formation of a mesoscopic skin barrier in mesoglobules of thermoresponsive polymers. J. Am. Chem. Soc. 2011, 133, 10832–10838. [Google Scholar] [CrossRef]
- Weda, P.; Trzebicka, B.; Dworak, A.; Tsvetanov, C.B. Thermosensitive nanospheres of low-density core—An approach to hollow nanoparticles. Polymer 2008, 49, 1467–1474. [Google Scholar] [CrossRef]
- Dimitrov, P.; Toncheva, N.; Weda, P.; Rangelov, S.; Trzebicka, B.; Dworak, A.; Tsvetanov, C.B. Nano-templates from thermoresponsive poly(ethoxytriethyleneglycol acrylate) for polymeric nano-capsules. Macromol. Symp. 2009, 278, 89–95. [Google Scholar] [CrossRef]
- Toncheva, N.; Tsvetanov, C.; Rangelov, S.; Trzebicka, B.; Dworak, A. Hydroxyl end-functionalized poly(2-isopropyl oxazoline)s used as nano-sized colloidal templates for preparation of hollow polymeric nanocapsules. Polymer 2013, 54, 5166–5173. [Google Scholar] [CrossRef]
- Trzebicka, B.; Haladjova, E.; Otulakowski, Ł.; Oleszko, N.; Wałach, W.; Libera, M.; Rangelov, S.; Dworak, A. Hybrid nanoparticles obtained from mixed mesoglobules. Polymer 2015, 68, 65–73. [Google Scholar] [CrossRef]
- Szweda, R.; Trzebicka, B.; Dworak, A.; Otulakowski, L.; Kosowski, D.; Hertlein, J.; Haladjova, E.; Rangelov, S.; Szweda, D. Smart Polymeric Nanocarriers of Met-enkephalin. Biomacromolecules 2016, 17, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Dworak, A.; Lipowska, D.; Szweda, D.; Suwinski, J.; Trzebicka, B.; Szweda, R. Degradable polymeric nanoparticles by aggregation of thermoresponsive polymers and “click” chemistry. Nanoscale 2015, 7, 16823–16833. [Google Scholar] [CrossRef] [Green Version]
- Lipowska-Kur, D.; Otulakowski, Ł.; Trzebicka, B.; Utrata-Wesołek, A.; Dworak, A. Thermoresponsive nanogels of modified poly(di(ethylene glycol) methyl ether methacrylate)-co-(2-aminoethyl methacrylate)s. Polymers 2020, 12, 1645. [Google Scholar] [CrossRef]
- Lipowska-Kur, D.; Szweda, R.; Trzebicka, B.; Dworak, A. Preparation and characterization of doxorubicin nanocarriers based on thermoresponsive oligo(ethylene glycol) methyl ether methacrylate polymer-drug conjugates. Eur. Polym. J. 2018, 109, 391–401. [Google Scholar] [CrossRef]
- Otulakowski, L.; Kasprow, M.; Dworak, A.; Trzebicka, B. Effect of sodium dodecyl sulfate on solution behavior of thermoresponsive polymers and their mixtures. Polimery 2019, 64, 469–479. [Google Scholar] [CrossRef]
- Otulakowski, Ł.; Kasprów, M.; Strzelecka, A.; Dworak, A.; Trzebicka, B. Thermal behaviour of common thermoresponsive polymers in phosphate buffer and in its salt solutions. Polymers 2021, 13, 90. [Google Scholar] [CrossRef]
- Kasprów, M.; Machnik, J.; Otulakowski, Ł.; Dworak, A.; Trzebicka, B. Thermoresponsive P(HEMA-co-OEGMA) copolymers: Synthesis, characteristics and solution behavior. RSC Adv. 2019, 9, 40966–40974. [Google Scholar] [CrossRef] [Green Version]
- Toncheva-Moncheva, N.; Dimitrov, P.; Tsvetanov, C.B.; Robak, B.; Trzebicka, B.; Dworak, A.; Rangelov, S. Formation of mesoglobules in aqueous media from thermo-sensitive poly(ethoxytriethyleneglycol acrylate). Polym. Bull. 2011, 67, 1335–1346. [Google Scholar] [CrossRef]
- Trzcinska, R.; Szweda, D.; Rangelov, S.; Suder, P.; Silberring, J.; Dworak, A.; Trzebicka, B. Bioactive mesoglobules of poly(di(ethylene glycol) monomethyl ether methacrylate)-peptide conjugate. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 3104–3115. [Google Scholar] [CrossRef]
- Trzebicka, B.; Robak, B.; Trzcinska, R.; Szweda, D.; Suder, P.; Silberring, J.; Dworak, A. Thermosensitive PNIPAM-peptide conjugate-Synthesis and aggregation. Eur. Polym. J. 2013, 49, 499–509. [Google Scholar] [CrossRef]
- Dworak, A.; Walach, W.; Trzebicka, B. Cationic polymerization of glycidol. Polymer structure and polymerization mechanism. Macromol. Chem. Phys. 1995, 196, 1963–1970. [Google Scholar] [CrossRef]
