Kinetic Monte-Carlo Simulation of Methane Steam Reforming over a Nickel Surface


Abstract
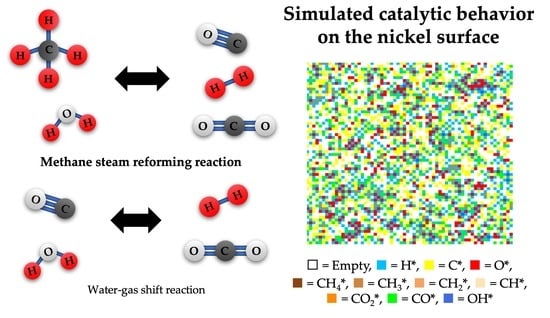
:
1. Introduction
2. Results
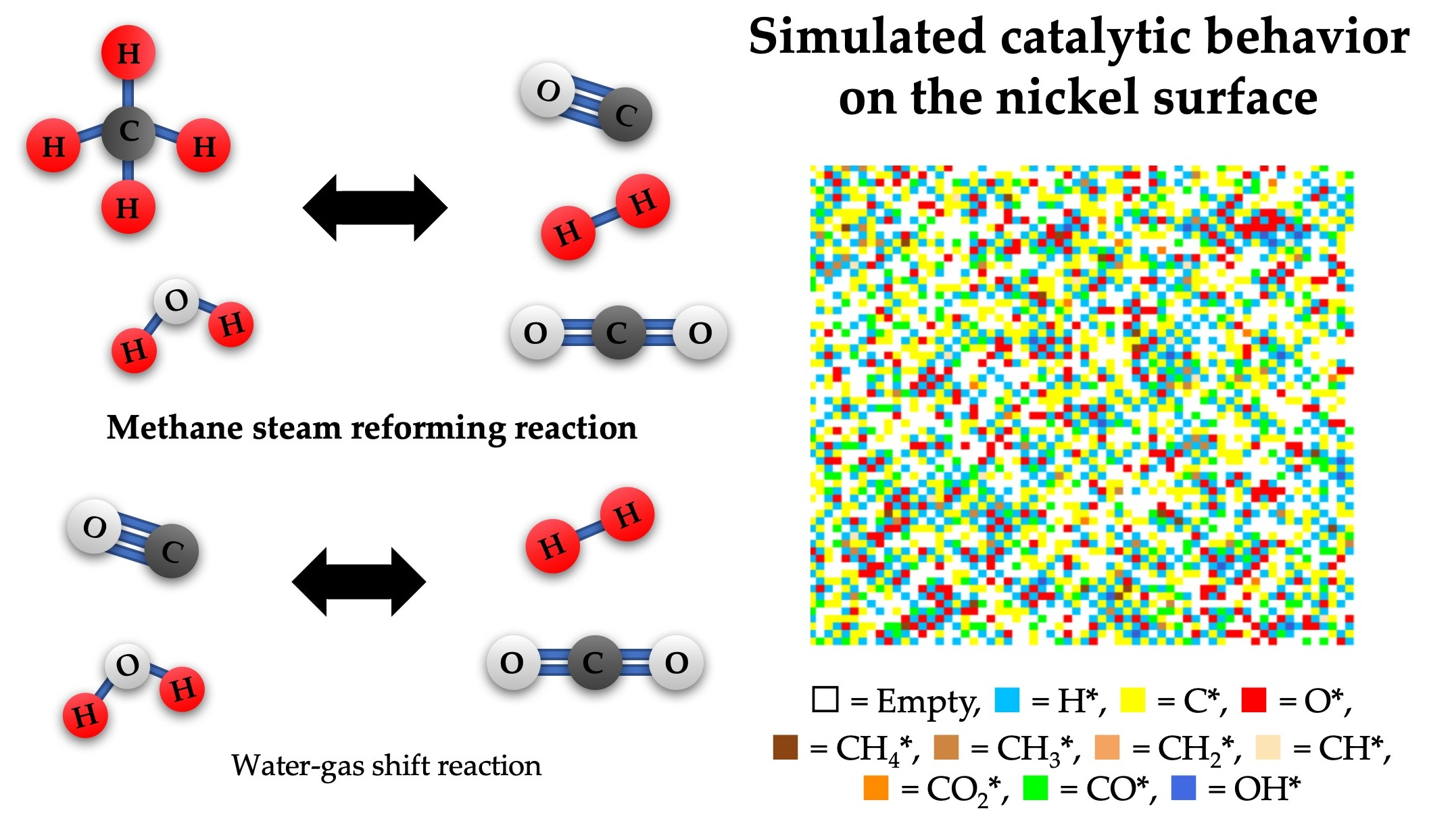
