Intermolecular Interactions in Polyelectrolyte and Surfactant Complexes in Solution


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
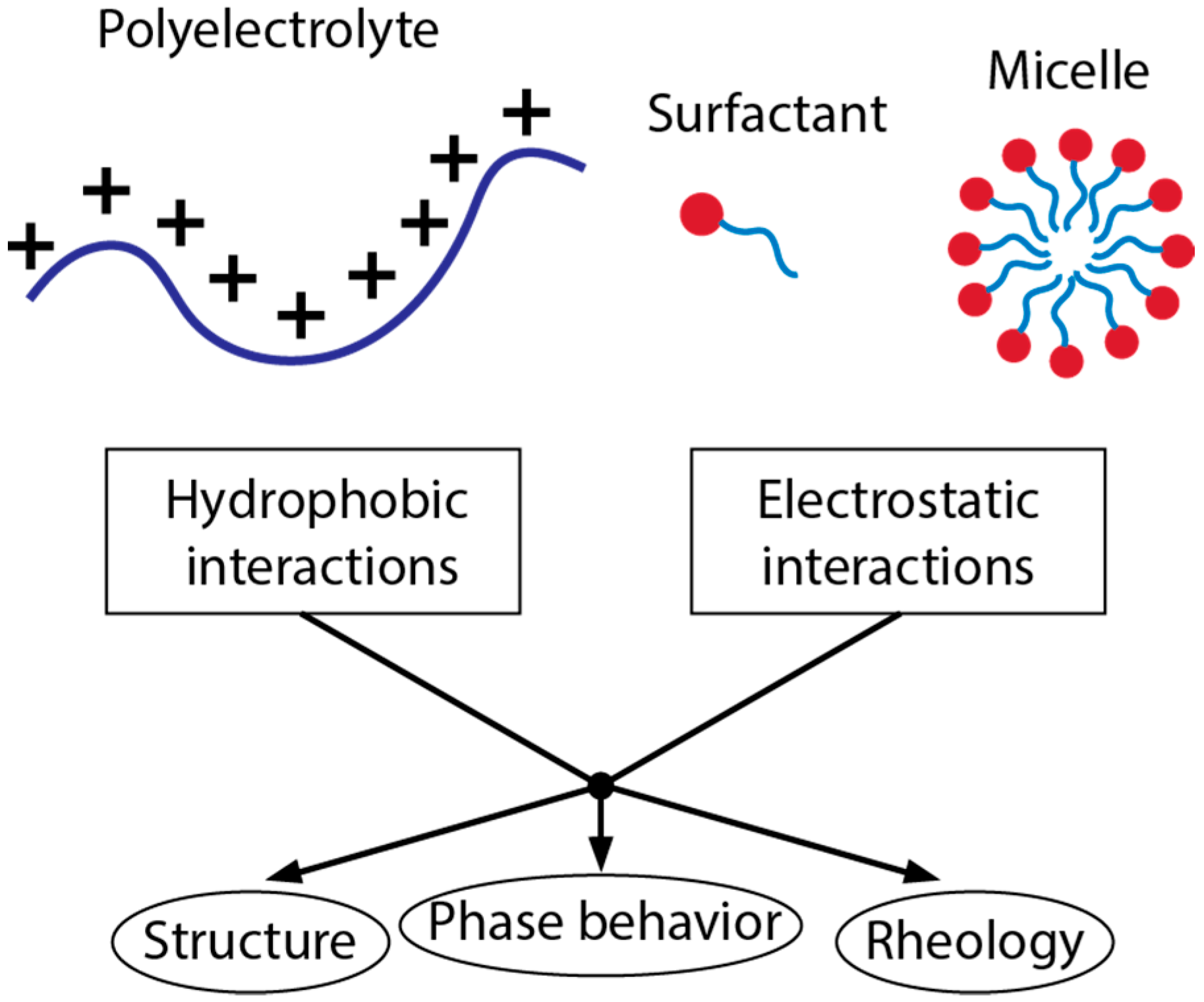
1. Introduction
2. Intermolecular Interactions
2.1. Hydrophobic
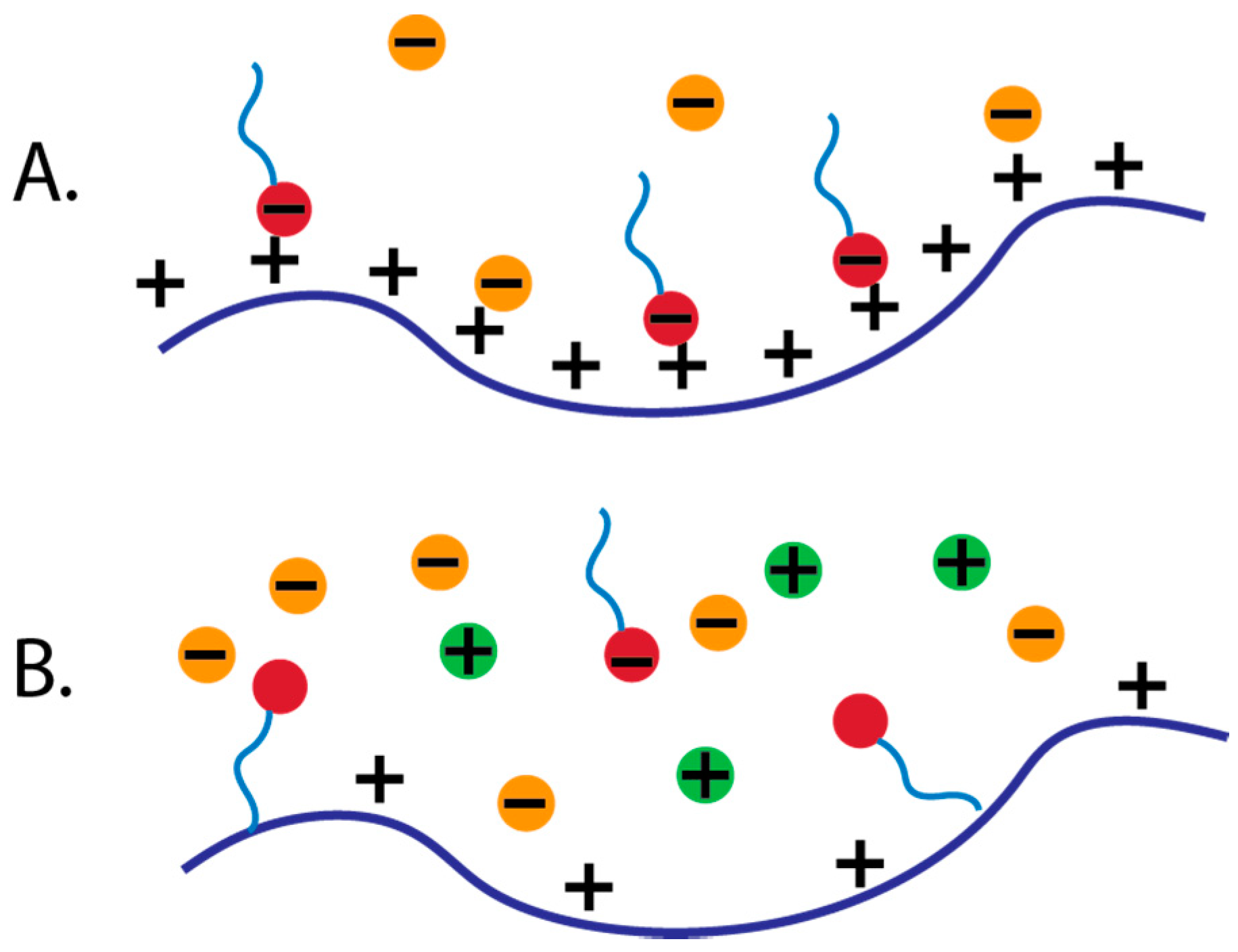
2.2. Electrostatic
2.3. Balance of Electrostatic and Hydrophobic Interactions
3. Interactions of Polyelectrolytes and Surfactants in the Bulk Solution
3.1. Formation of Complexes
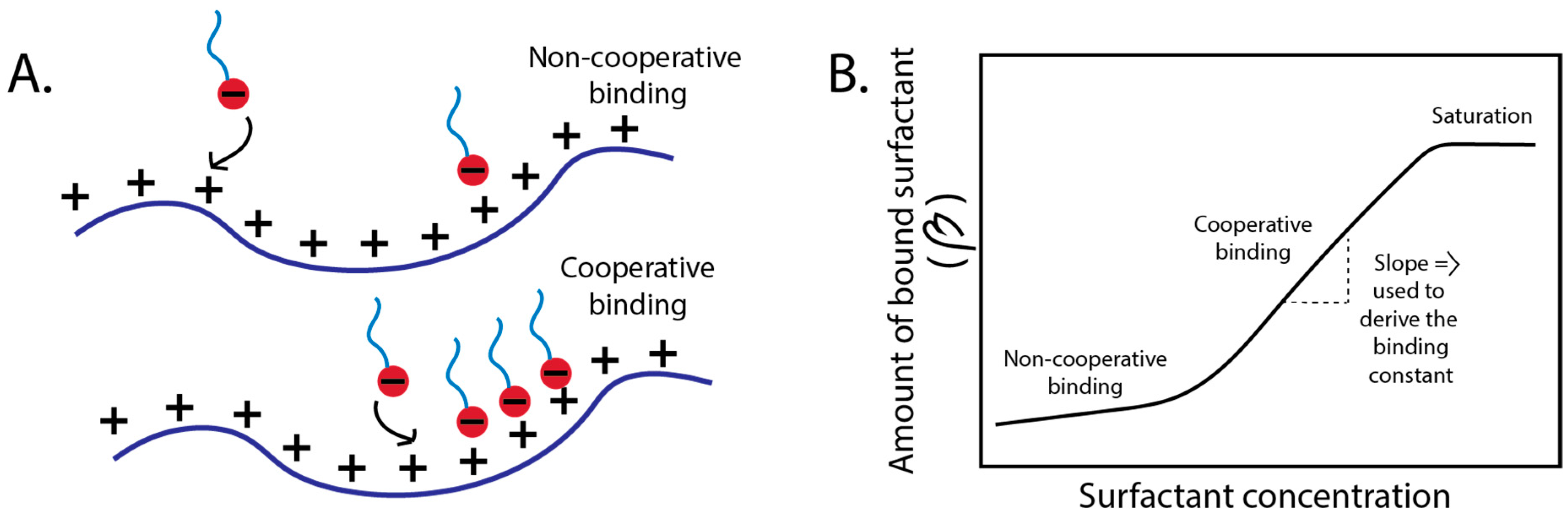
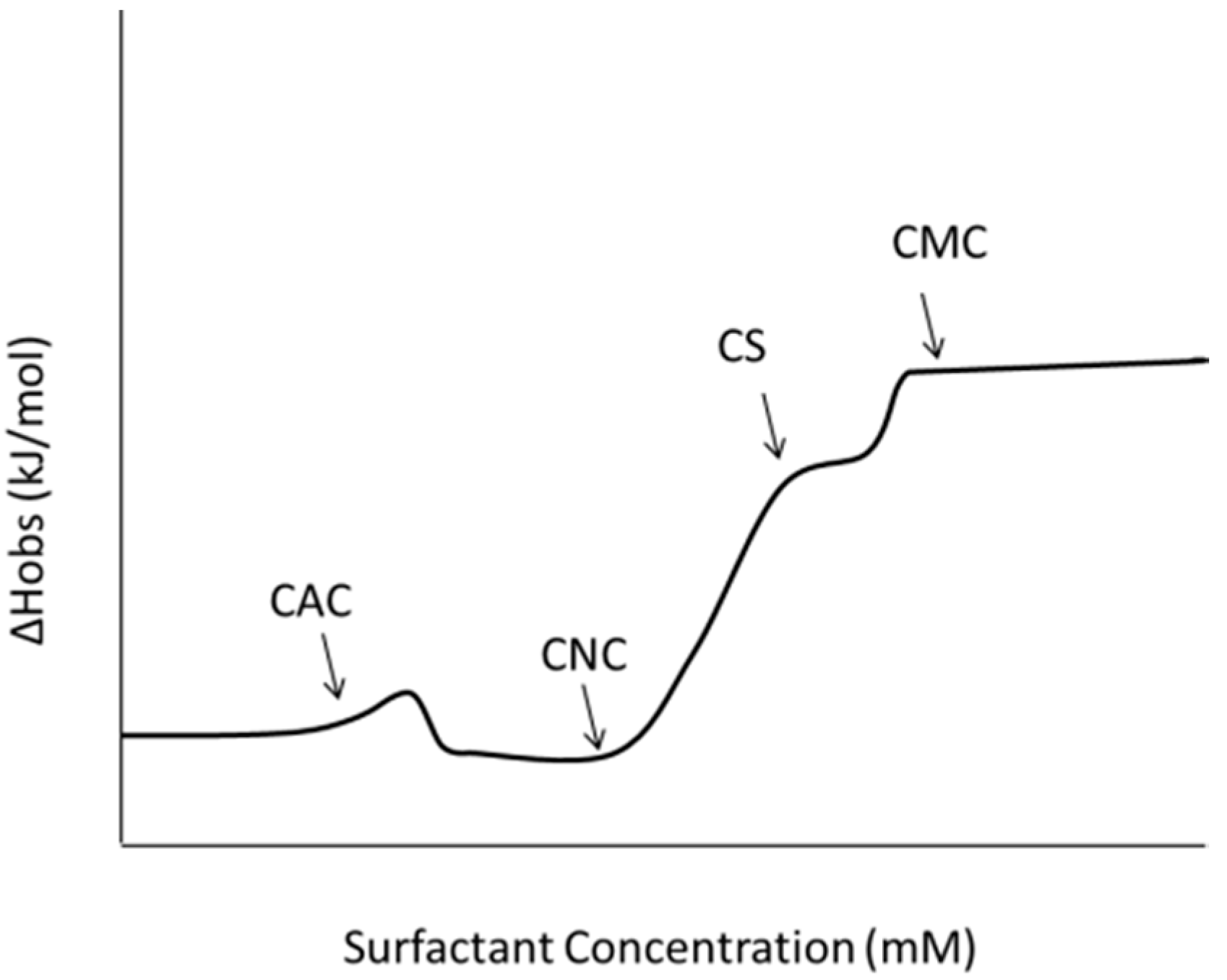
3.1.1. Binding Mechanisms—Cooperative, and Non-Cooperative
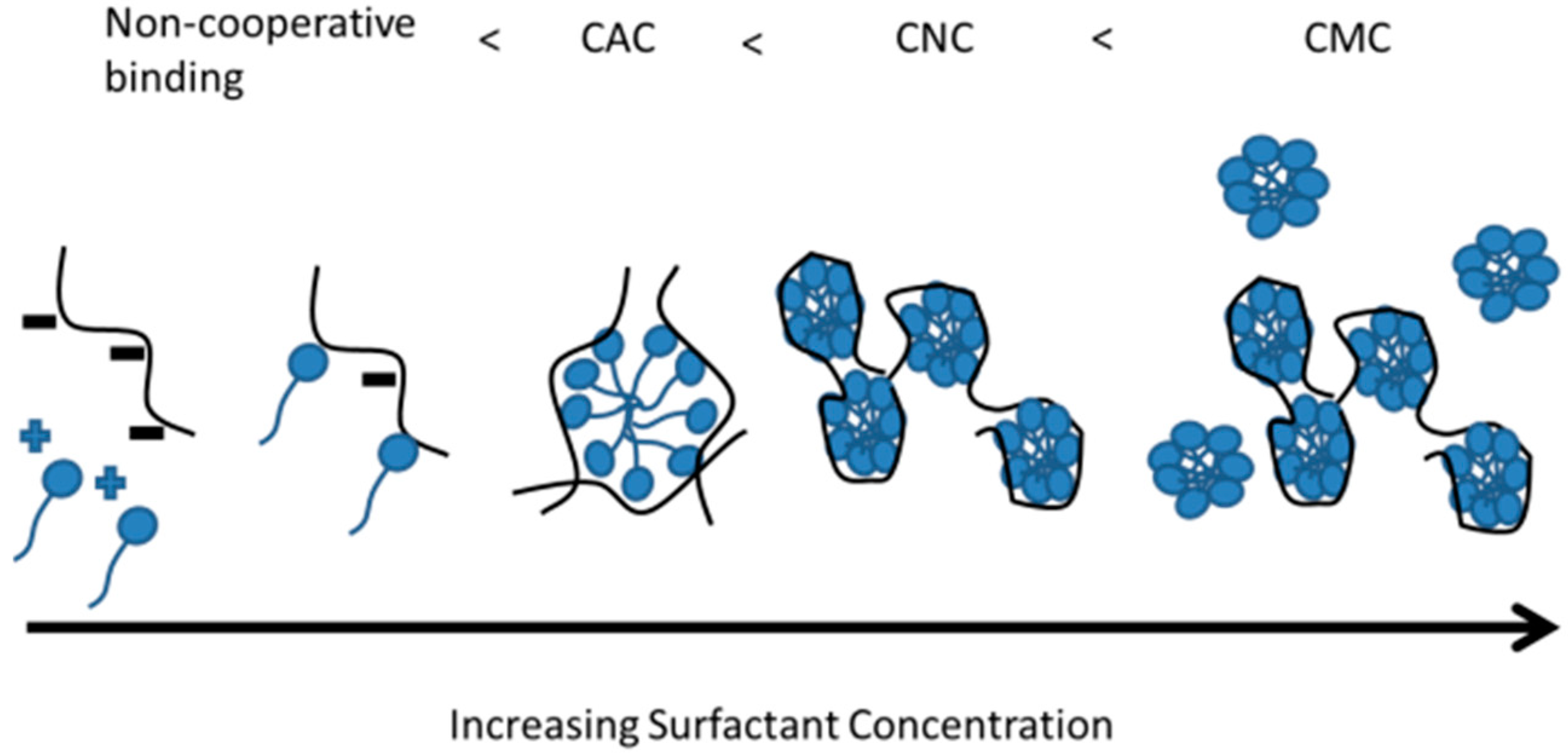
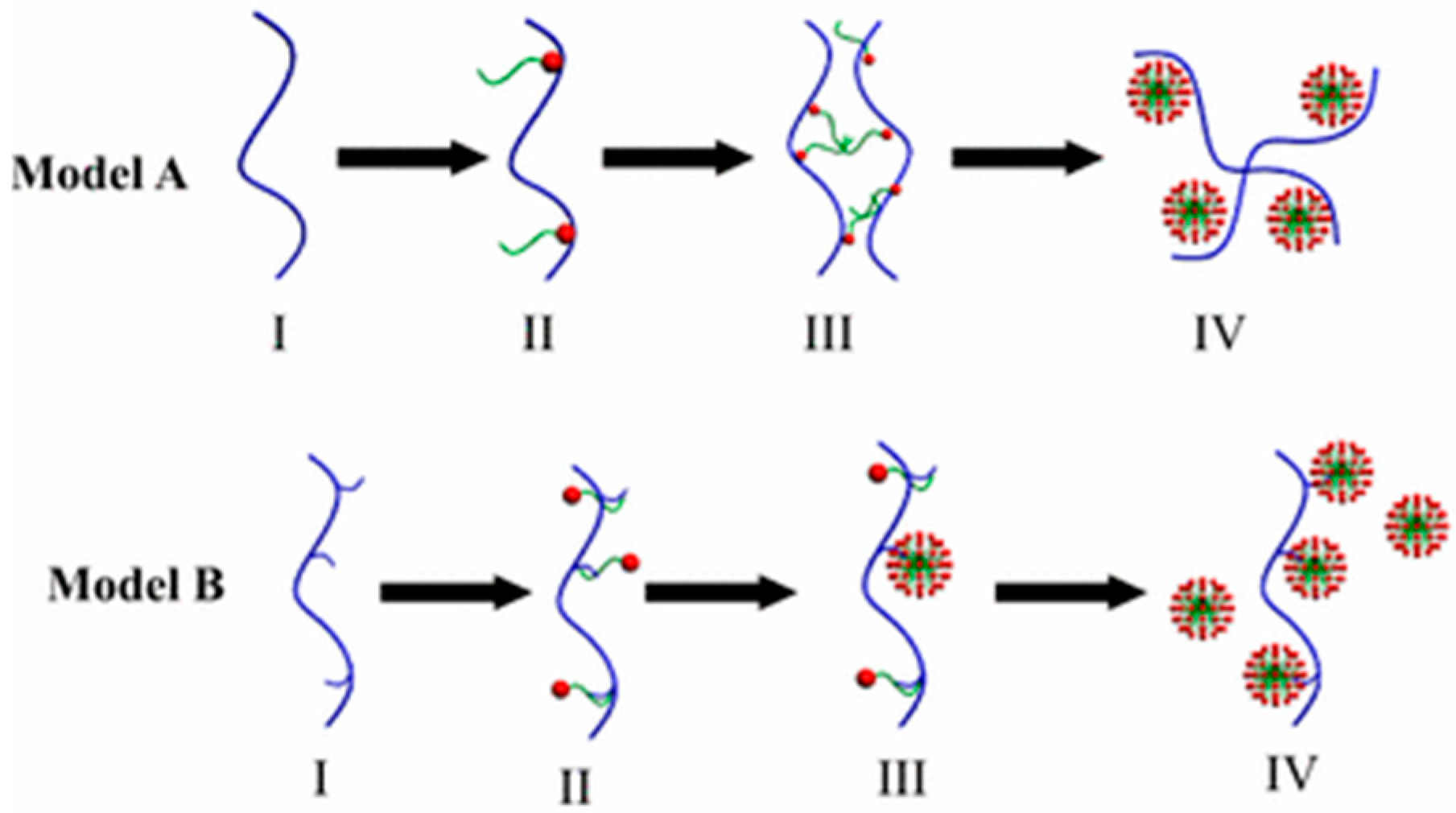
3.1.2. Formation of Micelle-Like Structures
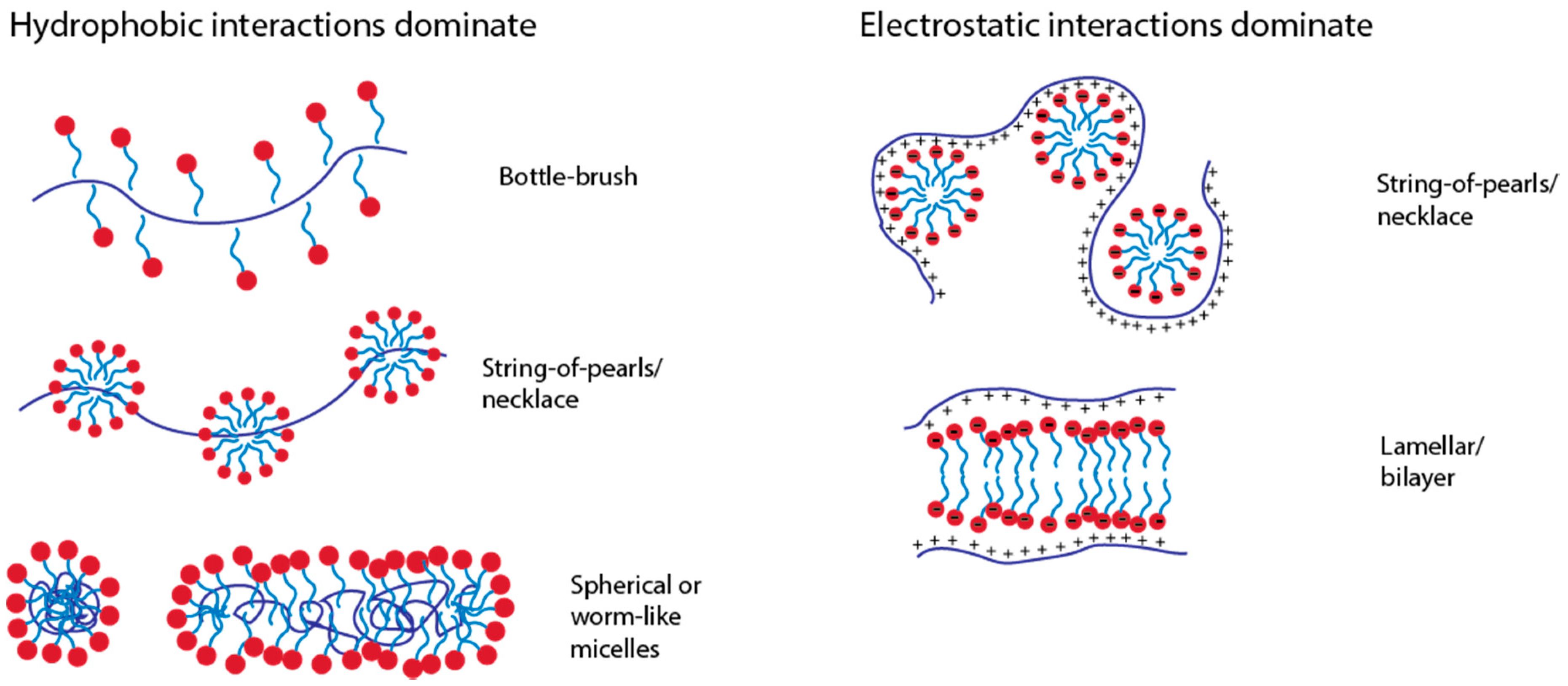
3.1.3. Types of Structures Formed and PE-Micelle Interactions
3.2. Experimental Factors Affecting Complexation
3.2.1. Concentration and Stoichiometry
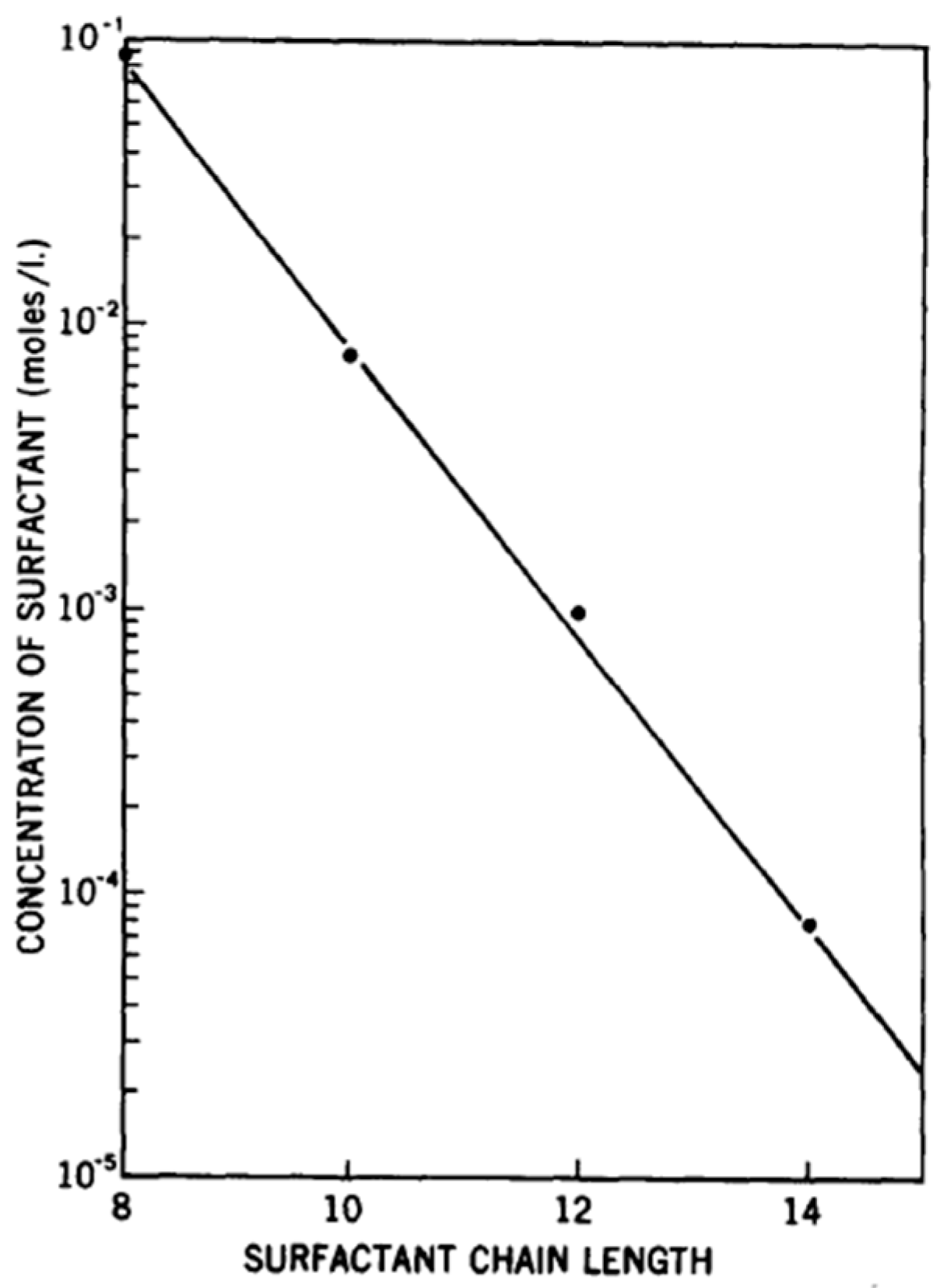
3.2.2. Surfactant Tail Length and Polyelectrolyte Molecular Weight
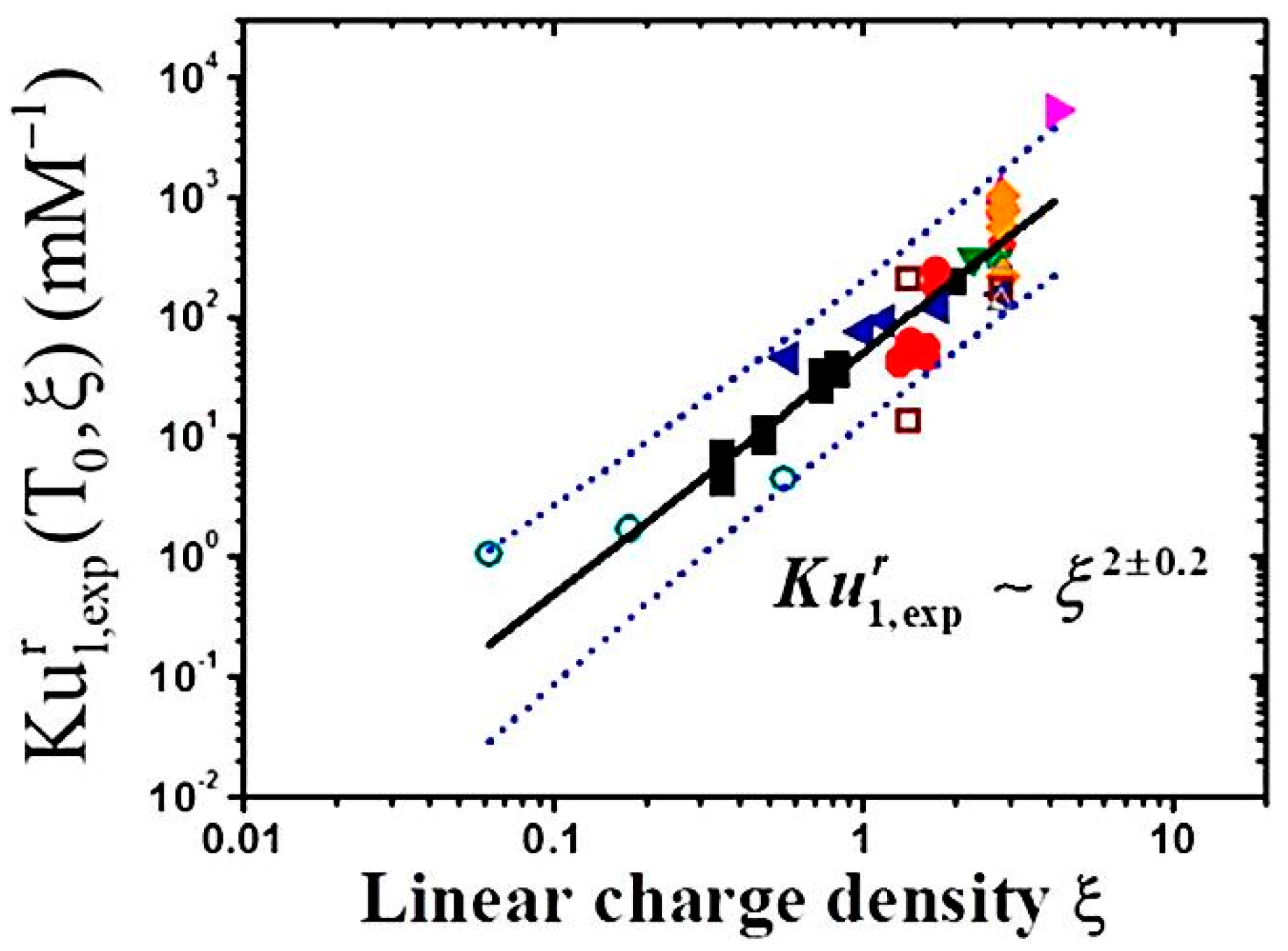
3.2.3. Linear Charge Density and Chain Flexibility
3.2.4. Effect of pH
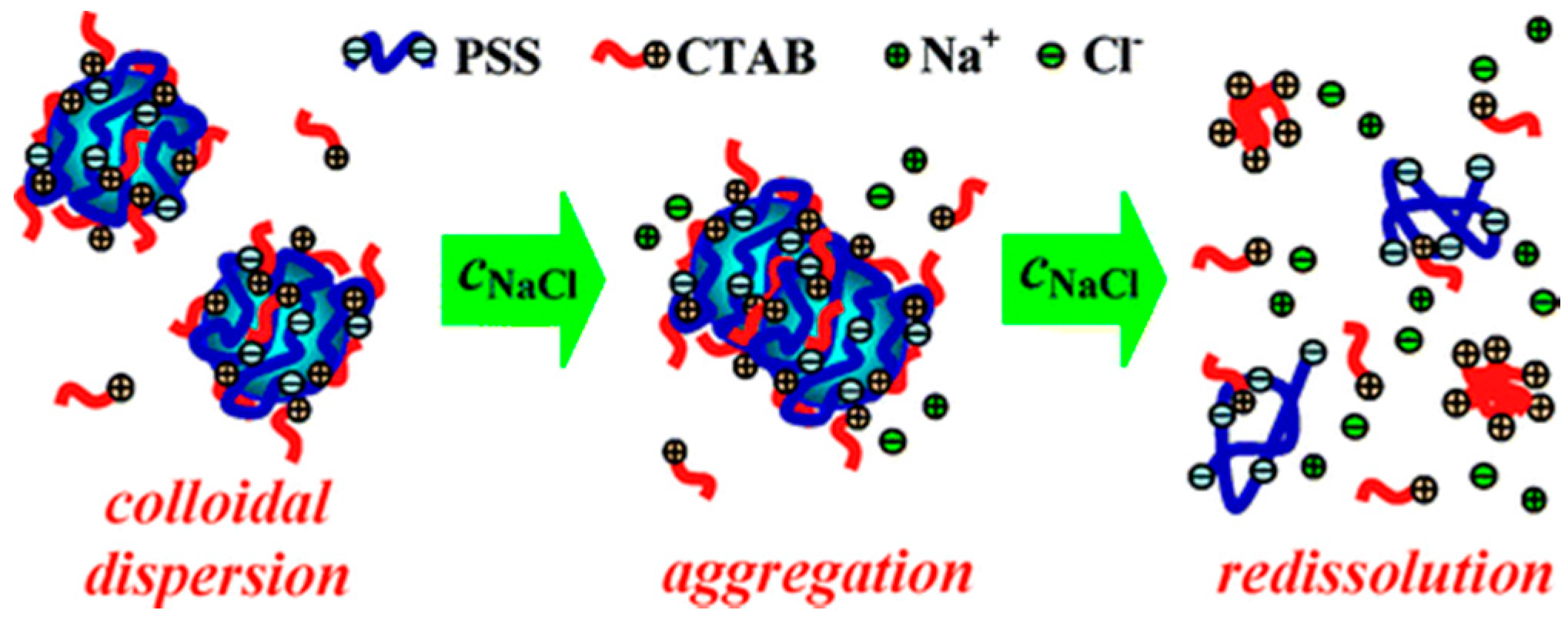
3.2.5. Ionic Strength
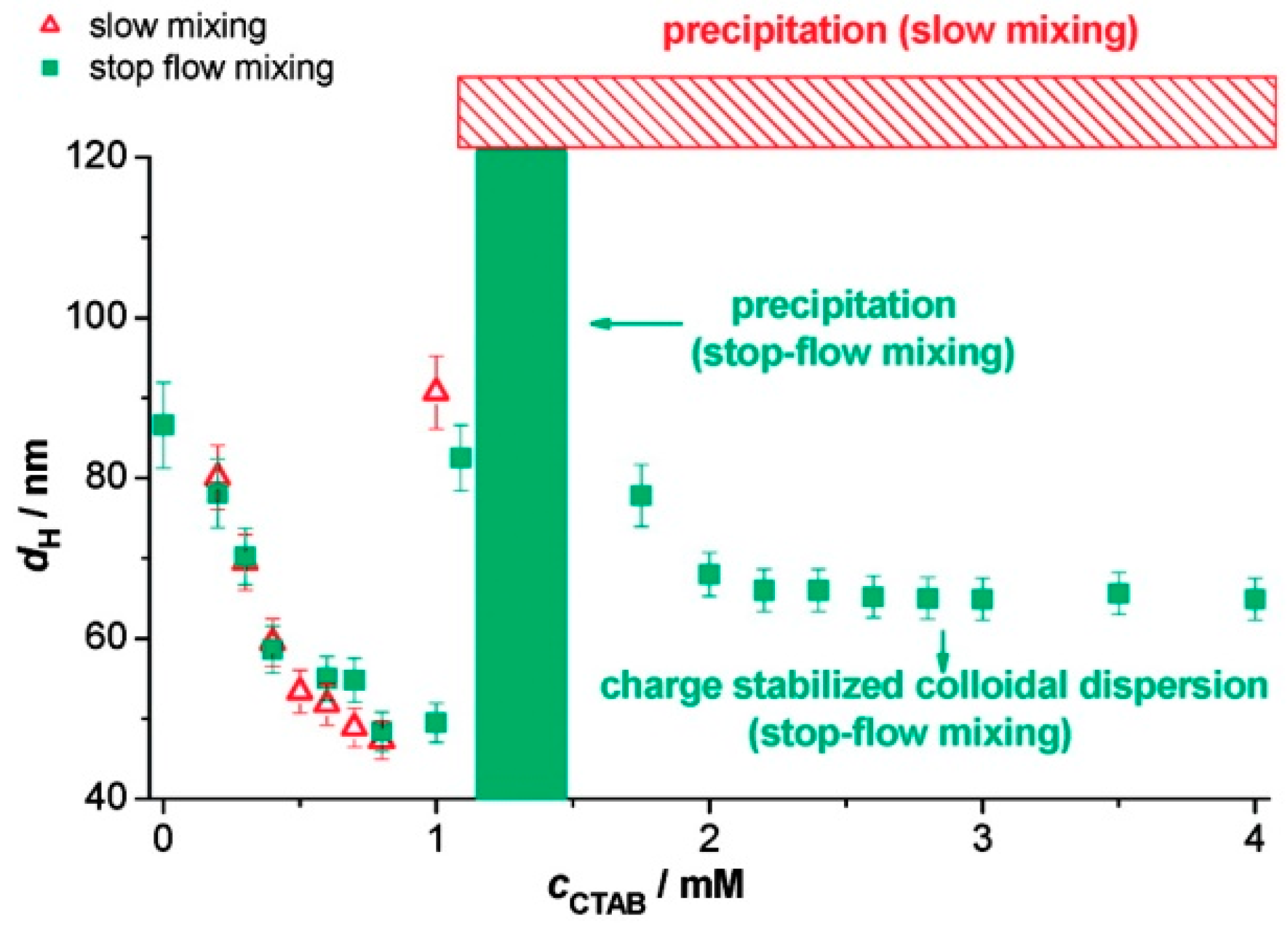
3.2.6. Mixing Procedure
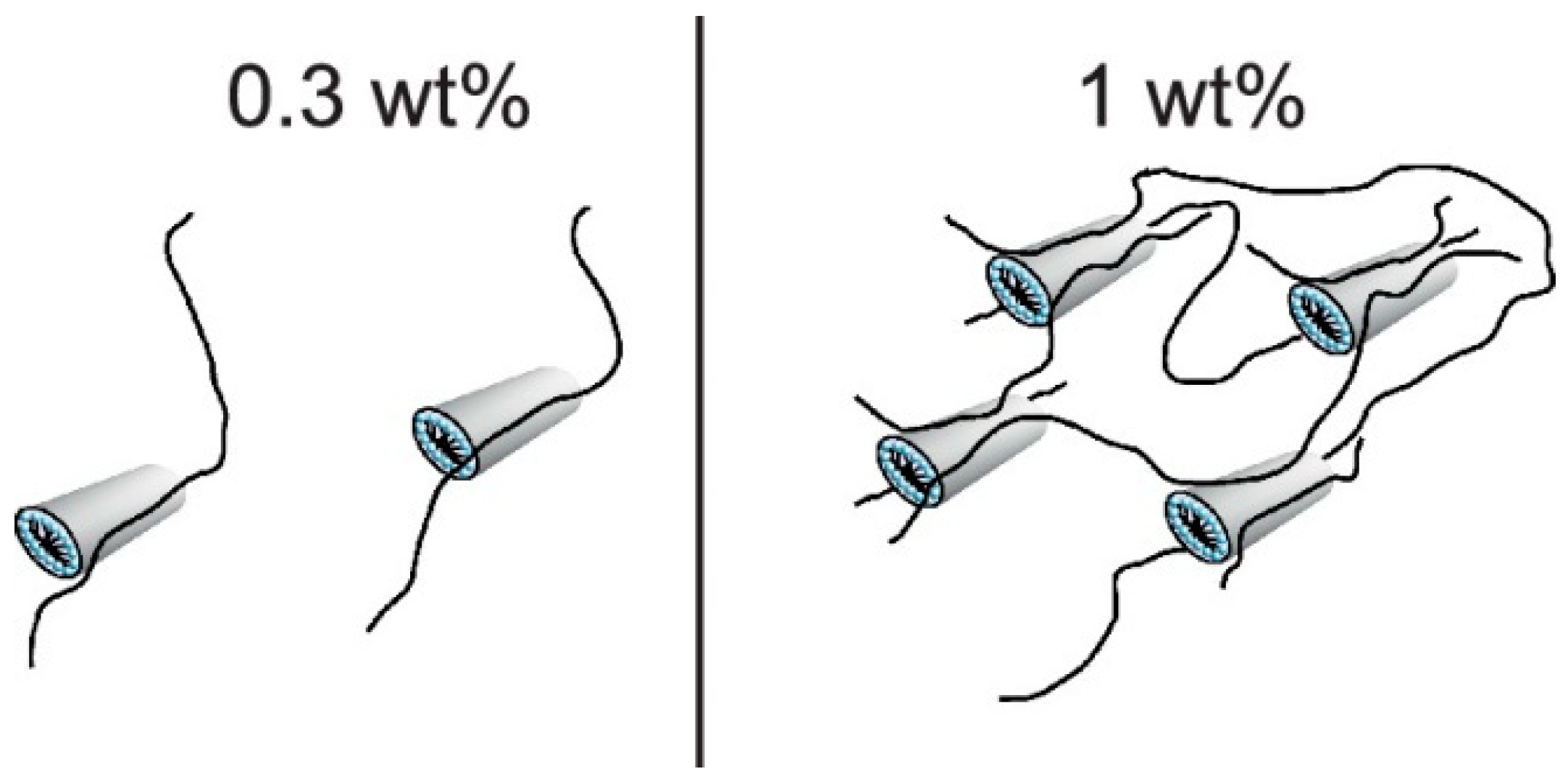
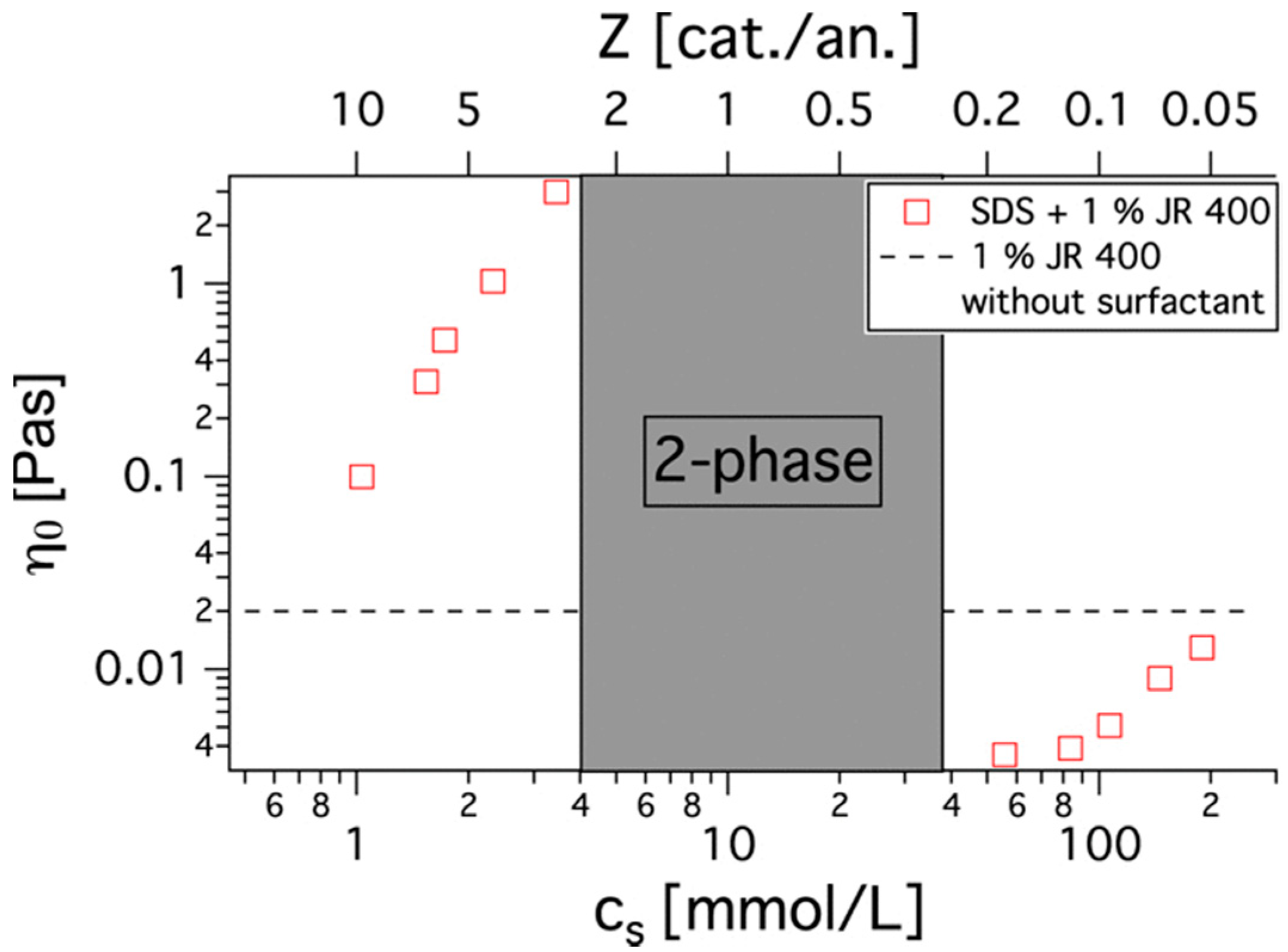
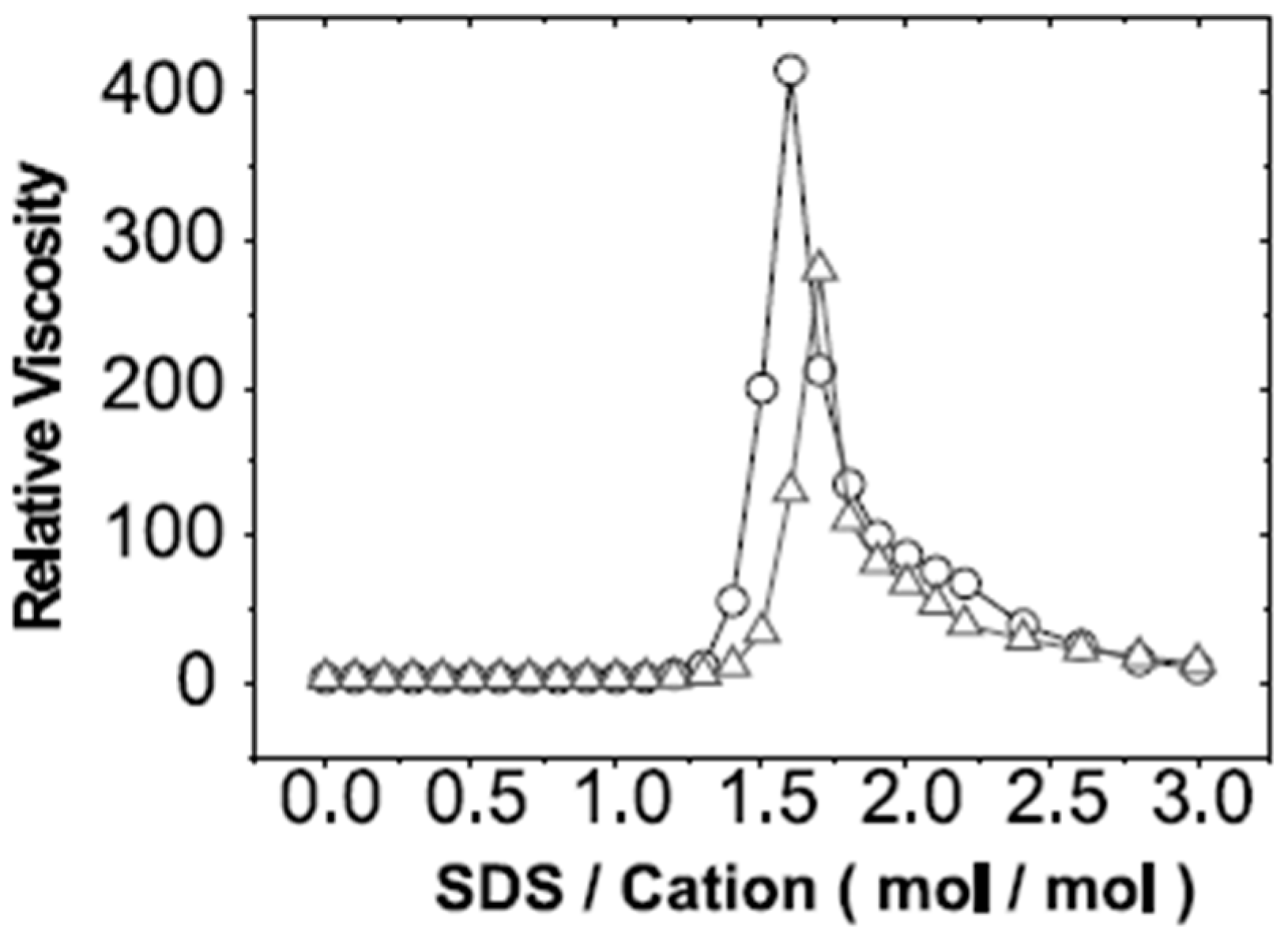
4. Outcomes of Complexation-Rheology
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tadros, T.F. An Introduction to Surfactants; Walter de Gruyter: Wokingham, UK, 2014; ISBN 978-3-11-031213-3. [Google Scholar]
- Liu, S.; Hu, C.; Huang, J.; Yan, Y. Fluorescent polyion complex for the detection of sodium dodecylbenzenesulfonate. Polymers 2018, 10, 657. [Google Scholar] [CrossRef]
- Rosen, M.; Kunjappu, J. Surfactants and Interfacial Phenomena Index, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Rhein, L.D.; Schlossman, M.; O’Lenick, A.; Somasundaran, P. Surfactants in Personal Care Products and Decorative Cosmetics, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2006; ISBN 9781574445312. [Google Scholar]
- Van der Meeren, P.; Verstraetet, W. Surfactants in relation to bioremediation and wastewater treatment. Curr. Opin. Colloid Interface Sci. 1996, 1, 624–634. [Google Scholar] [CrossRef]
- Witten, T.A.; Pincus, P.A. Structured Fluids: Polymers, Colloids, Surfactants; Oxford University Press: New York, NY, USA, 2004; ISBN 0198526881. [Google Scholar]
- Han, Y.; Wei, Y.; Wang, H.; Mei, Y.; Zhou, H. Contrasting the effects of hydrophobicity and counterion size on anionic wormlike micelle growth. J. Surfactants Deterg. 2013, 16, 139–145. [Google Scholar] [CrossRef]
- Dautzenberg, H. Polyelectrolyte complex formation in highly aggregating systems. 1. Effect of salt: polyelectrolyte complex formation in the presence of NaCl. Macromolecules 1997, 9297, 7810–7815. [Google Scholar] [CrossRef]
- Lindman, B.; Antunes, F.; Aidarova, S.; Miguel, M.; Nylander, T. Polyelectrolyte-surfactant association—from fundamentals to applications. Colloid J. 2014, 76, 585–594. [Google Scholar] [CrossRef]
- Pinotti, A.; Bevilacqua, A.; Zaritzky, N. Comparison of the performance of chitosan a cationic polyacrylamide as flocculants of emulsion systems. J. Surfactants Deterg. 2001, 4, 57–63. [Google Scholar] [CrossRef]
- Shulevich, Y.V.; Nguyen, T.H.; Tutaev, D.S.; Navrotskii, A.V.; Novakov, I.A. Purification of fat-containing wastewater using polyelectrolyte-surfactant complexes. Sep. Purif. Technol. 2013, 113, 18–23. [Google Scholar] [CrossRef]
- Petzold, G.; Mende, M.; Kochurova, N. Polymer-surfactant complexes as flocculants. Colloids Surf. A Physicochem. Eng. Asp. 2007, 298, 139–144. [Google Scholar] [CrossRef]
- Goddard, E.D. Polymer/surfactant interaction-Its relevance to detergent systems. J. Am. Oil Chem. Soc. 1994, 71, 1–16. [Google Scholar] [CrossRef]
