{CeO2/Bi2Mo1−xRuxO6} and {Au/Bi2Mo1−xRuxO6} Catalysts for Low-Temperature CO Oxidation

 , , ,
, , ,
Abstract
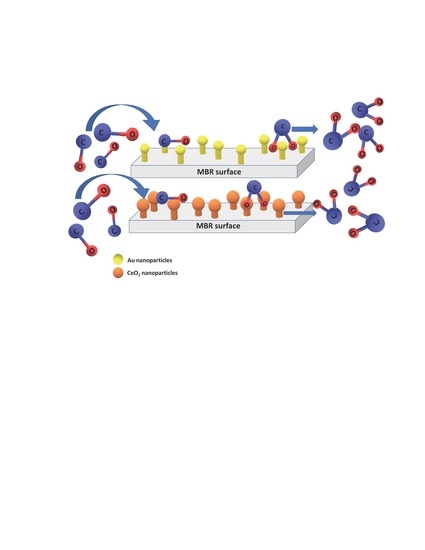
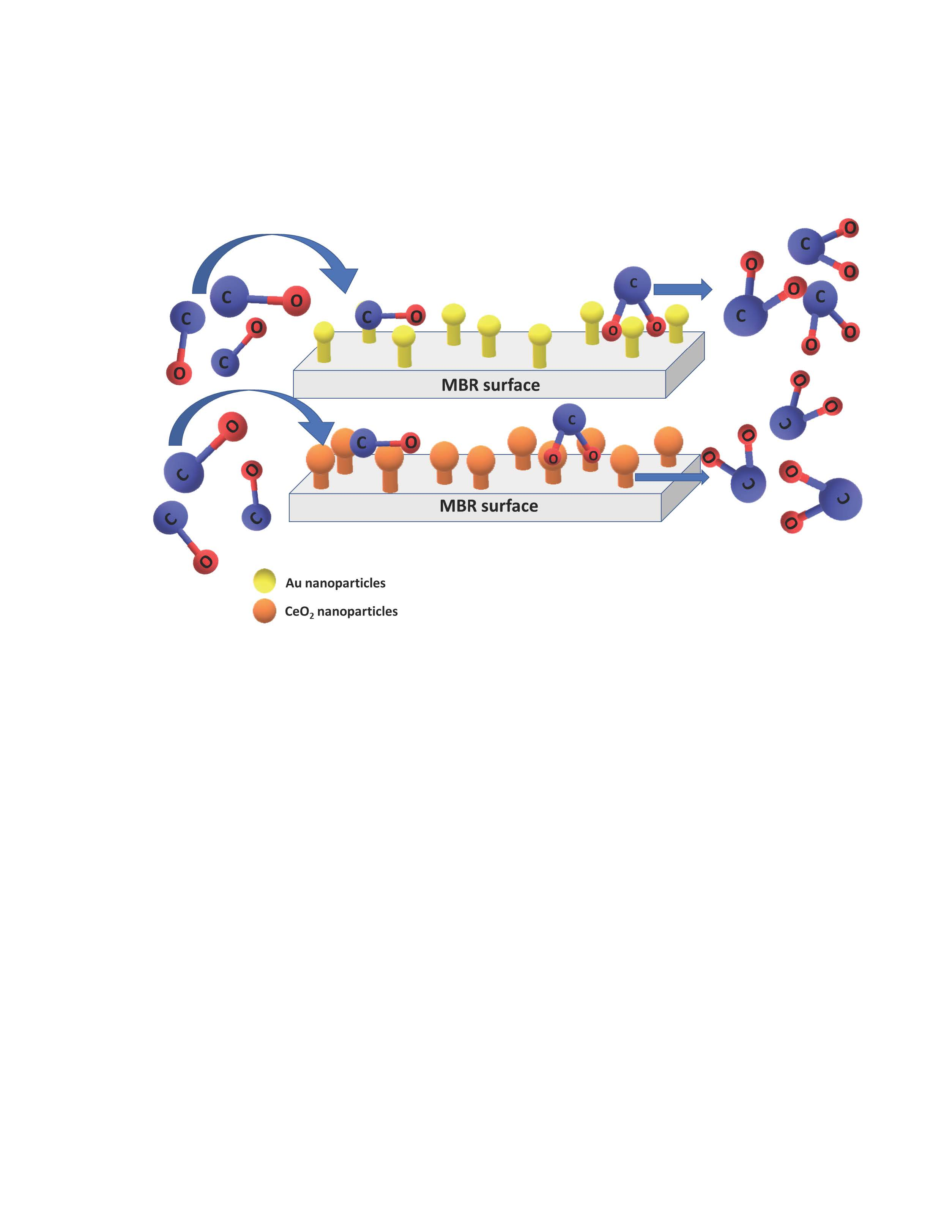