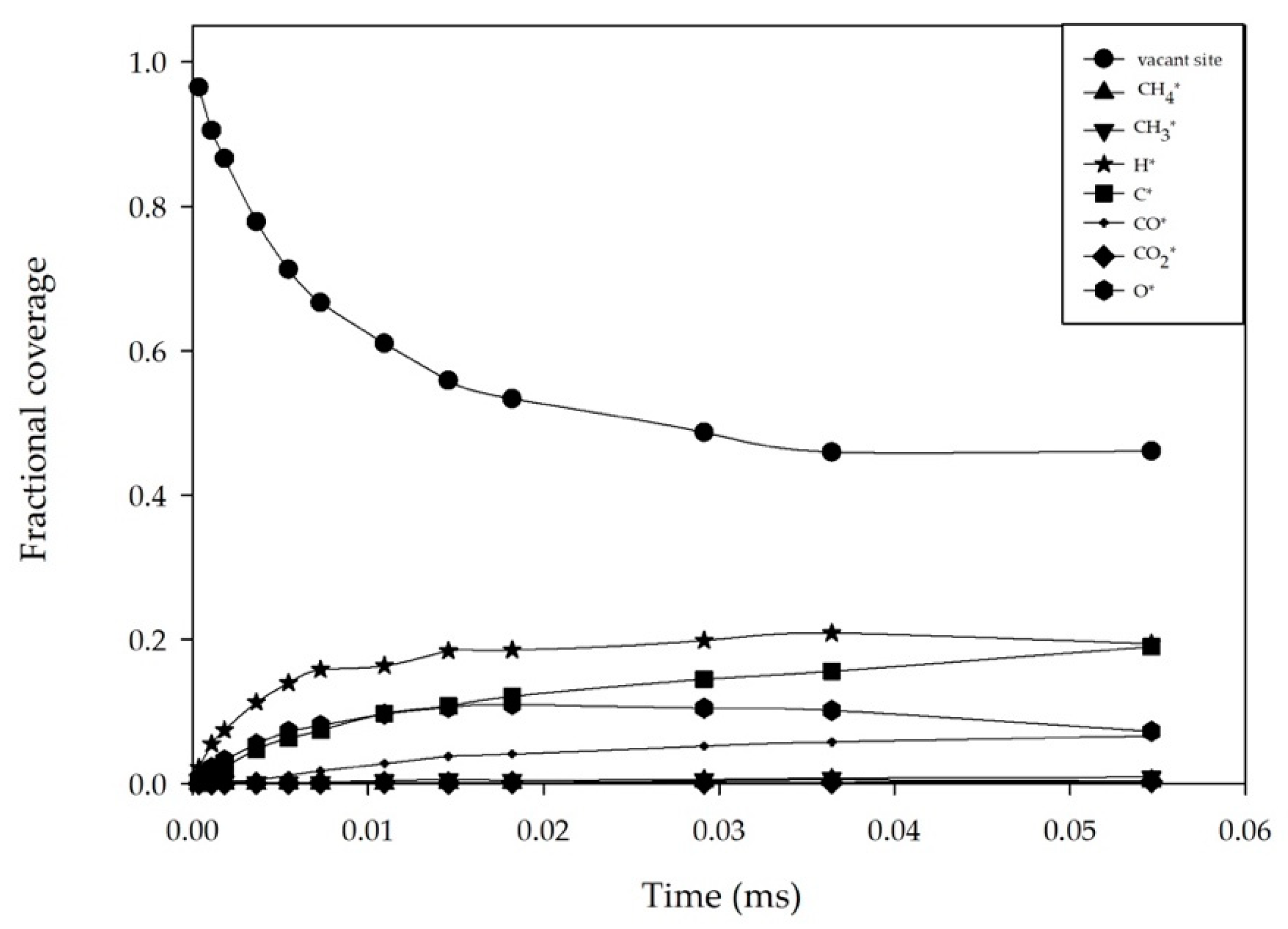
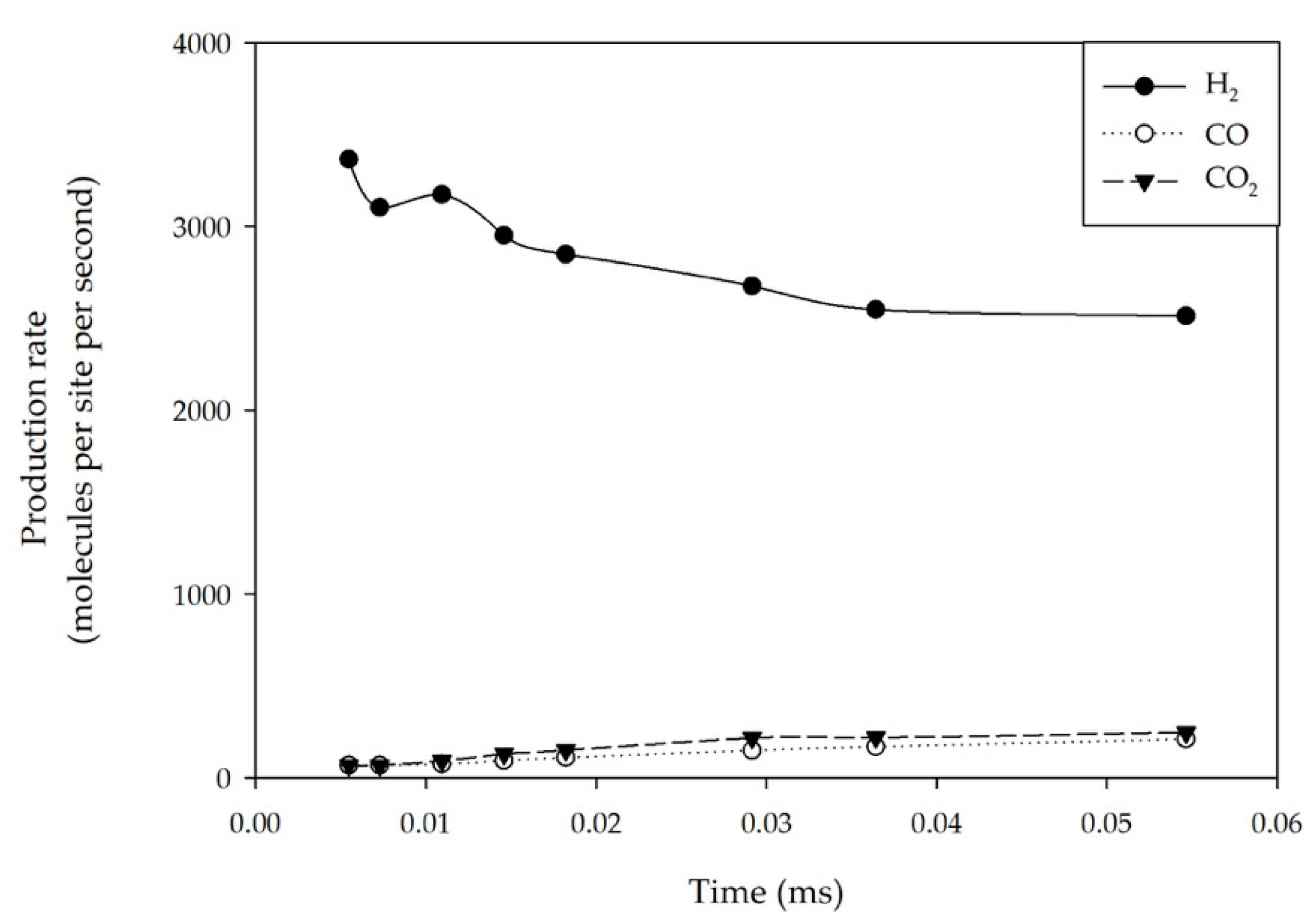
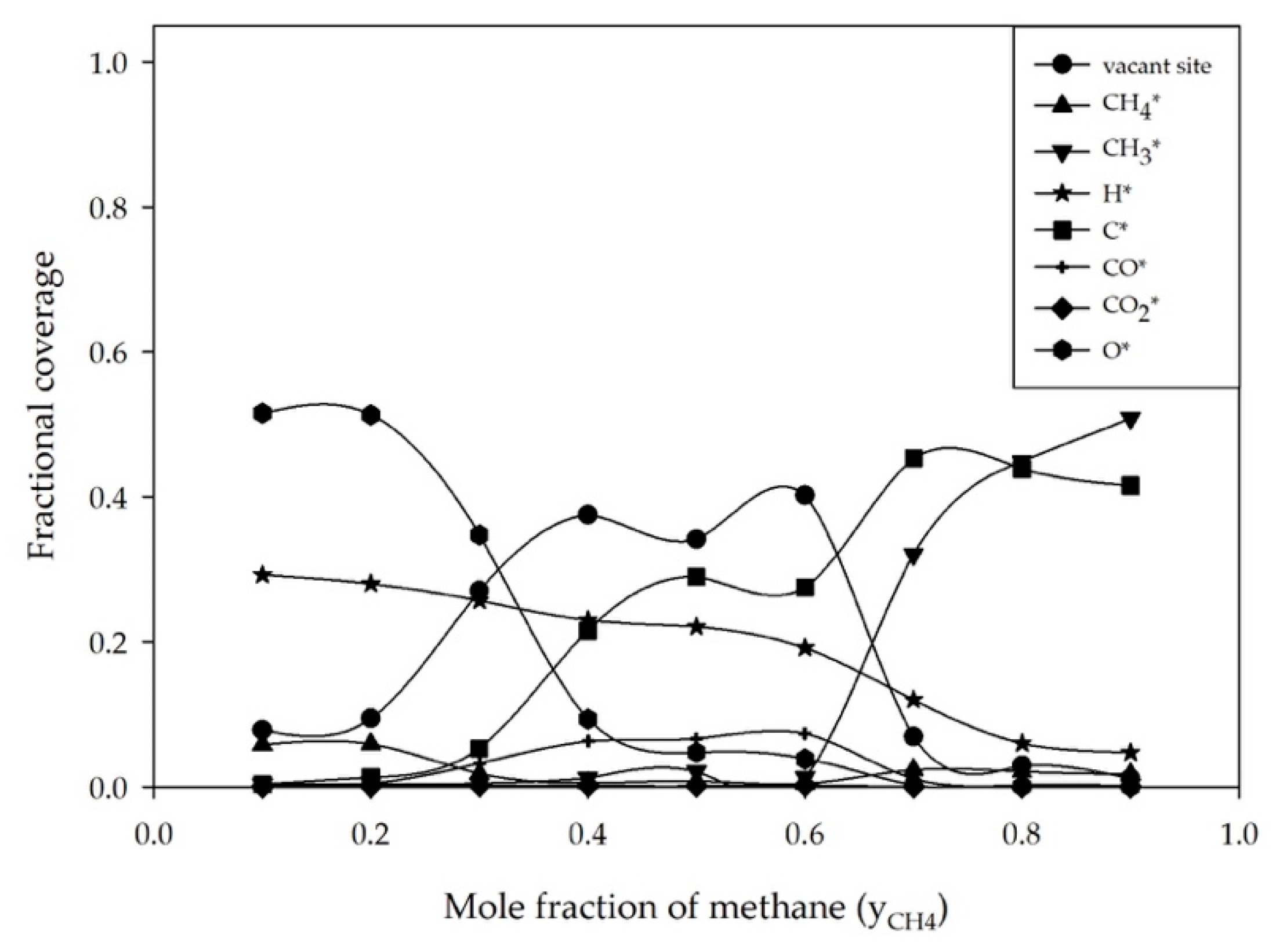
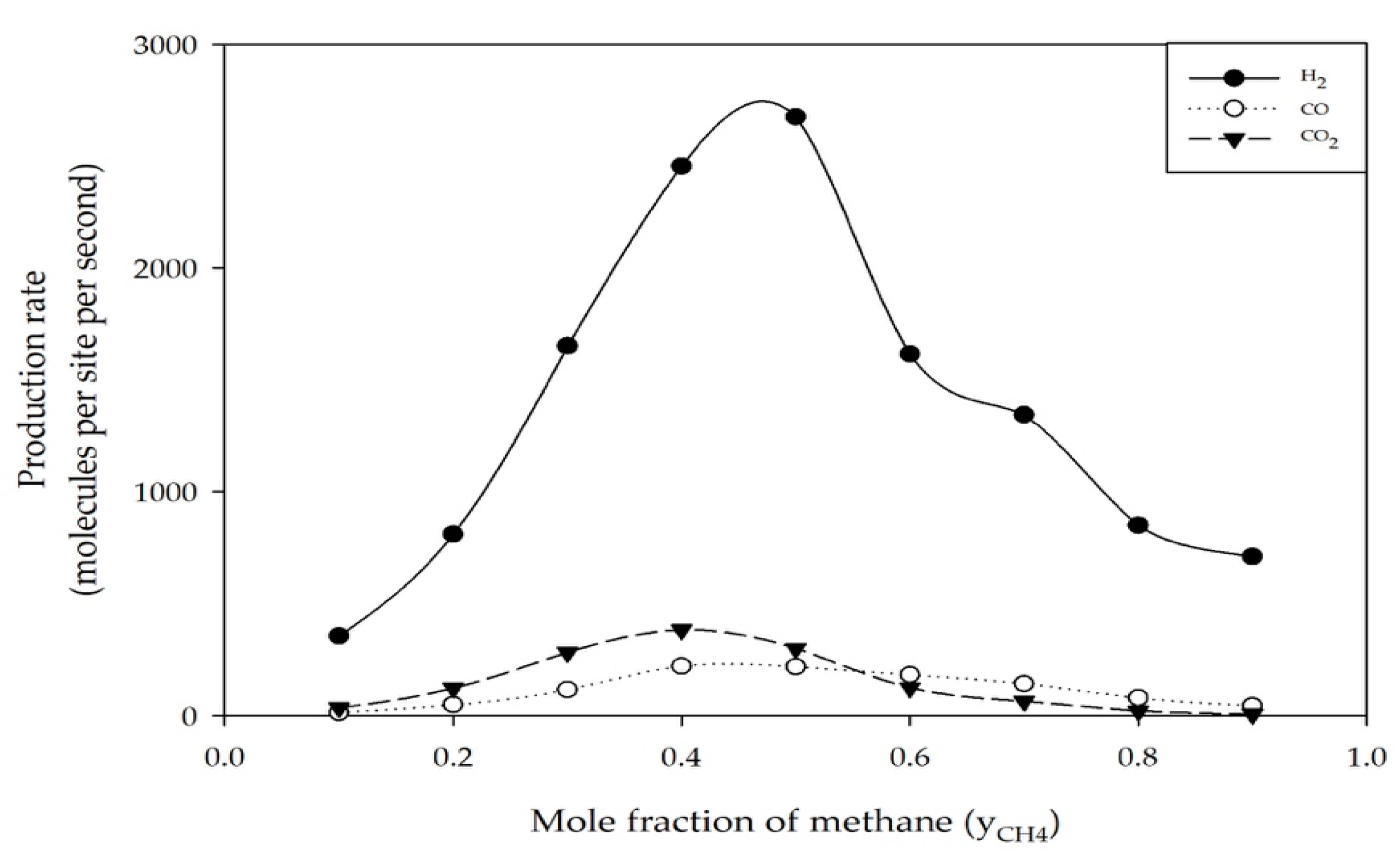
2.1. Effect of Feed Concentration