- Goddard, E.D. Polymer/surfactant interaction: Interfacial aspects. J. Colloid Interface Sci. 2002, 256, 228–235. [Google Scholar] [CrossRef]
- Llamas, S.; Guzmán, E.; Ortega, F.; Baghdadli, N.; Cazeneuve, C.; Rubio, R.G.; Luengo, G.S. Adsorption of polyelectrolytes and polyelectrolytes-surfactant mixtures at surfaces: A physico-chemical approach to a cosmetic challenge. Adv. Colloid Interface Sci. 2015, 222, 461–487. [Google Scholar] [CrossRef] [PubMed]
- Penfold, J.; Thomas, R.K.; Bradbury, R.; Tucker, I.; Petkov, J.T.; Jones, C.W.; Webster, J.R.P. Probing the surface of aqueous surfactant-perfume mixed solutions during perfume evaporation. Colloids Surf. A Physicochem. Eng. Asp. 2017, 520, 178–183. [Google Scholar] [CrossRef]
- Bradbury, R.; Penfold, J.; Thomas, R.K.; Tucker, I.M.; Petkov, J.T.; Jones, C. Manipulating perfume delivery to the interface using polymer-surfactant interactions. J. Colloid Interface Sci. 2016, 466, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zheng, P.-Y.; Zhao, F.-Y.; An, Q.-F.; Gao, C.-J. Preparation and pervaporation characteristics of novel ethanol permselective polyelectrolyte–surfactant complex membranes. RSC Adv. 2015, 5, 63545–63552. [Google Scholar] [CrossRef]
- Budhathoki, M.; Barnee, S.H.R.; Shiau, B.J.; Harwell, J.H. Improved oil recovery by reducing surfactant adsorption with polyelectrolyte in high saline brine. Colloids Surf. A Physicochem. Eng. Asp. 2016, 498, 66–73. [Google Scholar] [CrossRef]
- ShamsiJazeyi, H.; Verduzco, R.; Hirasaki, G.J. Reducing adsorption of anionic surfactant for enhanced oil recovery: Part I. Competitive adsorption mechanism. Colloids Surf. A Physicochem. Eng. Asp. 2014, 453, 162–167. [Google Scholar] [CrossRef]
- Schwartz, A.M.; Perry, J.W.; Bartell, F.E. Surface Active Agents. J. Phys. Colloid Chem. 1949, 53, 1467. [Google Scholar] [CrossRef]
- Chu, D.; Thomas, J.K. Effect of Cationic Surfactants. J. Am. Chem. Soc. 1986, 108, 6270–6276. [Google Scholar] [CrossRef]
- Nagarajan, R. Thermodynamics association of nonionic polymer-micelle. Colloids Surf. 1985, 13, 1–17. [Google Scholar] [CrossRef]
- Wallin, T.; Linse, P. Monte Carlo simulations of polyelectrolytes at charged micelles. 1. Effects of chain flexibility. Langmuir 1996, 12, 305–314. [Google Scholar] [CrossRef]
- Goddard, E.D. Polymer-surfactant interaction part II. Polymer and surfactant of opposite charge. Colloids Surf. 1986, 19, 301–329. [Google Scholar] [CrossRef]
- Guzmán, E.; Llamas, S.; Maestro, A.; Fernández-Peña, L.; Akanno, A.; Miller, R.; Ortega, F.; Rubio, R.G. Polymer-surfactant systems in bulk and at fluid interfaces. Adv. Colloid Interface Sci. 2016, 233, 38–64. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.D.; Claesson, P.M.; Langevin, D.; Meszaros, R.; Nylander, T.; Stubenrauch, C.; Titmuss, S.; von Klitzing, R. Complexes of surfactants with oppositely charged polymers at surfaces and in bulk. Adv. Colloid Interface Sci. 2010, 155, 32–49. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.N. The interaction of sodium dodecyl sulfate with polyethylene oxide. J. Colloid Interface Sci. 1967, 23, 36–42. [Google Scholar] [CrossRef]
- Baldwin, R.L. The new view of hydrophobic free energy. FEBS Lett. 2013, 587, 1062–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, R.L.; Rose, G.D. How the hydrophobic factor drives protein folding. Proc. Natl. Acad. Sci. USA 2016, 113, 12462–12466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Naim, A. The Rise and Fall of the Hydrophobic Effect in Protein Folding and Protein-Protein Association, and Molecular Recognition. Open J. Biophys. 2011, 01, 1–7. [Google Scholar] [CrossRef]