:
1. Introduction
2. Results
2.1. Scanning Electron Microscopy (SEM)
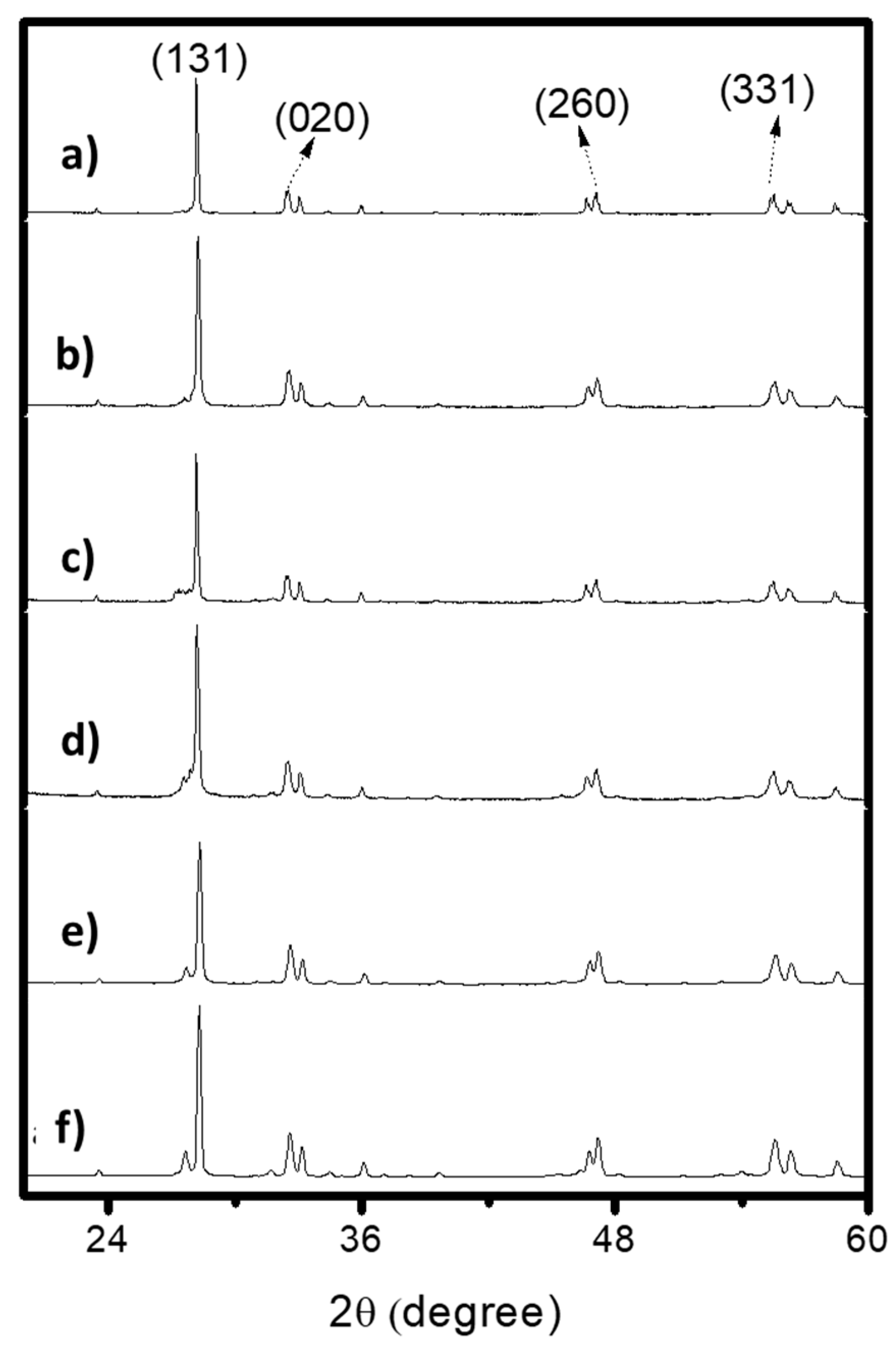
2.2. X-Ray Diffraction (XRD)
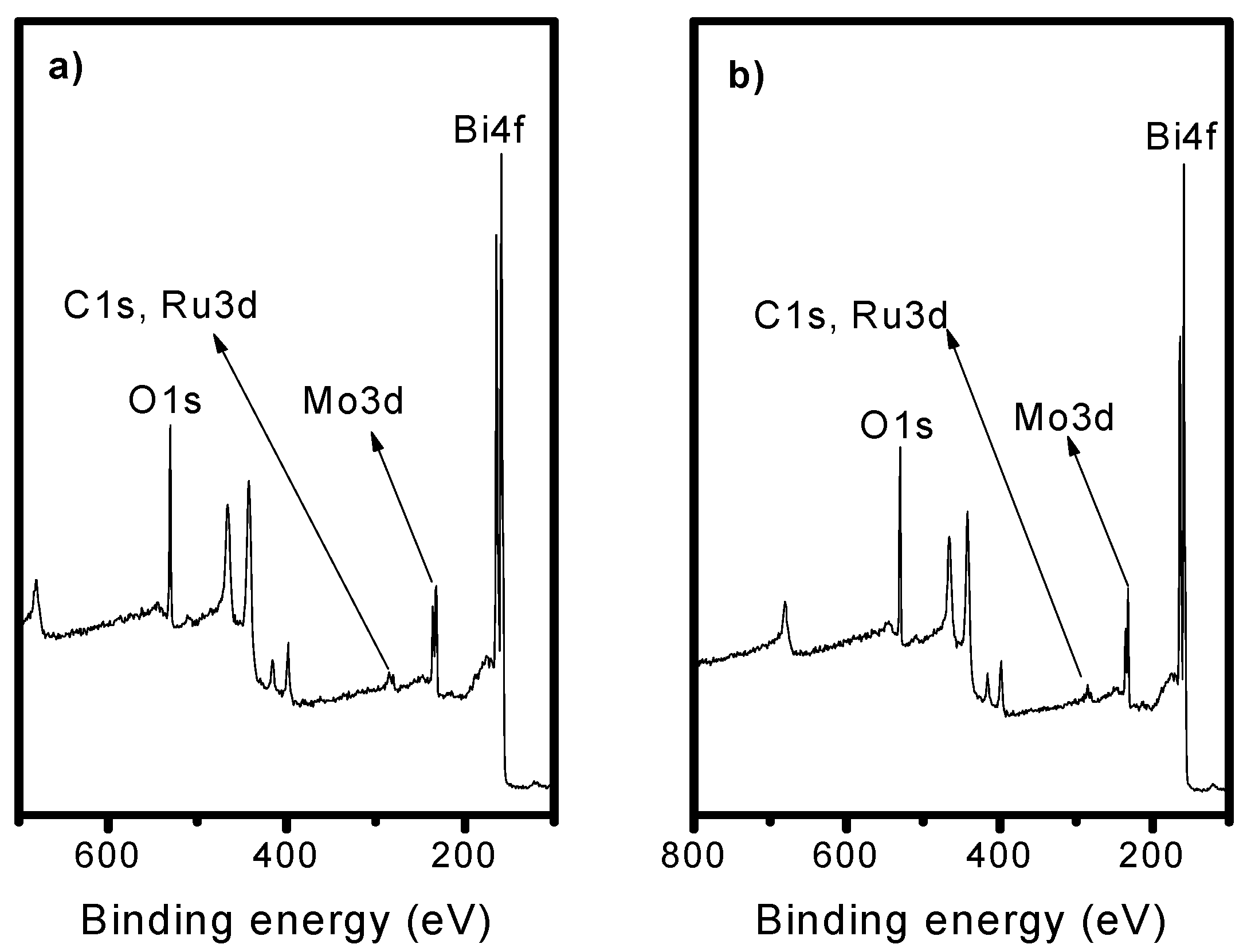