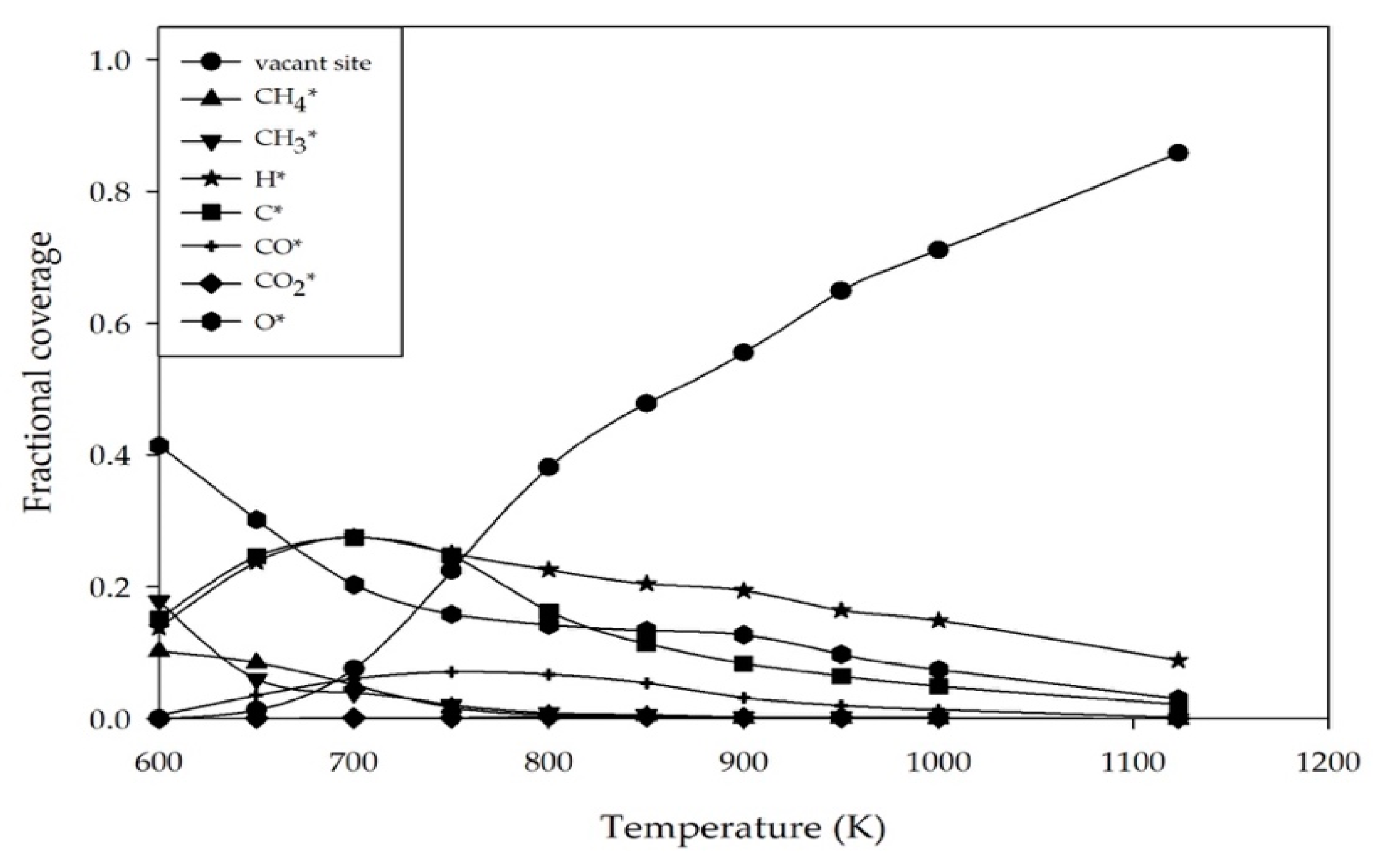
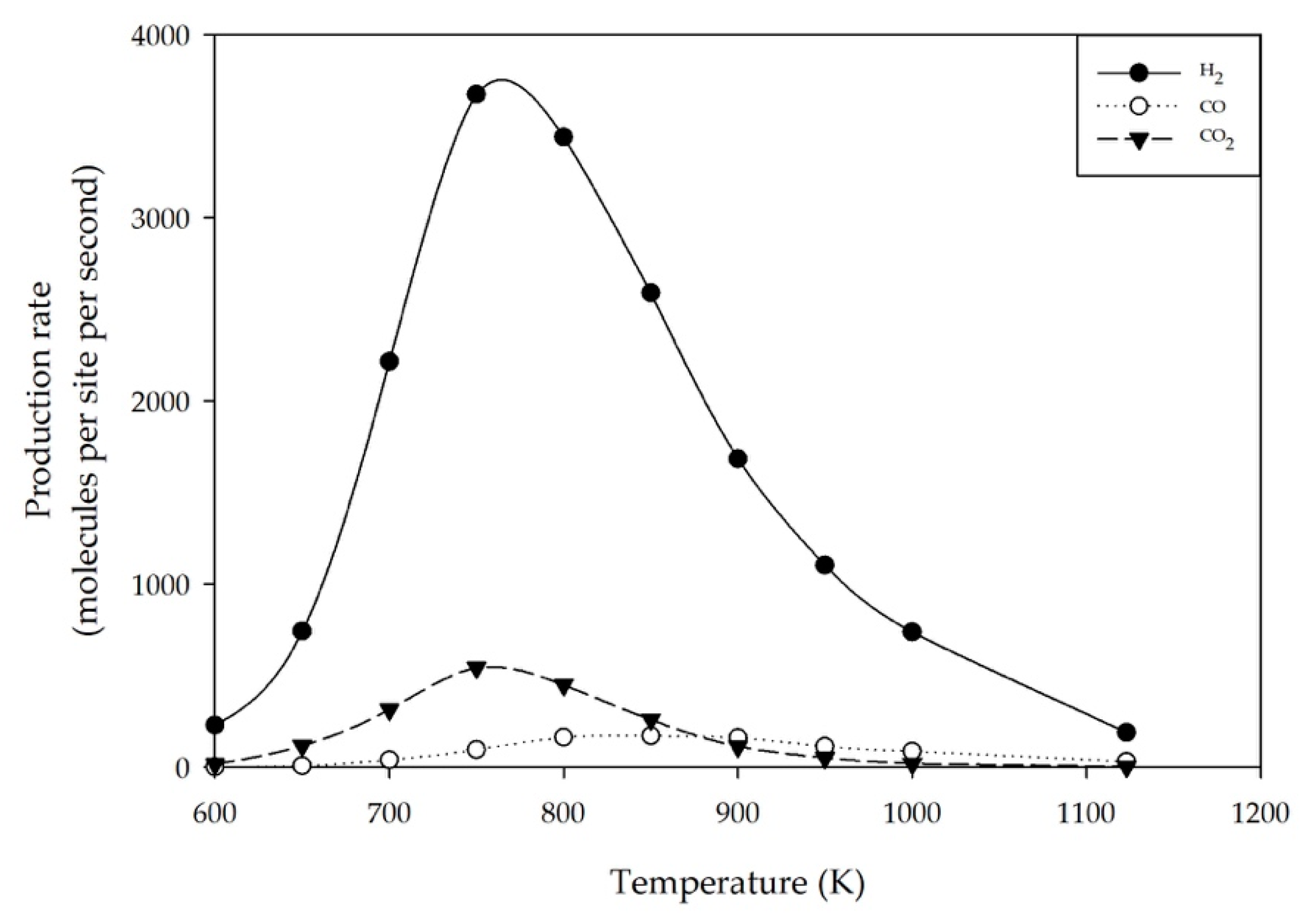
2.2. Effect of Reaction Temperature
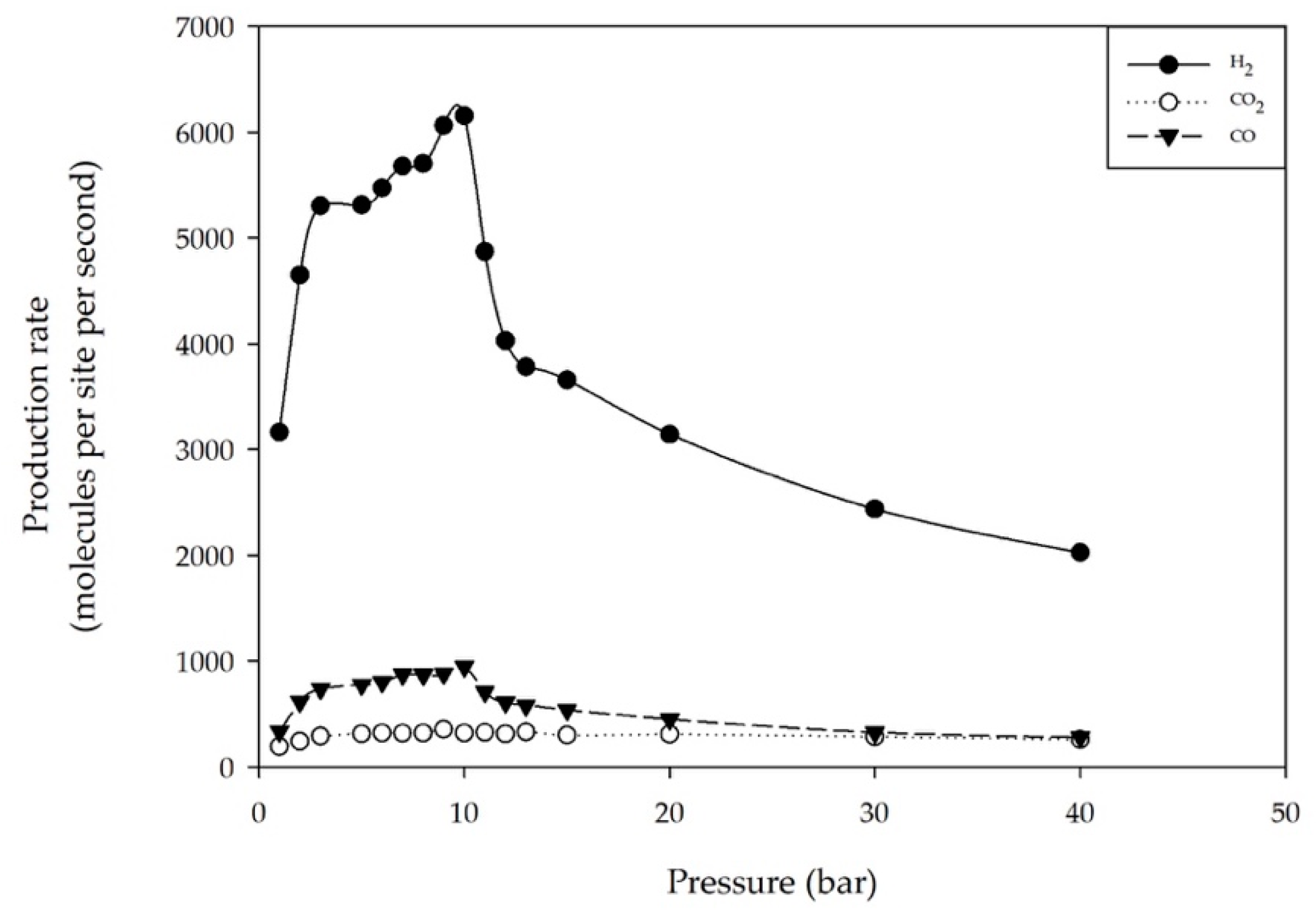
2.3. Effect of the Total Pressure of Reactants
3. Model and Simulation Procedure
3.1. Model
3.2. Simulation Procedure
- (a)
- Set the lattice site and initial configuration for the simulation.
- (b)
- Select one of the lattice sites randomly.
- (c)
- Perform the instantaneous event (steps 3, 4, 5, 6, 7, 8, 9, 11, 12, 14, 17, 20, 22, 24, 25, 28, 30, 31, and 34). If the conditions for the selected site and its neighboring are satisfied, the surface reaction will spontaneously take place.