- Ben-Naim, A. Hydrophobic interactions. Plenum Press: New York, NY, USA, 1980; ISBN 978-1-4684-3545-0. [Google Scholar]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A view of the hydrophobic effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Tanford, C. Interfacial free energy and the hydrophobic effect. Proc. Natl. Acad. Sci. USA 1979, 76, 4175–4176. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, Y.; Tanford, C. The solubility of amino in aqueous ethanol acids and two glycine dioxane solutions peptides. J. Biol. Chem. 1971, 246, 2211–2217. [Google Scholar]
- Kogej, K. Thermodynamic analysis of the conformational transition in aqueous solutions of isotactic and atactic poly(methacrylic acid) and the hydrophobic effect. Polymers 2016, 8. [Google Scholar] [CrossRef]
- Sugai, S.; Ebert, G. Conformations of hydrophobic polyelectrolytes. Adv. Colloid Interface Sci. 1985, 24, 247–282. [Google Scholar] [CrossRef]
- Baldwin, R.L. Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl. Acad. Sci. USA 1986, 83, 8069–8072. [Google Scholar] [CrossRef]
- MacKnight, W.J.; Ponomarenko, E.; Tirrell, D.A. Self-assembled polyelectrolyte-surfactant complexes in nonaqueous solvents and in the solid state. Acc. Chem. Res. 1998, 31, 781–788. [Google Scholar] [CrossRef]
- Mya, K.Y.; Jamieson, A.M.; Sirivat, A. Effect of temperature and molecular weight on binding between poly (ethylene oxide) and cationic surfactant in aqueous solutions. Langmuir 2000, 16, 6131–6135. [Google Scholar] [CrossRef]
- Priftis, D.; Laugel, N.; Tirrell, M. Thermodynamic characterization of polypeptide complex coacervation. Langmuir 2012, 28, 15947–15957. [Google Scholar] [CrossRef] [PubMed]
- Anthony, O.; Zana, R. Effect of Temperature on the Interactions between Neutral Polymers and a Cationic and a Nonionic Surfactant in Aqueous Solutions. Langmuir 1994, 10, 4048–4052. [Google Scholar] [CrossRef]
- Kizilay, E.; MacCarrone, S.; Foun, E.; Dinsmore, A.D.; Dubin, P.L. Cluster formation in polyelectrolyte—Micelle complex coacervation. J. Phys. Chem. B 2011, 115, 7256–7263. [Google Scholar] [CrossRef]
- Chollakup, R.; Smitthipong, W.; Eisenbach, C.D.; Tirrell, M. Phase behavior and coacervation of aqueous poly(acrylic acid)-poly(allylamine) solutions. Macromolecules 2010, 43, 2518–2528. [Google Scholar] [CrossRef]
- Santerre, J.P.; Hayakawa, K.; Kwak, J.C.T. A study of the temperature dependence of the binding of a cationic surfactant to an anionic polyelectrolyte. Colloids Surf. 1985, 13, 35–45. [Google Scholar] [CrossRef]
- Nilsson, S.; Blokhus, A.M.; Saure, A. Influence of Hydrophobic Cosolutes on Aqueous Two-Phase Polymer-Surfactant Systems. Langmuir 1998, 14, 6082–6085. [Google Scholar] [CrossRef]
- Nilsson, S.; Blokhus, A.M.; Hellebust, S.; Glomm, W.R. Influence of hydrophobic cosolutes on the associative/segregative phase separation of aqueous cationic surfactant-polymer systems. Langmuir 2002, 18, 6504–6506. [Google Scholar] [CrossRef]
- Piculell, L.; Lindman, B. Association and Segregation in Aqueous Polymer/Polymer, Polymer Surfactant, and Surfactant Surfactant Mixtures - Similarities and Differences. Adv. Colloid Interface Sci. 1992, 41, 149–178. [Google Scholar] [CrossRef]
- Lindman, S.; Lynch, I.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Systematic Investigation of the Thermodynamics of HSA Adsorption to N-tert-Butylacrylamide Copolymer Nanoparticles. Effects of Particle Size and Hydrophobicity. Nano Lett. 2007, 7, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Martelo, L.M.; Fonseca, S.M.; Marques, A.T.; Burrows, H.D.; Valente, A.J.M.; Justino, L.L.G.; Scherf, U.; Pradhan, S.; Song, Q. Effects of charge density on photophysics and aggregation behavior of anionic fluorene-arylene conjugated polyelectrolytes. Polymers 2018, 10. [Google Scholar] [CrossRef]
- Essafi, W.; Spiteri, M.N.; Williams, C.; Boue, F. Hydrophobic polyelectrolytes in better polar solvent. structure and chain conformation as seen by SAXS and SANS. Macromolecules 2009, 42, 9568–9580. [Google Scholar] [CrossRef]
- Dill, K.A.; Bromberg, S. Molecular Driving Forces, 2nd ed.; CRC Press: New York, NY, USA, 2011; ISBN 9780815344308. [Google Scholar]