- Dworak, A.; Baran, G.; Trzebicka, B.; Wałach, W. Polyglycidol-block-poly(ethylene oxide)-block-polyglycidol: Synthesis and swelling properties. React. Funct. Polym. 1999, 42, 31–36. [Google Scholar] [CrossRef]
- Dworak, A.; Panchev, I.; Trzebicka, B.; Walach, W. Hydrophilic and amphiphilic copolymers of 2,3-epoxypropanol-1. Macromol. Symp. 2000, 153, 233–242. [Google Scholar] [CrossRef]
- Dworak, A.; Walach, W.; Trzebicka, B.; Kowalczuk, A.; Nowicka, M.; Filak, J. Amphiphilic polyethers of controlled chain architecture. In Advances Macromolecular and Supramolecular Materials and Processes; Geckler, K., Ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2003; pp. 219–227. [Google Scholar]
- Kowalczuk-Bleja, A.; Trzebicka, B.; Komber, H.; Voit, B.; Dworak, A. Controlled radical polymerization of p-(iodomethyl)styrene—A route to branched and star-like structures. Polymer 2004, 45, 9–18. [Google Scholar] [CrossRef]
- Walach, W.; Kowalczuk, A.; Trzebicka, B.; Dworak, A. Synthesis of high-molar mass arborescent-branched polyglycidol via sequential grafting. Macromol. Rapid Commun. 2001, 22, 1272–1277. [Google Scholar] [CrossRef]
- Walach, W.; Trzebicka, B.; Justynska, J.; Dworak, A. High molecular arborescent polyoxyethylene with hydroxyl containing shell. Polymer 2004, 45, 1755–1762. [Google Scholar] [CrossRef]
- Dworak, A.; Wałach, W. Synthesis, characterization and properties of functional star and dendritic block copolymers of ethylene oxide and glycidol with oligoglycidol branching units. Polymer 2009, 50, 3440–3447. [Google Scholar] [CrossRef]
- Klajnert, B.; Walach, W.; Bryszewska, M.; Dworak, A.; Shcharbin, D. Cytotoxicity, haematotoxicity and genotoxicity of high molecular mass arborescent polyoxyethylene polymers with polyglycidol-block-containing shells. Cell Biol. Int. 2006, 30, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Basinska, T.; Slomkowski, S.; Dworak, A.; Panchev, I.; Chehimi, M.M. Synthesis and characterization of poly(styrene/α-t-butoxy-ω-vinylbenzyl-polyglycidol) microspheres. Colloid Polym. Sci. 2001, 279, 916–924. [Google Scholar] [CrossRef]
- Mendrek, A.; Mendrek, S.; Trzebicka, B.; Kuckling, D.; Walach, W.; Adler, H.J.; Dworak, A. Polyether core-shell cylinder-polymerization of polyglycidol macromonomers. Macromol. Chem. Phys. 2005, 206, 2018–2026. [Google Scholar] [CrossRef]
- Mendrek, A.; Mendrek, S.; Adler, H.J.; Dworak, A.; Kuckling, D. Amphiphilic behaviour of poly(glycidol)-based macromonomers and its influence on homo-polymerisation in water and in water/benzene mixture. Polymer 2010, 51, 342–354. [Google Scholar] [CrossRef]
- Kowalczuk, A.; Kronek, J.; Bosowska, K.; Trzebicka, B.; Dworak, A. Star poly(2-ethyl-2-oxazoline)s-synthesis and thermosensitivity. Polym. Int. 2011, 60, 1001–1009. [Google Scholar] [CrossRef]
- Libera, M.; Wałach, W.; Trzebicka, B.; Rangelov, S.; Dworak, A. Thermosensitive dendritic stars of tert-butyl-glycidylether and glycidol—Synthesis and encapsulation properties. Polymer 2011, 52, 3526–3536. [Google Scholar] [CrossRef]
- Libera, M.; Trzebicka, B.; Kowalczuk, A.; Wałach, W.; Dworak, A. Synthesis and thermoresponsive properties of four arm, amphiphilic poly(tert-butyl-glycidylether)-block-polyglycidol stars. Polymer 2011, 52, 250–257. [Google Scholar] [CrossRef]
- Libera, M.; Formanek, P.; Schellkopf, L.; Trzebicka, B.; Dworak, A.; Stamm, M. Amphiphilic dendritic copolymers of tert-butyl-glycidylether and glycidol as a nanocontainer for an anticancer ruthenium complex. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 3488–3497. [Google Scholar] [CrossRef]
- Mendrek, B.; Trzebicka, B. Synthesis and characterization of well-defined poly(tert-butyl acrylate) star polymers. Eur. Polym. J. 2009, 45, 1979–1993. [Google Scholar] [CrossRef]
- Mendrek, B.; Trzebicka, B.; Wałach, W.; Dworak, A. Solution behavior of 4-arm poly(tert-butyl acrylate) star polymers. Eur. Polym. J. 2010, 46, 2341–2351. [Google Scholar] [CrossRef]