- (d)
- Calculate the possibility of an event i (), as defined by Equation (6), where corresponds to the rate constant of step i. An event i is chosen from the possible events, except for the instantaneous event (steps 1, 2, 10, 13, 15, 16, 18, 19, 21, 23, 27, 29, 32, and 33). The possibility of each event is between 0 and 1. This procedure is known in different sources as the Bortz–Kalos–Lebowitz (BKL), the Gillespie algorithm, or the Random Selection method [50,51].
- (e)
- Perform the reaction event i selected in step (d) according to the following processes:
- (e-1)
- Adsorption
- If the adsorption of H2O (step 1) selected with a random number is and the selected site is vacant (*), the event is successful, and steam then adsorbs into the site (H2O*). If the site is occupied, the attempt is terminated.
- If the adsorption of CH4 (step 2) selected with a random number is , the procedure will be similar to that of the adsorption of H2O.
- (e-2)
- Surface reaction
- If the surface reaction (steps 10, 13, 15, 16, 18, 19, 21, 23, 27, and 29) is selected with a selection number is , and the selected site in step (b) is occupied by one of the reactants, a neighboring site next to the first site is then chosen randomly. The event is successful when the latter site is occupied by the other species of the same reaction. After that, the corresponding reaction is carried out, and both sites are then replaced by the products or one other is empty. If both sites are not occupied by the appropriate reactants, the attempt is terminated
- (e-3)
- Desorption
- If the desorption of H2, CO, or CO2 (step 31, 32, or 33, respectively) is selected with a random number is and the selected site in step (b) is occupied by the product, the event is successful and the product then desorbs from the site. The site is empty again because of the leaving of the product. If the site is not occupied by product, the attempt is terminated.
- (f)
- Update the time from t to by using Equation (7), where r is a uniformly distributed random number between 0 and 1, L is lattice length, and is the summation of all of the reaction constants, excluding the instantaneous events.
- (g)
- Repeat the algorithm from step (b) until the steady state is obtained.
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ishaq, H.; Dincer, I. Analysis and optimization for energy, cost and carbon emission for a solar driven steam-autothermal hybrid methane reforming for hydrogen, ammonia and power production. J. Clean. Prod. 2019, 234, 242–257. [Google Scholar] [CrossRef]
- Jokar, S.M.; Parvasi, P.; Basile, A. The performance evaluation of an industrial membrane reformer with catalyst-deactivation for a domestic methanol production plant. Int. J. Hydrogen Energy 2019, 44, 25730–25739. [Google Scholar] [CrossRef]
- Liu, C.; Li, S.; Dong, C.; Xiao, Y.; Li, T.; Wang, W. Hydrogen-rich syngas production by chemical looping steam reforming of acetic acid as bio-oil model compound over Fe-doped LaNiO3 oxygen carriers. Int. J. Hydrogen Energy 2019, 44, 17732–17741. [Google Scholar] [CrossRef]
- Noureldin, M.M.B.; Elbashir, N.O.; El-Halwagi, M.M. Optimization and selection of reforming approaches for syngas generation from natural/shale gas. Ind. Eng. Chem. Res. 2014, 53, 1841–1855. [Google Scholar] [CrossRef]
- Rezaei, E.; Dzuryk, S. Techno-economic comparison of reverse water gas shift reaction to steam and dry methane reforming reactions for syngas production. Chem. Eng. Res. Des. 2019, 144, 354–369. [Google Scholar] [CrossRef]
- Iulianelli, A.; Liguori, S.; Wilcox, J.; Basile, A. Advances on methane steam reforming to produce hydrogen through membrane reactors tecgnology: A review. Catal. Rev. 2016, 58, 1–35. [Google Scholar] [CrossRef]
- Park, H.-G.; Han, S.-Y.; Jun, K.-W.; Woo, Y.; Park, M.-J.; Kim, S.K. Bench-scale steam reforming of methane for hydrogen production. Catalysts 2019, 9, 615. [Google Scholar] [CrossRef]
- Navarro, M.V.; Plou, J.; López, J.M.; Grasa, G.; Murillo, R. Effect of oxidation-reduction cycles on steam-methane reforming kinetics over a nickel-based catalyst. Int. J. Hydrogen Energy 2019, 44, 12617–12627. [Google Scholar] [CrossRef]
- Bhat, S.A.; Sadhukhan, J. Process intensification aspects for steam methane reforming: An overview. AIChE J. 2009, 55, 408–422. [Google Scholar] [CrossRef]
- Okada, S.; Manabe, R.; Inagaki, R.; Ogo, S.; Sekine, Y. Methane dissociative adsorption in catalytic steam reforming of methane over Pd/CeO2 in an electric field. Catal. Today 2018, 307, 272–276. [Google Scholar] [CrossRef]
- Pashchenko, D. Experimental investigation of reforming and flow characteristics of a steam methane reformer filled with nickel catalyst of various shapes. Energy Convers. Manag. 2019, 185, 465–472. [Google Scholar] [CrossRef]
- De Castro, T.P.; Silveira, E.B.; Rabelo-Neto, R.C.; Borges, L.E.P.; Noronha, F.B. Study of the performance of Pt/Al2O3 and Pt/CeO2/Al2O3 catalysts for steam reforming of toluene, methane and mixtures. Catal. Today 2018, 299, 251–262. [Google Scholar] [CrossRef]