- Dautzenberg, H. Polyelectrolytes: Formation, Characterization and Application; Hanser Publishers: Munich, Germany, 1994; ISBN 1569901279. [Google Scholar]
- Harris, F.E.; Rice, S.A. A chain model for polyelectrolytes. I. J. Phys. Chem. 1954, 58, 725–732. [Google Scholar] [CrossRef]
- Norwood, D.P.; Minatti, E.; Reed, W.F. Surfactant/polymer assemblies. 1. Surfactant binding properties. Macromolecules 1998, 31, 2957–2965. [Google Scholar] [CrossRef]
- Manning, G.S. Counterion Binding in Polyelectrolyte Theory. Acc. Chem. Res. 1979, 12, 443–449. [Google Scholar] [CrossRef]
- Xu, A.Y.; Kizilay, E.; Madro, S.P.; Vadenais, J.Z.; McDonald, K.W.; Dubin, P.L. Dilution induced coacervation in polyelectrolyte-micelle and polyelectrolyte-protein systems. Soft Matter 2018, 14, 2391–2399. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Yan, H.; Zhang, J.; Thomas, R.K. Binding of sodium dodecyl sulfate with linear and branched polyethyleneimines in aqueous solution at different pH values. Langmuir 2006, 22, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Santos, L.M.N.B.F.; Nichifor, M.; Lopes, A.; Bastos, M. Thermodynamics of the Interaction between a Hydrophobically Modified Polyelectrolyte and Sodium Dodecyl Sulfate in Aqueous Solution. J. Phys. Chem. B 2004, 108, 405–413. [Google Scholar] [CrossRef]
- Marciel, A.; Chung, E.J.; Brettmann, B.K.; Leon, L. Bulk and nanoscale polypeptide based polyelectrolyte complexes. Adv. Colloid Interface Sci. 2018, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Sing, C.E. Development of the modern theory of polymeric complex coacervation. Adv. Colloid Interface Sci. 2017, 239, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.; Rosa, M.; Pais, A.C.; Miguel, M.; Lindman, B. DNA-surfactant interactions. Compaction, condensation, decompaction and phase separation. J. Chin. Chem. Soc. 2004, 51, 447–469. [Google Scholar] [CrossRef]
- Lapitsky, Y.; Parikh, M.; Kaler, E.W. Calorimetric determination of surfactant/polyelectrolyte binding isotherms. J. Phys. Chem. B 2007, 111, 8379–8387. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Muthukumar, M. Entropy and enthalpy of polyelectrolyte complexation: Langevin dynamics simulations. J. Chem. Phys. 2006, 124. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fan, Y.; Wang, Y. Thermodynamic association behaviors of sodium dodecyl sulfate (SDS) with poly(4-vinylpyridine N-oxide) (PVPNO) at different pH values and ionic strengths. J. Surfactants Deterg. 2017, 20, 647–657. [Google Scholar] [CrossRef]
- Bahadur, P.; Dubin, P.L.; Rao, Y.K. Complex Formation between Sodium Dodecyl Sulfate and Poly(4-vinylpyridine N-oxide). Langmuir 1995, 1951–1955. [Google Scholar] [CrossRef]
- Aoki, K.; Hort, J.; Sakurai, K. Interaction between surface active agents and proteins III. Precipitation Curve of the System Sodium Dodecyl Sulfate-Egg Albumin at Various pH ’ s and the determination of the concentration of protein by the titration using surfactant. Bull. Chem. Soc. Jpn. 1956, 29, 758–761. [Google Scholar] [CrossRef]
- Dos Santos, S.; Gustavsson, C.; Gudmundsson, C.; Linse, P.; Piculell, L. When do water-insoluble polyion-surfactant ion complex salts “redissolve” by added excess surfactant? Langmuir 2011, 27, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Anghel, D.F.; Saito, S.; Iovescu, A.; Bǎran, A.; Stîngǎ, G. Counterion effect of cationic surfactants upon the interaction with poly(methacrylic acid). J. Surfactants Deterg. 2011, 14, 91–101. [Google Scholar] [CrossRef]
- Goddard, E.D.; Hannan, R.B. Cationic polymer/anionic surfactant interactions. J. Colloid Interface Sci. 1976, 55, 73–79. [Google Scholar] [CrossRef]
- Koetz, J.; Kosmella, S. Polyelectrolytes and Nanoparticles; Springer Laboratory: Berlin/Heidelberg, Germany, 2007; ISBN 978-3-540-46382-5. [Google Scholar]