- Dworak, A.; Kowalczuk-Bleja, A.; Trzebicka, B.; Walach, W. Amphiphilic core-shell PEO stars by Williamson etherification reaction. Polym. Bull. 2002, 49, 9–16. [Google Scholar] [CrossRef]
- Kowalczuk-Bleja, A.; Sierocka, B.; Trzebicka, B.; Dworak, A. Polimery gwiaździste z rozgałęzionymi rdzeniami poli[p-(halogenometylo)styrenowymi]. Polimery 2005, 50, 555–561. [Google Scholar] [CrossRef]
- Kowalczuk-Bleja, A.; Sierocka, B.; Muszynski, J.; Trzebicka, B.; Dworak, A. Core-shell polyacrylate and polystyrene-block-polyacrylate stars. Polymer 2005, 46, 8555–8564. [Google Scholar] [CrossRef]
- Kowalczuk, A.; Stoyanova, E.; Mitova, V.; Shestakova, P.; Momekov, G.; Momekova, D.; Koseva, N. Star-shaped nano-conjugates of cisplatin with high drug payload. Int. J. Pharm. 2011, 404, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Kowalczuk, A.; Mendrek, B.; Zymełka-Miara, I.; Libera, M.; Marcinkowski, A.; Trzebicka, B.; Smet, M.; Dworak, A. Solution behavior of star polymers with oligo(ethylene glycol) methyl ether methacrylate arms. Polymer 2012, 53, 5619–5631. [Google Scholar] [CrossRef]
- Mendrek, B.; Sieroń, L.; Libera, M.; Smet, M.; Trzebicka, B.; Sieroń, A.L.; Dworak, A.; Kowalczuk, A. Polycationic star polymers with hyperbranched cores for gene delivery. Polymer 2014, 55, 4551–4562. [Google Scholar] [CrossRef]
- Mendrek, B.; Sieroń, L.; Zymełka-Miara, I.; Binkiewicz, P.; Libera, M.; Smet, M.; Trzebicka, B.; Sieroń, A.L.; Kowalczuk, A.; Dworak, A. Nonviral Plasmid DNA Carriers Based on N,N′-Dimethylaminoethyl Methacrylate and Di(ethylene glycol) Methyl Ether Methacrylate Star Copolymers. Biomacromolecules 2015, 16, 3275–3285. [Google Scholar] [CrossRef]
- Mendrek, B. Behavior of methacrylate star copolymers in solutions. Polimery 2016, 61, 413–420. [Google Scholar] [CrossRef]
- Mendrek, B.; Fus, A.; Klarzyńska, K.; Sieroń, A.L.; Smet, M.; Kowalczuk, A.; Dworak, A. Synthesis, characterization and cytotoxicity of novel thermoresponsive star copolymers of N,N’-dimethylaminoethyl methacrylate and hydroxyl-bearing oligo(ethylene glycol) methacrylate. Polymers 2018, 10, 1255. [Google Scholar] [CrossRef] [Green Version]
- Kowalczuk, A.; Vandendriessche, A.; Trzebicka, B.; Mendrek, B.; Szeluga, U.; Cholewiński, G.; Smet, M.; Dworak, A.; Dehaen, W. Core-shell nanoparticles with hyperbranched poly(arylene-oxindole) interiors. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 1120–1135. [Google Scholar] [CrossRef]
- Dworak, A.; Kowalczuk, A.; Mendrek, B.; Trzebicka, B. Star-like polymers of tert-butyl acrylate via controlled radical polymerization—Synthesis and properties. Macromol. Symp. 2011, 308, 93–100. [Google Scholar] [CrossRef]
- Kowalczuk, A.; Trzebicka, B.; Rangelov, S.; Smet, M.; Dworak, A. Star macromolecules with hyperbranched poly(arylene oxindole) cores and polyacid arms: Synthesis and solution behavior. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 5074–5086. [Google Scholar] [CrossRef]
- Dworak, A.; Trzebicka, B.; Kowalczuk, A.; Utrata-Wesołek, A.; Wałach, W.; Libera, M.; Kronek, J. Termoczułe polimery gwieździste—Synteza i właściwości. Polimery 2012, 57, 441–448. [Google Scholar] [CrossRef]
- Mendrek, B.; Zymełka-Miara, I.; Sieroń, Ł.; Fus, A.; Balin, K.; Kubacki, J.; Smet, M.; Trzebicka, B.; Sieroń, A.L.; Kowalczuk, A. Stable star polymer nanolayers and their thermoresponsiveness as a tool for controlled culture and detachment of fibroblast sheets. J. Mater. Chem. B 2018, 6, 641–655. [Google Scholar] [CrossRef]