- Jakobsen, J.G.; Jørgensen, T.L.; Chorkendorff, I.; Sehested, J. Steam and CO2 reforming of methane over a Ru/ZrO2 catalyst. Appl. Catal. A Gen. 2010, 377, 158–166. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Colby, J.L.; Schmidt, L.D. Effects of biomass inorganics on rhodium catalysts: I. Steam methane reforming. Appl. Catal. B Environ. 2011, 107, 88–94. [Google Scholar] [CrossRef]
- Watanabe, F.; Kaburaki, I.; Shimoda, N.; Satokawa, S. Influence of nitrogen impurity for steam methane reforming over noble metal catalysts. Fuel Process. Technol. 2016, 152, 15–21. [Google Scholar] [CrossRef]
- Nieva, M.A.; Villaverde, M.M.; Monzón, A.; Garetto, T.F.; Marchi, A.J. Steam-methane reforming at low temperature on nickel-based catalysts. Chem. Eng. J. 2014, 235, 158–166. [Google Scholar] [CrossRef]
- Sebai, I.; Boulahaouache, A.; Trari, M.; Salhi, N. Preparation and characterization of 5%Ni/γ-Al2O3 catalysts by complexation with NH3 derivatives active in methane steam reforming. Int. J. Hydrogen Energy 2019, 44, 9949–9958. [Google Scholar] [CrossRef]
- Liu, C.-J.; Ye, J.; Jiang, J.; Pan, Y. Progresses in the preparation of coke resistant Ni-based catalyst for steam and CO2 reforming of methane. ChemCatChem 2011, 3, 529–541. [Google Scholar] [CrossRef]
- Zhou, L.; Li, L.; Wei, N.; Li, J.; Basset, J.-M. Effect of NiAl2O4 formation on Ni/Al2O3 stability during dry reforming of methane. ChemCatChem 2015, 7, 2508–2516. [Google Scholar] [CrossRef]
- Xu, J.; Froment, G.F. Methane steam reforming, methanation and water-gas shift: I. Intrinsic kinetics. AIChE J. 1989, 35, 88–96. [Google Scholar] [CrossRef]
- Chen, D.; Lødeng, R.; Svendsen, H.; Holmen, A. Hierarchical multiscale modeling of methane steam reforming reactions. Ind. Eng. Chem. Res. 2011, 50, 2600–2612. [Google Scholar] [CrossRef]
- Hou, K.; Hughes, R. The kinetics of methane steam reforming over a Ni/α-Al2O3 catalyst. Chem. Eng. J. 2001, 82, 311–328. [Google Scholar] [CrossRef]
- Sprung, C.; Arstad, B.; Olsbye, U. Methane steam reforming over a Ni/NiAl2O4 model catalyst-Kinetics. ChemCatChem 2014, 6, 1969–1982. [Google Scholar] [CrossRef]
- Delgado, K.H.; Maier, L.; Tischer, S.; Zellner, A.; Stotz, H.; Deutschmann, O. Surface reaction kinetics of steam- and CO2-reforming as well as oxidation of methane over nickel-based catalysts. Catalysts 2015, 5, 871–904. [Google Scholar] [CrossRef]
- Hess, F.; Over, H. Kinetic Monte Carlo simulations of heterogeneously catalyzed oxidation reactions. Catal. Sci. Technol. 2014, 4, 583–598. [Google Scholar] [CrossRef]
- Andersen, M.; Panosetti, C.; Reuter, K. A practical guide to surface kinetic Monte Carlo simulations. Front. Chem. 2019, 7, 202. [Google Scholar] [CrossRef]
- Schulze, T.P. Efficient kinetic monte carlo simulation. J. Comput. Phys. 2008, 227, 2455–2462. [Google Scholar] [CrossRef]
- Jansen, A.P.J.; Lukkien, J.J. Dynamic Monte-Carlo simulations of reactions in heterogeneous catalysis. Catal. Today 1999, 53, 259–271. [Google Scholar] [CrossRef]
- Ziff, R.; Gulari, E.; Barshad, Y. Kinetic phase transitions in an irreversible surface-reaction model. Phys. Rev. Lett. 1986, 56, 2553–2556. [Google Scholar] [CrossRef]
- Loscar, E.S.; Albano, E.V. Hysteretic effects in the first-order irreversible phase transition of the ZGB model. Comput. Phys. Commun. 2009, 180, 488–492. [Google Scholar] [CrossRef]
- Sinha, I.; Mukherjee, A.K. Monte Carlo simulation of a surface oxide model of CO oxidation. Chem. Phys. Lett. 2012, 553, 30–35. [Google Scholar] [CrossRef]
- Buendía, G.M.; Rikvold, P.A. A model for the catalytic oxidation of CO that includes CO desorption and diffusion, O repulsion, and impurities in the gas phase. Phys. A Stat. Mech. Appl. 2015, 424, 217–224. [Google Scholar] [CrossRef] [Green Version]
- De Andrade, M.F.; Figueiredo, W. Dynamical critical behavior of the Ziff–Gulari–Barshad model with quenched impurities. Phys. Lett. A 2016, 380, 2628–2631. [Google Scholar] [CrossRef]
- Pruksawan, S.; Kitiyanan, B.; Ziff, R.M. Partial oxidation of methane on a nickel catalyst: Kinetic Monte-Carlo simulation study. Chem. Eng. Sci. 2016, 147, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Prats, H.; Álvarez, L.; Illas, F.; Sayós, R. Kinetic Monte Carlo simulations of the water gas shift reaction on Cu(111) from density functional theory based calculations. J. Catal. 2016, 333, 217–226. [Google Scholar] [CrossRef]
- Fichthorn, K.; Gulari, E.; Ziff, R. Self-sustained oscillations in a heterogeneous catalytic reaction: A Monte Carlo simulation. Chem. Eng. Sci. 1989, 44, 1403–1411. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.J.; Matera, S.; Reuter, K. kmos: A lattice kinetic Monte Carlo framework. Comput. Phys. Commun. 2014, 185, 2138–2150. [Google Scholar] [CrossRef] [Green Version]
- Leetmaa, M.; Skorodumova, N.V. KMCLib: A general framework for lattice kinetic Monte Carlo (KMC) simulations. Comput. Phys. Commun. 2014, 185, 2340–2349. [Google Scholar] [CrossRef] [Green Version]
- Sprung, C.; Kechagiopoulos, P.N.; Thybaut, J.W.; Arstad, B.; Olsbye, U.; Marin, G.B. Microkinetic evaluation of normal and inverse kinetic isotope effects during methane steam reforming to synthesis gas over a Ni/NiAl2O4 model catalyst. Appl. Catal. A Gen. 2015, 492, 231–242. [Google Scholar] [CrossRef]