- Liu, Z.H.; Lv, W.J.; Zhao, S.L.; Shang, Y.Z.; Peng, C.J.; Wang, H.L.; Liu, H.L. Effects of the hydrophilicity or hydrophobicity of the neutral block on the structural formation of a block polyelectrolyte/surfactant complex: A molecular dynamics simulation study. Comput. Condens. Matter 2015, 2, 16–24. [Google Scholar] [CrossRef]
- Jackson, N.E.; Brettmann, B.K.; Vishwanath, V.; Tirrell, M.; Pablo, J.J. De Comparing Solvophobic and Multivalent Induced Collapse in Polyelectrolyte Brushes. ACS Macro Lett. 2017, 6, 155–160. [Google Scholar] [CrossRef]
- Lee, N.; Thirumalai, D. Dynamics of Collapse of Flexible Polyelectrolytes in Poor Solvents. Macromolecules 2001, 34, 3446–3457. [Google Scholar] [CrossRef]
- Tsuchida, E.; Abe, K. Interactions Between Macromolecules in Solution and Intermacromolecular Complexes. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1982; Volume 45, ISBN 978-3-540-39443-3. [Google Scholar]
- Anthony, O.; Zana, R. Fluorescence Investigation of the Binding of Pyrene to Hydrophobic Microdomains in Aqueous Solutions of Polysoaps. Macromolecules 1994, 27, 3885–3891. [Google Scholar] [CrossRef]
- Nizri, G.; Lagerge, S.; Kamyshny, A.; Major, D.T.; Magdassi, S. Polymer-surfactant interactions: Binding mechanism of sodium dodecyl sulfate to poly(diallyldimethylammonium chloride). J. Colloid Interface Sci. 2008, 320, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Santerre, J.P.; Kwak, J.C.T. Study of surfactant-polyelectrolyte interactions. binding of dodecyl- and tetradecyltrimethylammonium bromide by some carboxylic polyelectrolytes. Macromolecules 1983, 16, 1642–1645. [Google Scholar] [CrossRef]
- Kogej, K.; Evmenenko, G.; Theunissen, E.; Berghmans, H.; Reynaers, H. Investigation of Structures in Polyelectrolyte / Surfactant Complexes by X-ray Scattering. Langmuir 2001, 17, 3175–3184. [Google Scholar] [CrossRef]
- Tseng, H.W.; Chen, P.C.; Tsui, H.W.; Wang, C.H.; Hu, T.Y.; Chen, L.J. Effect of molecular weight of poly(acrylic acid) on the interaction of oppositely charged ionic surfactant–polyelectrolyte mixtures. J. Taiwan Inst. Chem. Eng. 2018, 1–8. [Google Scholar] [CrossRef]
- Wallin, T.; Linse, P. Monte Carlo simulations of polyelectrolytes at charged micelles. 2. Effects of linear charge density. J. Phys. Chem. 1996, 100, 17873–17880. [Google Scholar] [CrossRef]
- Delisavva, F.; Uchman, M.; Štěpánek, M.; Kereïche, S.; Hordyjewicz-Baran, Z.; Appavou, M.S.; Procházka, K. Coassembly of Gemini Surfactants with Double Hydrophilic Block Polyelectrolytes Leading to Complex Nanoassemblies. Macromolecules 2017, 50, 8745–8754. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Li, J.; Wang, J.; Wang, Y.; Guo, Z.; Yan, H. Salt effect on the complex formation between polyelectrolyte and oppositely charged surfactant in aqueous solution. J. Phys. Chem. B 2005, 109, 10807–10812. [Google Scholar] [CrossRef] [PubMed]
- Wallin, T.; Linse, P. Monte Carlo simulations of polyelectrolytes at charged micelles. 3. Effects of surfactant tail. J. Phys. Chem. B 1997, 101, 5506–5513. [Google Scholar] [CrossRef]
- Li, D.; Kelkar, M.S.; Wagner, N.J. Phase behavior and molecular thermodynamics of coacervation in oppositely charged polyelectrolyte/surfactant systems: A cationic polymer JR 400 and anionic surfactant SDS mixture. Langmuir 2012, 28, 10348–10362. [Google Scholar] [CrossRef]
- Anthony, O.; Zana, R. Interactions between Water-Soluble Polymers and Surfactants: Effect of the Polymer Hydrophobicity. 1. Hydrophilic Polyelectrolytes. Langmuir 1996, 12, 1967–1975. [Google Scholar] [CrossRef]
- Satake, I.; Yang, J.E.N.T.S.I. Interaction of Sodium Decyl Sulfate with Poly (L- ornithine) and Poly (L-lysine) in Aqueous Solution. Biopolymers 1976, 15, 2263–2275. [Google Scholar] [CrossRef]
- Arnold, G.B.; Breuer, M.M. Adsorption of Polymer-JR-sodium-dodecyl-sulfate complex on the surface of alumina. Colloids Surf. 1985, 13, 103–112. [Google Scholar] [CrossRef]