- Donev, R.; Koseva, N.; Petrov, P.; Kowalczuk, A.; Thome, J. Characterisation of different nanoparticles with a potential use for drug delivery in neuropsychiatric disorders. World J. Biol. Psychiatry 2011, 12, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, E.; Mitova, V.; Shestakova, P.; Kowalczuk, A.; Momekov, G.; Momekova, D.; Marcinkowski, A.; Koseva, N. Reversibly PEGylated nanocarrier for cisplatin delivery. J. Inorg. Biochem. 2013, 120, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Antonova, N.; Koseva, N.; Kowalczuk, A.; Riha, P.; Ivanov, I. Rheological and electrical properties of polymeric nanoparticle solutions and their influence on RBC suspensions. Appl. Rheol. 2014, 24, 25–31. [Google Scholar] [CrossRef]
- Georgiou, T.K. Star polymers for gene delivery. Polym. Int. 2014, 63, 1130–1133. [Google Scholar] [CrossRef] [Green Version]
- Fus-Kujawa, A.; Teper, P.; Botor, M.; Klarzyńska, K.; Sieroń, Ł.; Verbelen, B.; Smet, M.; Sieroń, A.L.; Mendrek, B.; Kowalczuk, A. Functional star polymers as reagents for efficient nucleic acids delivery into HT-1080 cells. Int. J. Polym. Mater. Polym. Biomater. 2021, 70, 356–370. [Google Scholar] [CrossRef]
- Teper, P.; Sotirova, A.; Mitova, V.; Oleszko-Torbus, N.; Utrata-Wesołek, A.; Koseva, N.; Kowalczuk, A.; Mendrek, B. Antimicrobial Activity of Hybrid Nanomaterials Based on Star and Linear Polymers of N,N′-Dimethylaminoethyl Methacrylate with In Situ Produced Silver Nanoparticles. Materials 2020, 13, 3037. [Google Scholar] [CrossRef]
- Mendrek, B.; Fus-Kujawa, A.; Teper, P.; Botor, M.; Kubacki, J.; Sieroń, A.L.; Kowalczuk, A. Star polymer-based nanolayers with immobilized complexes of polycationic stars and DNA for deposition gene delivery and recovery of intact transfected cells. Int. J. Pharm. 2020, 589. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, P.; Utrata-Wesołek, A.; Rangelov, S.; Wałach, W.; Trzebicka, B.; Dworak, A. Synthesis and self-association in aqueous media of poly(ethylene oxide)/poly(ethyl glycidyl carbamate) amphiphilic block copolymers. Polymer 2006, 47, 4905–4915. [Google Scholar] [CrossRef]
- Dimitrov, P.; Jamróz-Piegza, M.; Trzebicka, B.; Dworak, A. The influence of hydrophobic substitution on self-association of poly(ethylene oxide)-b-poly(n-alkyl glycidyl carbamate)s-b-poly(ethylene oxide) triblock copolymers in aqueous media. Polymer 2007, 48, 1866–1874. [Google Scholar] [CrossRef]
- Jamróz-Piegza, M.; Wałach, W.; Dworak, A.; Trzebicka, B. Polyether nanoparticles from covalently crosslinked copolymer micelles. J. Colloid Interface Sci. 2008, 325, 141–148. [Google Scholar] [CrossRef]
- Trzebicka, B.; Koseva, N.; Mitova, V.; Dworak, A. Organization of poly(2-ethyl-2-oxazoline)-block-poly(2-phenyl-2-oxazoline) copolymers in water solution. Polymer 2010, 51, 2486–2493. [Google Scholar] [CrossRef]
- Otulakowski, Ł.; Gadzinowski, M.; Slomkowski, S.; Basinska, T.; Forys, A.; Dworak, A.; Trzebicka, B. Micellisation of polystyrene-b-polyglycidol copolymers in water solution. Eur. Polym. J. 2018, 99, 72–79. [Google Scholar] [CrossRef]
- Otulakowski, L.; Dworak, A.; Forys, A.; Gadzinowski, M.; Slomkowski, S.; Basinska, T.; Trzebicka, B. Micellization of polystyrene-b-polyglycidol in dioxane and water/dioxane solutions. Polymers 2020, 12, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendrek, S.; Mendrek, A.; Adler, H.-J.; Walach, W.; Dworak, A.; Kuckling, D. Synthesis of poly(glycidol)-block-poly(N -isopropylacrylamide) copolymers using new hydrophilic poly(glycidol) macroinitiator. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 2488–2499. [Google Scholar] [CrossRef]
- Mendrek, S.; Mendrek, A.; Adler, H.J.; Dworak, A.; Kuckling, D. Temperature-sensitive behaviour of poly(glycidol)-b-poly(N-isopropylacrylamide) block copolymers. Colloid Polym. Sci. 2010, 288, 777–786. [Google Scholar] [CrossRef]
- Mendrek, S.; Mendrek, A.; Adler, H.J.; Dworak, A.; Kuckling, D. Preparation of temperature-sensitive core-shell poly(glycidol)/poly(N-isopropylacrylamide) nanohydrogels under surfactant-free conditions. Macromolecules 2009, 42, 9161–9169. [Google Scholar] [CrossRef]