- Maier, L.; Schädel, B.; Delgado, K.H.; Tischer, S.; Deutschmann, O. Steam reforming of methane over nickel: Development of a multi-step surface reaction mechanism. Top. Catal. 2011, 54, 845–858. [Google Scholar] [CrossRef]
- Osman, A.I.; Meudal, J.; Laffir, F.; Thompsom, J.; Rooney, D. Enhanced catalytic activity of Ni on η-Al2O3 and ZSM-5 on addition of ceria zirconia for the partial oxidation of methane. Appl. Catal. B Environ. 2017, 212, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-B.; Gau, G.-Y.; Gau, S.-J.; Tang, C.-W.; Bi, J.-L. Preparation and characterization of nanosized nickel oxide. Catal. Lett. 2005, 101, 241–247. [Google Scholar] [CrossRef]
- Hacarlioglu, P.; Gu, Y.; Oyama, S.T. Studies of the methane steam reforming reaction at high pressure in a ceramic membrane reactor. J. Nat. Gas Chem. 2006, 15, 73–81. [Google Scholar] [CrossRef]
- Al-Sayari, S.A. Recent developments in the partial oxidation of methane to syngas. Open Catal. J. 2013, 6, 17–28. [Google Scholar] [CrossRef]
- Fichthorn, K.A.; Weinberg, W.H. Theoretical foundations of dynamic Monte Carlo simulations. J. Chem. Phys. 1991, 95, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Gelten, R.J.; van Santen, R.A.; Jansen, A.P.J. Chapter 18 Dynamic Monte Carlo simulations of oscillatory heterogeneous catalytic reactions. Theor. Comput. Chem. 1999, 7, 737–784. [Google Scholar]
- Raimondeau, S.; Vlachos, D.G. Recent developments on multiscale, hierarchical modeling of chemical reactors. Chem. Eng. J. 2002, 90, 3–23. [Google Scholar] [CrossRef]
- Cortés, J.; Valencia, E.; Araya, P. Monte Carlo simulations in the preferential oxidation of carbon monoxide on a copper-ceria catalyst. Chem. Phys. Lett. 2014, 612, 97–100. [Google Scholar] [CrossRef]
- Cortés, J.; Valencia, E.; Araya, P. Monte Carlo simulation studies of the catalytic combustion of methane. Catal. Lett. 2006, 112, 121–128. [Google Scholar] [CrossRef]
- Bortz, A.B.; Kalos, M.H.; Lebowitz, J.L. A new algorithm for Monte Carlo simulation of Ising spin systems. J. Comput. Phys. 1975, 17, 10–18. [Google Scholar] [CrossRef]
- Gillespie, D.T. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976, 22, 403–434. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Steps (i) | Elementary Reaction | β | ||
|---|---|---|---|---|
| 1 | H2O(g) + [*] → [H2O*] | 2.70 103 () | 0 | 0 |
| 2 | CH4(g) + [*] → [CH4*] | 2.87 103 () | 0 | 0 |
| 3 b | [CH4*] + [*] → [CH3*] + [H*] | 4.10 1012 | 0.087 | 55.8 |
| 4 b | [CH3*] + [H*] → [CH4*] + [*] | 3.83 1013 | −0.087 | 63.4 |
| 5 b | [CH3*] + [*] → [CH2*] + [H*] | 4.10 1015 | 0.087 | 98.1 |
| 6 b | [CH2*] + [H*] → [CH3*] + [*] | 8.22 1014 | −0.087 | 57.2 |
| 7 b | [CH2*] + [*] → [CH*] + [H*] | 9.84 1015 | 0.087 | 95.2 |
| 8 b | [CH*] + [H*] → [CH2*] + [*] | 2.60 1016 | −0.087 | 81.0 |
| 9 b | [CH*] + [*] → [C*] + [H*] | 2.63 1012 | 0.500 | 21.9 |
| 10 | [C*] + [H*] → [CH*] + [*] | 4.52 1015 | −0.500 | 157.9 |
| 11 b | [CH4*] + [O*] → [CH3*] + [OH*] | 1.49 1016 | −0.101 | 92.7 |
| 12 b | [CH3*] + [OH*] → [CH4*] + [O*] | 7.93 1013 | 0.101 | 25.8 |
| 13 | [CH3*] + [O*] → [CH2*] + [OH*] | 3.25 1016 | −0.101 | 134.6 |
| 14 b | [CH2*] + [OH*] → [CH3*] + [O*] | 3.70 1012 | 0.101 | 19.0 |
| 15 | [CH2*] + [O*] → [CH*] + [OH*] | 3.25 1016 | −0.101 | 131.3 |
| 16 | [CH*] + [OH*] → [CH2*] + [O*] | 1.17 1014 | 0.101 | 42.4 |
| 17 b | [CH*] + [O*] → [C*] + [OH*] | 6.57 1012 | 0.312 | 57.7 |
| 18 | [C*] + [OH*] → [CH*] + [O*] | 6.46 1012 | −0.312 | 118.9 |
| 19 | [H2O*] + [*] → [H*] + [OH*] | 9.76 1012 | −0.086 | 92.9 |
| 20 b | [H*] + [OH*] → [H2O*] + [*] | 4.92 1011 | 0.086 | 41.5 |
| 21 | [H*] + [O*] → [OH*] + [*] | 1.05 1015 | −0.188 | 104.3 |
| 22 b | [OH*] + [*] → [H*] + [O*] | 5.99 1011 | 0.188 | 29.6 |
| 23 | [C*] + [O*] → [CO*] + [*] | 9.04 1014 | 0 | 148.1 |
| 24 b | [C*] + [OH*] → [H*] + [CO*] | 1.03 1017 | 0.188 | 62.5 |
| 25 b | [CO*] + [O*] → [CO2*] + [*] | 5.32 1010 | 0 | 123.6 |
| 26 b | [CO2*] + [*] → [CO*] + [O*] | 1.23 1015 | −1.000 | 89.3 |
| 27 | [CO*] + [OH*] → [COOH*] + [*] | 1.60 1013 | 0.213 | 97.6 |
| 28 b | [COOH*] + [*] → [CO*] + [OH*] | 3.88 1015 | −0.213 | 54.3 |
| 29 | [CO2*] + [H*] → [COOH*] + [*] | 1.66 1016 | −0.475 | 117.2 |
| 30 b | [COOH*] + [*] → [CO2*] + [H*] | 9.92 1011 | 0.475 | 33.6 |
| 31 b | [H*] + [H*] → H2(g) + [*] + [*] | 6.76 1011 | 0 | 95.2 |
| 32 | [CO2*] → CO2(g) + [*] | 1.71 10−1 | 0 | 25.9 |
| 33 | [CO*] → CO(g) + [*] | 9.47 102 | 0 | 111.2 |
| 34 b | [H2O*] → H2O(g) + [*] | 9.92 103 | 0 | 60.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unruean, P.; Plianwong, T.; Pruksawan, S.; Kitiyanan, B.; Ziff, R.M. Kinetic Monte-Carlo Simulation of Methane Steam Reforming over a Nickel Surface. Catalysts 2019, 9, 946. https://doi.org/10.3390/catal9110946
Unruean P, Plianwong T, Pruksawan S, Kitiyanan B, Ziff RM. Kinetic Monte-Carlo Simulation of Methane Steam Reforming over a Nickel Surface. Catalysts. 2019; 9(11):946. https://doi.org/10.3390/catal9110946
Chicago/Turabian StyleUnruean, Palawat, Teetuch Plianwong, Sirawit Pruksawan, Boonyarach Kitiyanan, and Robert M. Ziff. 2019. "Kinetic Monte-Carlo Simulation of Methane Steam Reforming over a Nickel Surface" Catalysts 9, no. 11: 946. https://doi.org/10.3390/catal9110946