- Kwak, J.C.T. Polymer-Surfactant Systems; CRC Press: New York, NY, USA, 1998; ISBN 9780824702328. [Google Scholar]
- Monteux, C.; Williams, C.E.; Meunier, J.; Anthony, O.; Bergeron, V. Adsorption of Oppositely Charged Polyelectrolyte/Surfactant Complexes at the Air/Water Interface: Formation of Interfacial Gels. Langmuir 2004, 20, 57–63. [Google Scholar] [CrossRef]
- Jain, N.J.; Albouy, P.A.; Langevin, D. Study of adsorbed monolayers of a cationic surfactant and an anionic polyelectrolyte at the air-water interface. Role of the polymer charge density. Langmuir 2003, 19, 8371–8379. [Google Scholar] [CrossRef]
- Okuzaki, H.; Osada, Y. Effects of Hydrophobic Interaction on the Cooperative Binding of a Surfactant to a Polymer Network. Macromolecules 1994, 27, 502–506. [Google Scholar] [CrossRef]
- Brackman, J.C.; Engberts, J.B.F.N. The effect of surfactant headgroup charge on polymer-micelle interaction. J. Colloid Interface Sci. 1989, 132, 250–255. [Google Scholar] [CrossRef]
- Dubin, P.L.; Rigsbee, D.R.; Gan, L.; Fallon, M.A. Equilibrium Binding of Mixed Micelles to Oppositely Charged Polyelectrolytes. Macromolecules 1988, 21, 2555–2559. [Google Scholar] [CrossRef]
- Bakshi, M.S. Polymer-Induced Incompatibility in the Mixed Micelle Formation of Cationic Surfactants with Bulky Polar Head Groups. J. Surfactants Deterg. 2001, 4, 297–302. [Google Scholar] [CrossRef]
- Barbosa, A.M.; Santos, I.J.B.; Ferreira, G.M.D.; Hespanhol Da Silva, M.D.C.; Teixeira, Á.V.N.D.C.; Da Silva, L.H.M. Microcalorimetric and SAXS determination of PEO-SDS interactions: The effect of cosolutes formed by ions. J. Phys. Chem. B 2010, 114, 11967–11974. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Kwak, J.C.T. Surfactant-polyelectrolyte interactions. 1. Binding of dodecyltrimethylammonium ions by sodium dextran sulfate and sodium poly(styrenesulfonate) in aqueous solution in the presence of sodium chloride. J. Phys. Chem. 1982, 86, 3866–3870. [Google Scholar] [CrossRef]
- Li, D.; Wagner, N.J. Universal binding behavior for ionic alkyl surfactants with oppositely charged polyelectrolytes. J. Am. Chem. Soc. 2013, 135, 17547–17555. [Google Scholar] [CrossRef]
- Passos, C.B.; Kuhn, P.S.; Diehl, A. Flexible polyelectrolyte conformation in the presence of cationic and anionic surfactants. Phys. A Stat. Mech. Appl. 2015, 438, 436–446. [Google Scholar] [CrossRef]
- Lam, V.D.; Walker, L.M. A pH-induced transition of surfactant-polyelectrolyte aggregates from cylindrical to string-of-pearls structure. Langmuir 2010, 26, 10489–10496. [Google Scholar] [CrossRef]
- Macdonald, P.M.; Strashko, V. A Thermotropic Phase Transition in Polyelectrolyte -Surfactant Complexes As Characterized by Deuterium NMR. Langmuir 1998, 14, 4758–4764. [Google Scholar] [CrossRef]
- Lapitsky, Y.; Kaler, E.W. Formation and structural control of surfactant and polyelectrolyte gels. Colloids Surf. A Physicochem. Eng. Asp. 2006, 282–283, 118–128. [Google Scholar] [CrossRef]
- Lapitsky, Y.; Eskuchen, W.J.; Kaler, E.W. Surfactant and Polyelectrolyte Gel Particles that Swell Reversibly. Langmuir 2006, 22, 6375–6379. [Google Scholar] [CrossRef]
- Langevin, D. Complexation of oppositely charged polyelectrolytes and surfactants in aqueous solutions. A review. Adv. Colloid Interface Sci. 2009, 147–148, 170–177. [Google Scholar] [CrossRef]
- Antonietti, M.; Burger, C.; Effing, J. Mesomorphous polyelectrolyte-surfactant complexes. Adv. Mater. 1995, 7, 751–753. [Google Scholar] [CrossRef]
- Loh, W.; Brinatti, C.; Tam, K.C. Use of isothermal titration calorimetry to study surfactant aggregation in colloidal systems. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 999–1016. [Google Scholar] [CrossRef] [PubMed]
- Nizri, G.; Magdassi, S.; Schmidt, J.; Cohen, Y.; Talmon, Y. Microstructural characterization of micro- and nanoparticles formed by polymer-surfactant interactions. Langmuir 2004, 20, 4380–4385. [Google Scholar] [CrossRef]