- Mendrek, S.; Mendrek, A.; Adler, H.; Dworak, A.; Kuckling, D. Synthesis and characterization of pH sensitive poly(glycidol)- b -poly(4-vinylpyridine) block copolymers. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 1782–1794. [Google Scholar] [CrossRef]
- Oleszko-Torbus, N.; Bochenek, M.; Utrata-Wesołek, A.; Kowalczuk, A.; Marcinkowski, A.; Dworak, A.; Fus-Kujawa, A.; Sieroń, A.L.; Wałach, W. Poly(2-oxazoline) matrices with temperature-dependent solubility-interactions with water and use for cell culture. Materials 2020, 13, 2702. [Google Scholar] [CrossRef]
- Oleszko-Torbus, N.; Utrata-Wesołek, A.; Wałach, W.; Dworak, A. Solution behavior of thermoresponsive random and gradient copolymers of 2-n-propyl-2-oxazoline. Eur. Polym. J. 2017, 88, 613–622. [Google Scholar] [CrossRef]
- Adamus, A.; Komasa, J.; Kadłubowski, S.; Ulański, P.; Rosiak, J.M.; Kawecki, M.; Klama-Baryła, A.; Dworak, A.; Trzebicka, B.; Szweda, R. Thermoresponsive poly[tri(ethylene glycol) monoethyl ether methacrylate]-peptide surfaces obtained by radiation grafting-synthesis and characterisation. Colloids Surf. B Biointerfaces 2016, 145, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Dworak, A.; Utrata-Wesołek, A.; Szweda, D.; Kowalczuk, A.; Trzebicka, B.; Anioł, J.; Sieroń, A.L.; Klama-Baryła, A.; Kawecki, M. Poly[tri(ethylene glycol) ethyl ether methacrylate]-coated surfaces for controlled fibroblasts culturing. ACS Appl. Mater. Interfaces 2013, 5, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Kawecki, M.; Kraut, M.; Klama-Baryła, A.; Łabuś, W.; Kitala, D.; Nowak, M.; Glik, J.; Sieroń, A.L.; Utrata-Wesołek, A.; Trzebicka, B.; et al. Transfer of fibroblast sheets cultured on thermoresponsive dishes with membranes. J. Mater. Sci. Mater. Med. 2016, 27. [Google Scholar] [CrossRef] [Green Version]
- Lesiak, M.; Sieroń, Ł.; Gutmajster, E.; Kowalczuk, A.; Bochenek, M.; Utrata-Wesołek, A.; Dworak, A.; Trzebicka, B.; Klama-Baryła, A.; Glik, J.; et al. Fibroblast and keratinocyte crosstalk: The effect of a poly(tri[ethylene glycol] ethyl ether methacrylate) thermoresponsive surface on short-term co-culture. Eur. Dermatol. EJD 2019, 29, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Kitala, D.; Klama-Baryła, A.; Kraut, M.; Łabuś, W.; Glik, J.; Kawecki, M.; Trzebicka, B.; Dworak, A.; Adamus-Włodarczyk, A.; Komasa, J.; et al. Amniotic Stem Cells Cultured on Thermoresponsive Polymers Allow Obtaining a Full Cell Sheet. Transplant. Proc. 2020, 52, 2198–2203. [Google Scholar] [CrossRef]
- Utrata-Wesołek, A.; Wałach, W.; Anioł, J.; Sieroń, A.L.; Dworak, A. Multiple and terminal grafting of linear polyglycidol for surfaces of reduced protein adsorption. Polymer 2016, 97, 44–54. [Google Scholar] [CrossRef]
- Utrata-Wesołek, A.; Oleszko, N.; Trzebicka, B.; Anioł, J.; Zagdańska, M.; Lesiak, M.; Sieroń, A.; Dworak, A. Modified polyglycidol based nanolayers of switchable philicity and their interactions with skin cells. Eur. Polym. J. 2013, 49, 106–117. [Google Scholar] [CrossRef]
- Dworak, A.; Trzebicka, B.; Walach, W.; Utrata, A. Nowe termowrażliwe reaktywne polietery oparte na poliglicydolu. Polimery 2003, 48, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Dworak, A.; Trzebicka, B.; Utrata, A.; Wałach, W. Hydrophobically modified polyglycidol—The control of lower critical solution temperature. Polym. Bull. 2003, 50, 47–54. [Google Scholar] [CrossRef]
- Dworak, A.; Trzebicka, B.; Wałach, W.; Utrata, A.; Tsvetanov, C. Novel reactive thermosensitive polyethers—Control of transition point. Macromol. Symp. 2004, 210, 419–426. [Google Scholar] [CrossRef]
- Jamróz-Piegza, M.; Utrata-Wesołek, A.; Trzebicka, B.; Dworak, A. Hydrophobic modification of high molar mass polyglycidol to thermosensitive polymers. Eur. Polym. J. 2006, 42, 2497–2506. [Google Scholar] [CrossRef]