- Plazzotta, B.; Fegyver, E.; Mészáros, R.; Pedersen, J.S. Anisometric Polyelectrolyte/Mixed Surfactant Nanoassemblies Formed by the Association of Poly(diallyldimethylammonium chloride) with Sodium Dodecyl Sulfate and Dodecyl Maltoside. Langmuir 2015, 31, 7242–7250. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Borreguero, J.M.; Pincus, P.A.; Sumpter, B.G. Surfactant-Mediated Polyelectrolyte Self-Assembly in a Polyelectrolyte-Surfactant Complex. Macromolecules 2015, 48, 9050–9059. [Google Scholar] [CrossRef]
- Smitter, L.M.; Guédez, J.F.; Müller, A.J.; Sáez, A.E. Interactions between poly(ethylene oxide) and sodium dodecyl sulfate in elongational flows. J. Colloid Interface Sci. 2001, 236, 343–353. [Google Scholar] [CrossRef]
- Chiappisi, L.; Hoffmann, I.; Gradzielski, M. Complexes of oppositely charged polyelectrolytes and surfactants—Recent developments in the field of biologically derived polyelectrolytes. Soft Matter 2013, 9, 3896–3909. [Google Scholar] [CrossRef]
- Antonietti, M.; Maskos, M. Fine-Tuning of Phase Structures and Thermoplasticity of Polyelectrolyte-Surfactant Complexes: Copolymers of Ionic Monomers with N-Alkylacrylamides. Macromolecules 1996, 29, 4199–4205. [Google Scholar] [CrossRef]
- Antonietti, M.; Conrad, J. Synthesis of Very Highly Ordered Liquid Crystalline Phases by Complex Formation of Polyacrylic Acid with Cationic Surfactants. Angew. Chem. Int. Ed. 1994, 33, 1869–1870. [Google Scholar] [CrossRef]
- Nizri, G.; Magdassi, S. Solubilization of hydrophobic molecules in nanoparticles formed by polymer-surfactant interactions. J. Colloid Interface Sci. 2005, 291, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Pojjaźk, K.; Bertalanits, E.; Meźszaźros, R. Effect of salt on the equilibrium and nonequilibrium features of polyelectrolyte/surfactant association. Langmuir 2011, 27, 9139–9147. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Claesson, P.M.; Bergström, M.; Dedinaite, A. Trapped non-equilibrium states in aqueous solutions of oppositely charged polyelectrolytes and surfactants: Effects of mixing protocol and salt concentration. Colloids Surf. A Physicochem. Eng. Asp. 2005, 253, 83–93. [Google Scholar] [CrossRef]
- Goddard, E.D.; Hannan, R.B. Polymer/Surfactant Interactions. J. Am. Oil Chem. Soc. 1977, 54, 561–566. [Google Scholar] [CrossRef]
- Wang, Y.; Kimura, K.; Dubin, P.L.; Jaeger, W. Polyelectrolyte—Micelle Coacervation: Effects of Micelle Surface Charge Density, Polymer Molecular Weight, and Polymer/Surfactant Ratio. Macromolecules 2000, 33, 3324–3331. [Google Scholar] [CrossRef]
- Wang, Y.; Kimura, K.; Huang, Q.; Dubin, P.L.; Jaeger, W. Effects of Salt on Polyelectrolyte—Micelle Coacervation. Macromolecules 1999, 32, 7128–7134. [Google Scholar] [CrossRef]
- Wesley, R.D.; Dreiss, C.A.; Cosgrove, T.; Armes, S.P.; Thompson, L.; Baines, F.L.; Billingham, N.C. Structure of a hydrophilic-hydrophobic block copolymer and its interactions with salt and an anionic surfactant. Langmuir 2005, 21, 4856–4861. [Google Scholar] [CrossRef]
- Essafi, W.; Abdelli, A.; Bouajila, G.; Boué, F. Behavior of hydrophobic polyelectrolyte solution in mixed aqueous/organic solvents revealed by neutron scattering and viscosimetry. J. Phys. Chem. B 2012, 116, 13525–13537. [Google Scholar] [CrossRef]
- Borreguero, J.M.; Pincus, P.A.; Sumpter, B.G.; Goswami, M. Unraveling the Agglomeration Mechanism in Charged Block Copolymer and Surfactant Complexes. Macromolecules 2017, 50, 1193–1205. [Google Scholar] [CrossRef]
- Berret, J.-F.; Herve, P.; Aguerre-Chariol, O.; Oberdisse, J. Colloidal Complexes Obtained from Charged Block Copolymers and Surfactants: A Comparison between Small-Angle Neutron Scattering, Cryo-TEM, and Simulations. J. Phys. Chem. B 2003, 107, 8111–8118. [Google Scholar] [CrossRef]
- Asnacios, A.; Klitzing, R.; Langevin, D. Mixed monolayers of polyelectrolytes and surfactants at the air—Water interface. Colloids Surf. 2000, 167, 189–197. [Google Scholar] [CrossRef]