- Dworak, A.; Utrata-Wesołek, A.; Oleszko, N.; Wałach, W.; Trzebicka, B.; Anioł, J.; Sieroń, A.L.; Klama-Baryła, A.; Kawecki, M. Poly(2-substituted-2-oxazoline) surfaces for dermal fibroblasts adhesion and detachment. J. Mater. Sci. Mater. Med. 2014, 25, 1149–1163. [Google Scholar] [CrossRef]
- Oleszko, N.; Wałach, W.; Utrata-Wesołek, A.; Kowalczuk, A.; Trzebicka, B.; Klama-Baryła, A.; Hoff-Lenczewska, D.; Kawecki, M.; Lesiak, M.; Sieroń, A.L.; et al. Controlling the crystallinity of thermoresponsive poly(2-oxazoline)-based nanolayers to cell adhesion and detachment. Biomacromolecules 2015, 16, 2805–2813. [Google Scholar] [CrossRef]
- Güner, P.T.; Mikó, A.; Schweinberger, F.F.; Demirel, A.L. Self-assembled poly(2-ethyl-2-oxazoline) fibers in aqueous solutions. Polym. Chem. 2012, 3, 322–324. [Google Scholar] [CrossRef] [Green Version]
- Demirel, A.L.; Meyer, M.; Schlaad, H. Formation of polyamide nanofibers by directional crystallization in aqueous solution. Angew. Chem. Int. Ed. 2007, 46, 8622–8624. [Google Scholar] [CrossRef]
- Demirel, A.L.; Tatar Güner, P.; Verbraeken, B.; Schlaad, H.; Schubert, U.S.; Hoogenboom, R. Revisiting the crystallization of poly(2-alkyl-2-oxazoline)s. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 721–729. [Google Scholar] [CrossRef]
- Oleszko-Torbus, N.; Utrata-Wesołek, A.; Bochenek, M.; Lipowska-Kur, D.; Dworak, A.; Wałach, W. Thermal and crystalline properties of poly(2-oxazoline)s. Polym. Chem. 2020, 11, 15–33. [Google Scholar] [CrossRef]
- Oleszko, N.; Utrata-Wesołek, A.; Wałach, W.; Libera, M.; Hercog, A.; Szeluga, U.; Domański, M.; Trzebicka, B.; Dworak, A. Crystallization of poly(2-isopropyl-2-oxazoline) in organic solutions. Macromolecules 2015, 48, 1852–1859. [Google Scholar] [CrossRef]
- Oleszko-Torbus, N.; Mendrek, B.; Kowalczuk, A.; Utrata-Wesołek, A.; Dworak, A.; Wałach, W. Selective partial hydrolysis of 2-isopropyl-2-oxazoline copolymers towards decreasing the ability to crystallize. Materials 2020, 13, 3403. [Google Scholar] [CrossRef]
- Oleszko-Torbus, N.; Wałach, W.; Utrata-Wesołek, A.; Dworak, A. Control of the Crystalline Properties of 2-Isopropyl-2-oxazoline Copolymers in Condensed State and in Solution Depending on the Composition. Macromolecules 2017, 50, 7636–7645. [Google Scholar] [CrossRef]
- Wałach, W.; Klama-Baryła, A.; Sitkowska, A.; Kowalczuk, A.; Oleszko-Torbus, N. Alternative to poly(2-isopropyl-2-oxazoline) with a reduced ability to crystallize and physiological lcst. Int. J. Mol. Sci. 2021, 22, 2221. [Google Scholar] [CrossRef]
- Trzcinska, R.; Suder, P.; Bodzon-Kulakowska, A.; Skalska, M.; Marcinkowski, A.; Kubacki, J.; Pedrys, R.; Silberring, J.; Dworak, A.; Trzebicka, B. Synthesis and characterisation of PEG-peptide surfaces for proteolytic enzyme detection. Anal. Bioanal. Chem. 2013, 405, 9049–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzcinska, R.; Balin, K.; Kubacki, J.; Marzec, M.E.; Pedrys, R.; Szade, J.; Silberring, J.; Dworak, A.; Trzebicka, B. Relevance of the poly(ethylene glycol) linkers in peptide surfaces for proteases assays. Langmuir 2014, 30, 5015–5025. [Google Scholar] [CrossRef] [PubMed]
- Teper, P.; Chojniak-Gronek, J.; Hercog, A.; Oleszko-Torbus, N.; Płaza, G.; Kubacki, J.; Balin, K.; Kowalczuk, A.; Mendrek, B. Nanolayers of poly(N, N’-dimethylaminoethyl methacrylate) with a star topology and their antibacterial activity. Polymers 2020, 12, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utrata-Wesołek, A.; Wałach, W.; Bochenek, M.; Trzebicka, B.; Anioł, J.; Sieroń, A.L.; Kubacki, J.; Dworak, A. Branched polyglycidol and its derivatives grafted-from poly(ethylene terephthalate) and silica as surfaces that reduce protein fouling. Eur. Polym. J. 2018, 105, 313–322. [Google Scholar] [CrossRef]
- Christova, D.; Ivanova, S.; Trzebicka, B.; Walach, W.; Velichkova, R. Dwor Glutaraldehyde-crosslinked poly(glycidol-block-ethylene oxide- block- glycidol ) networks with temperature- responsive swelling behaviour. e-Polymers 2003, 42, 1–11. [Google Scholar]
- Utrata-Wesołek, A.; Trzebicka, B.; Dworak, A.; Ivanova, S.; Christova, D. Thermoresponsive hydrogels of hydrophobically modified polyglycidol. e-Polymers 2007, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Petrov, P.; Utrata-Wesołek, A.; Trzebicka, B.; Tsvetanov, C.B.; Dworak, A.; Anioł, J.; Sieroń, A. Biocompatible cryogels of thermosensitive polyglycidol derivatives with ultra-rapid swelling properties. Eur. Polym. J. 2011, 47, 981–988. [Google Scholar] [CrossRef]
- Utrata-Wesołek, A.; Żymełka-Miara, I.; Kowalczuk, A.; Trzebicka, B.; Dworak, A. Photocrosslinking of Polyglycidol and Its Derivative: Route to Thermoresponsive Hydrogels. Photochem. Photobiol. 2018, 94, 52–60. [Google Scholar] [CrossRef]
- Walach, W.; Oleszko-Torbus, N.; Utrata-Wesolek, A.; Bochenek, M.; Kijeńska-Gawrońska, E.; Górecka, Z.; Świȩszkowski, W.; Dworak, A. Processing of (Co)poly(2-oxazoline)s by electrospinning and extrusion from melt and the postprocessing properties of the (co)polymers. Polymers 2020, 12, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Abbreviation | Polymer Specification | Mn (g/mol) | CP (g/L) | Additive * s/p | Rh (nm) | Heating Mode | Ref. |
|---|---|---|---|---|---|---|---|
| POEGMA | P(T-ran-O475)_4.5 | 48,600 | 0.1 | - | 430 | gradual | [16] |
| 0.5 | - | 875 | |||||
| 0.1 | - | 95 | abrupt (54 °C) | ||||
| P(T-ran-O475)_30.5 | 46,800 | 0.1 | - | 800 | gradual | ||
| 0.5 | - | 1185 | |||||
| 0.1 | - | 101 | abrupt (81 °C) | ||||
| P(D-ran-O300)_37 | 32,000 | 0.1 | - | 465 | gradual | ||
| 0.5 | - | 1080 | |||||
| 0.1 | - | 96 | abrupt (81 °C) | ||||
| P(HEMA-OEGMA) | - | 33,000 | 0.5 | - | 740 | gradual | [29,30] |
| NaCl | 1425 | ||||||
| PBS | 1540 | ||||||
| - | 230 | abrupt (70 °C) | |||||
| NaCl | 1600 | ||||||
| PBS | 1330 | ||||||
| PETEGA | - | 7000–40,000 | 0.5 | - | 177 | abrupt (40 °C) | [21,31] |
| - | 91 | abrupt (70 °C) | |||||
| 0.5 | 0.1 | 110 | abrupt (40 °C) | ||||
| 0.1 | 70 | abrupt (70 °C) | |||||
| 0.5 | 0.4 | n.d | abrupt (40 °C) | ||||
| 0.4 | 10/71 | abrupt (70 °C) | |||||
| - | P(D-co-A_A) 7% A | 42,000 | 0.2 | - | 62 | gradual | [25,26] |
| 0.5 | - | 77 | |||||
| P(D-co-A_Pr) 7% Pr | 42,000 | 0.1 | - | 330 | gradual | ||
| 0.5 | - | 620 | |||||
| 0.1 | - | 80 | abrupt | ||||
| P(D-co-A_Az) 7% Az | 42,000 | 0.1 | - | 130 | gradual | ||
| 0.5 | - | 215 | |||||
| 0.1 | - | 20 | abrupt | ||||
| PIPOx | - | 3660 | 0.5 | 0.5 | 403 | abrupt | [22] |
| - | 494 | abrupt (80 °C) | |||||
| - | 700 | gradual | |||||
| 5540 | 0.5 | 0.5 | 370 | abrupt (70 °C) | |||
| - | 650 | abrupt | |||||
| - | 800 | gradual | |||||
| 8940 | 0.5 | 0.5 | 180 | abrupt (70 °C) | |||
| - | 800 | ||||||
| - | 1400 | gradual | |||||
| mPGL | P(G-co-EGC)-28 | 800,000 | 0.05 | 175 | abrupt (80 °C) | [17] | |
| 0.2 | 260 | ||||||
| 175 | dropwise (80 °C) | ||||||
| P(G-co-EGC)-35 | 0.05 | 112 | abrupt (80 °C) | ||||
| 0.2 | 100 | ||||||
| 100 | dropwise (80 °C) | ||||||
| PNIPAM | 84,000 | 1 | 0.05 | 45 | abrupt (70 °C) | [20] | |
| 0.5 | 10 |
| Core | Number of Arms | Arms | Properties/Application | Ref. |
|---|---|---|---|---|
| poly[p-(chloromethyl)styrene] | 12–26 | PEG | amphiphilic | [52] |
| poly[p-(iodomethyl)styrene] | 10 | PS, PtBuAc, PAA | PtBuAc stars: thermal properties PAA stars: amphiphilic/formation of reversible complexes between COOH groups of stars and model drug: cisplatin neurotoxicity evaluation electrical and rheological properties of star solutions | [38,53,54,55,66,67,68] |
| polyglycidol and dipentaerythritol | 6, 13 | PEOx | thermoresponsive | [46,64] |
| pentaerythritol derivatives | 4, 6 | tert-butyl glycidyl ether, glycidol | amphiphilic, thermoresponsive pyrene encapsulation formation of reversible complexes between OH groups of stars and Ru(NH3)3Cl3 | [47,48,49] |