- Kiefer, J.J.; Somasundaran, P.; Ananthapadmanabhan, K.P. Interaction of Tetradecyltrimethylammonium Bromide with Poly(acrylic acid) and Poly(methacrylic acid). Effect of Charge Density. Langmuir 1993, 9, 1187–1192. [Google Scholar] [CrossRef]
- Tummino, A.; Toscano, J.; Sebastiani, F.; Noskov, B.A.; Varga, I.; Campbell, R.A. Effects of Aggregate Charge and Subphase Ionic Strength on the Properties of Spread Polyelectrolyte/Surfactant Films at the Air/Water Interface under Static and Dynamic Conditions. Langmuir 2018, 34, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Mezei, A.; Meszaros, R.; Varga, I.; Gilany, T. Effect of Mixing on the Formation of Complexes of Hyperbranched Cationic Polyelectrolytes and Anionic Surfactants. Langmuir 2007, 4237–4247. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, I.; Farago, B.; Schweins, R.; Falus, P.; Sharp, M.; Gradzielski, M. Structure and dynamics of polyelectrolytes in viscous polyelectrolyte- surfactant complexes at the mesoscale. EPL 2013, 104. [Google Scholar] [CrossRef]
- Fuoss, R.; Strauss, U.P. Electrostatic Interaction of Polyelectrolytes and Simple Electrolytes. J. Polym. Sci. 1948, 636, 602–603. [Google Scholar] [CrossRef]
- Roshan Deen, G. Solution Properties of Water-Soluble “Smart” Poly(N-acryloyl-N′-ethyl piperazine-co-methyl methacrylate). Polymers 2012, 4, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, S.; Thuresson, K.; Hansson, P.; Lindman, B. Mixed Solutions of Surfactant and Hydrophobically Modified Polymer. Controlling Viscosity with Micellar Size. J. Phys. Chem. B 1998, 102, 7099–7105. [Google Scholar] [CrossRef]
- Noda, T.; Hashidzume, A.; Morishima, Y. Solution properties of micelle networks formed by nonionic surfactant moieties covalently bound to a polyelectrolyte: salt effects on rheological behavior. Langmuir 2000, 16, 5324–5332. [Google Scholar] [CrossRef]
- Hoffmann, I.; Simon, M.; Farago, B.; Schweins, R.; Falus, P.; Holderer, O.; Gradzielski, M. Structure and dynamics of polyelectrolyte surfactant mixtures under conditions of surfactant excess. J. Chem. Phys. 2016, 145. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, I.; Farago, B.; Schweins, R.; Falus, P.; Sharp, M.; Prévost, S.; Gradzielski, M. On the mesoscopic origins of high viscosities in some polyelectrolyte-surfactant mixtures. J. Chem. Phys. 2015, 143. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Anaya, L.; Roux, D.; Soltero Martínez, J.; Carvajal Ramos, F.; Pignon, F.; Mannix, O.; Rinaudo, M. Role of Electrostatic Interactions on Supramolecular Organization in Calf-Thymus DNA Solutions under Flow. Polymers 2018, 10, 1204. [Google Scholar] [CrossRef]
- Guillot, S.; Mcloughlin, D.; Jain, N.; Langevin, D. Polyelectrolyte – surfactant complexes at interfaces and in bulk. J. Phys. Condens. Matter 2003, 15, S219–S224. [Google Scholar] [CrossRef]
- Zheng, X.; Cao, W. Interaction of main chain cationic polyelectrolyte with sodium dodecyl sulfate. Eur. Polym. J. 2001, 37, 2259–2262. [Google Scholar] [CrossRef]
- Wang, R.; Yan, H.; Hu, W.; Li, Y.; Mei, Z. Micellization of Anionic Sulfonate Gemini Surfactants and Their Interactions With Anionic Polyacrylamide. J. Surfactants Deterg. 2018, 21, 81–90. [Google Scholar] [CrossRef] [Green Version]
















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, N.; Brettmann, B. Intermolecular Interactions in Polyelectrolyte and Surfactant Complexes in Solution. Polymers 2019, 11, 51. https://doi.org/10.3390/polym11010051
Khan N, Brettmann B. Intermolecular Interactions in Polyelectrolyte and Surfactant Complexes in Solution. Polymers. 2019; 11(1):51. https://doi.org/10.3390/polym11010051
Chicago/Turabian StyleKhan, Nasreen, and Blair Brettmann. 2019. "Intermolecular Interactions in Polyelectrolyte and Surfactant Complexes in Solution" Polymers 11, no. 1: 51. https://doi.org/10.3390/polym11010051