| aliphatic alcohols and calix[4]arenes | 3, 4, 6, 12, 16 | PtBuAc | branching parameters, scaling equations | [50,51] |
| PArOx | 20, 22 | P(DEGMA-co-OEGMA) | amphiphilic, thermoresponsive, nontoxic/encapsulation of fluorescent probe | [56,65] |
| PArOx | 28 | PtBuAc, PtBuMAc, PAA and PMA | polyacid stars: pH responsive | [61,62,63] |
| PArOx | 28 | PDMAEMA | thermo- and pH responsive, nontoxic/gene delivery | [57,59] |
| PArOx | 28 | PDMAEMA with AgNPs | antibacterial agents | [71] |
| PArOx | 28 | P(DMAEMA-co-DEGMA) P(DMAEMA-co-OEGMA-OH) | thermo- and pH responsive, nontoxic, gene delivery | [58,59,60,70,72] |
| Polymer | Mn [g/mol] | Support | Type of the Layer | Thickness of the Layer | Grafting Density | Application | Ref. |
|---|---|---|---|---|---|---|---|
| grafting to | |||||||
| PGL | 1,900,000 | silica | interpenetrating polymer chains | 15–120 nm | - | reduce fibrinogen adsorption | [90] |
| PGL | 8000 | silica | interpenetrating polymer chains | 7–140 nm | - | reduce fibrinogen adsorption | [90] |
| PGL | 8000 | silica | brushes | 1.1–1.5 nm (+/−0.3 nm) | 0.083–0.113 chains/nm2 | reduce fibrinogen adsorption | [90] |
| P(Gl-EO) | 6000 | silica | brushes | 1.7–2.3 nm (+/−0.3 nm) | 0.17–0.23 chains/nm2 | reduce fibrinogen adsorption | [90] |
| mPGL | 2,200,000 | silica, glass | interpenetrating polymer chains | 20–60 nm | - | the growth of fibroblasts and keratinocytes | [91] |
| P(EtOx-NonOx) | 14,000–21,800 | glass | brushes | 4–8 nm | 0.19–0.22 chains/nm2 | the growth and harvesting of fibroblasts | [96] |
| PIPOx | 20,800–42,000 | silica, glass | brushes | 5–11 nm | 0.16–0.26 chains/nm2 | the growth and harvesting of fibroblasts | [96,97] |
| PEG | 576–1545 | silica | brushes | - | 2.88–3.54 pmol/mm2 | proteolytic enzyme detection | [106,107] |
| P(DEGMA-ran-OEGMA-ran-GMA) | 380,000 | silica, glass | star polymer | 58 nm | - | the growth and harvesting of fibroblasts | [65] |
| P(DEGMA-OEGMA) | 417,000 | silica | star polymer | 30 nm | - | deposition gene delivery and the growth and harvesting of HT-1080 | [72] |
| PDMAEMA | 9000–1,000,000 | silica, glass | linear and star polymer | 3–100 nm | - | antibacterial | [108] |
| grafting from | |||||||
| P(TEGMA-EE) | 23,000–189,000 | silica, glass | brushes | 3–18 nm | 0.1 chain/nm2 | the growth and harvesting of fibroblasts and coculture of fibroblasts and keratinocytes | [86,87,88] |
| P(TEGMA-EE) | - | polypropylene | crosslinked layer | >10 μm | 1.15 +/−0.03 mg/cm2 | the growth and harvesting of fibroblasts and amniotic stem cells | [85,89] |
| PGL | 1600–290,000 | silica, PET | hyperbranched | 4–5 nm | reduce fibrinogen adsorption | [109] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oleszko-Torbus, N.; Mendrek, B.; Kowalczuk, A.; Wałach, W.; Trzebicka, B.; Utrata-Wesołek, A. The Role of Polymer Structure in Formation of Various Nano- and Microstructural Materials: 30 Years of Research in the Laboratory of Nano- and Microstructural Materials at the Centre of Polymer and Carbon Materials PAS. Polymers 2021, 13, 2892. https://doi.org/10.3390/polym13172892
Oleszko-Torbus N, Mendrek B, Kowalczuk A, Wałach W, Trzebicka B, Utrata-Wesołek A. The Role of Polymer Structure in Formation of Various Nano- and Microstructural Materials: 30 Years of Research in the Laboratory of Nano- and Microstructural Materials at the Centre of Polymer and Carbon Materials PAS. Polymers. 2021; 13(17):2892. https://doi.org/10.3390/polym13172892
Chicago/Turabian StyleOleszko-Torbus, Natalia, Barbara Mendrek, Agnieszka Kowalczuk, Wojciech Wałach, Barbara Trzebicka, and Alicja Utrata-Wesołek. 2021. "The Role of Polymer Structure in Formation of Various Nano- and Microstructural Materials: 30 Years of Research in the Laboratory of Nano- and Microstructural Materials at the Centre of Polymer and Carbon Materials PAS" Polymers 13, no. 17: 2892. https://doi.org/10.3390/polym13172892