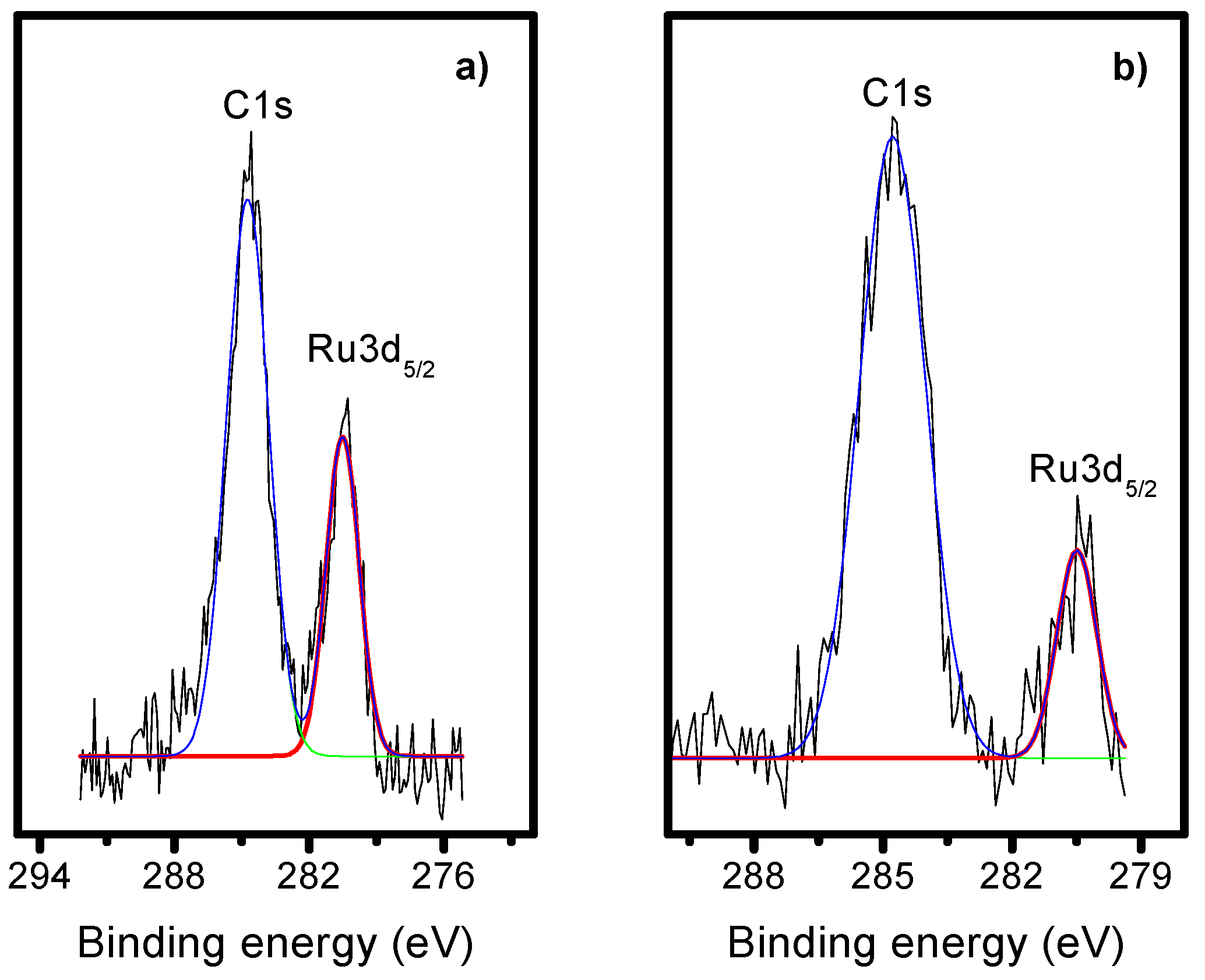
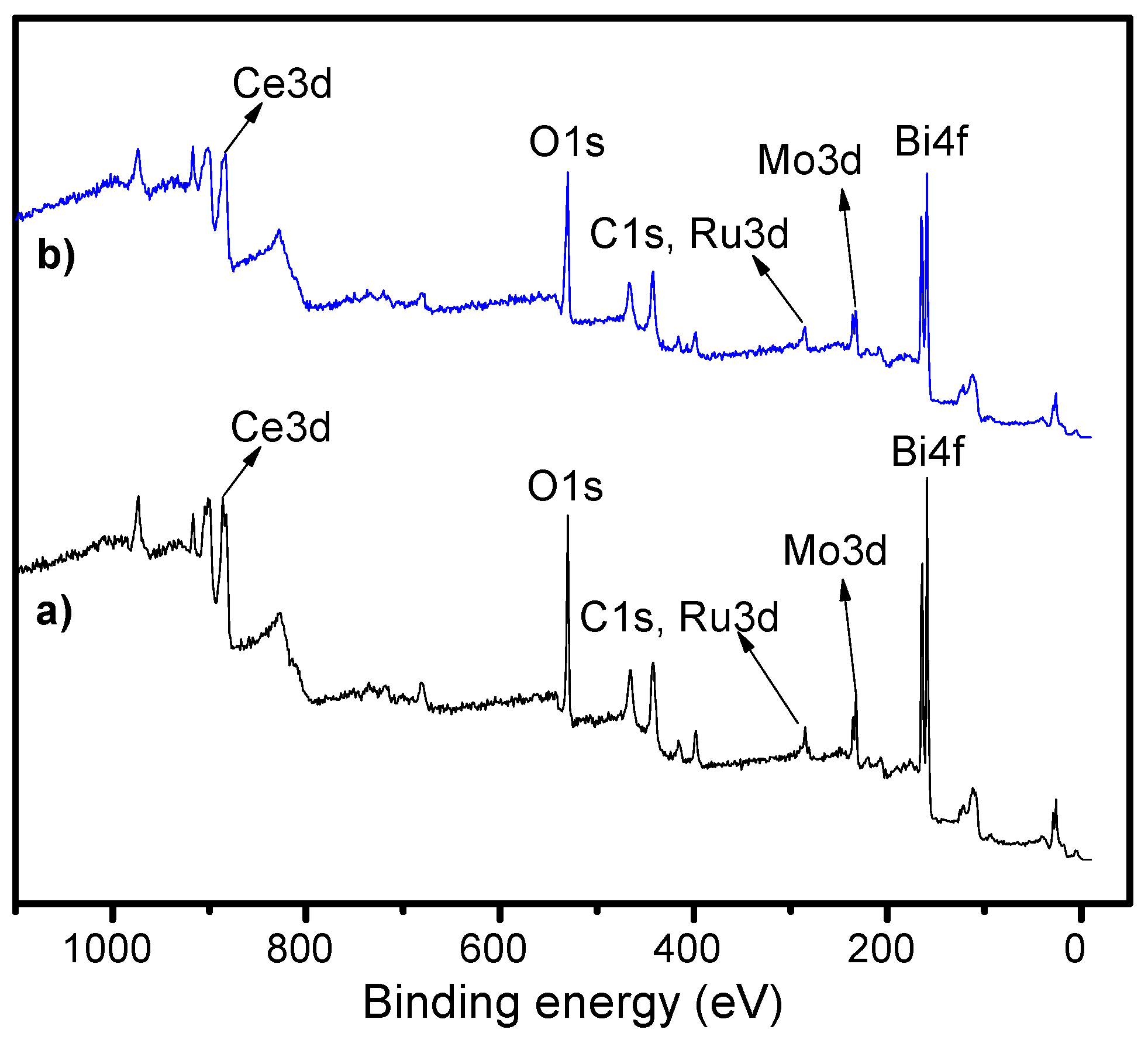
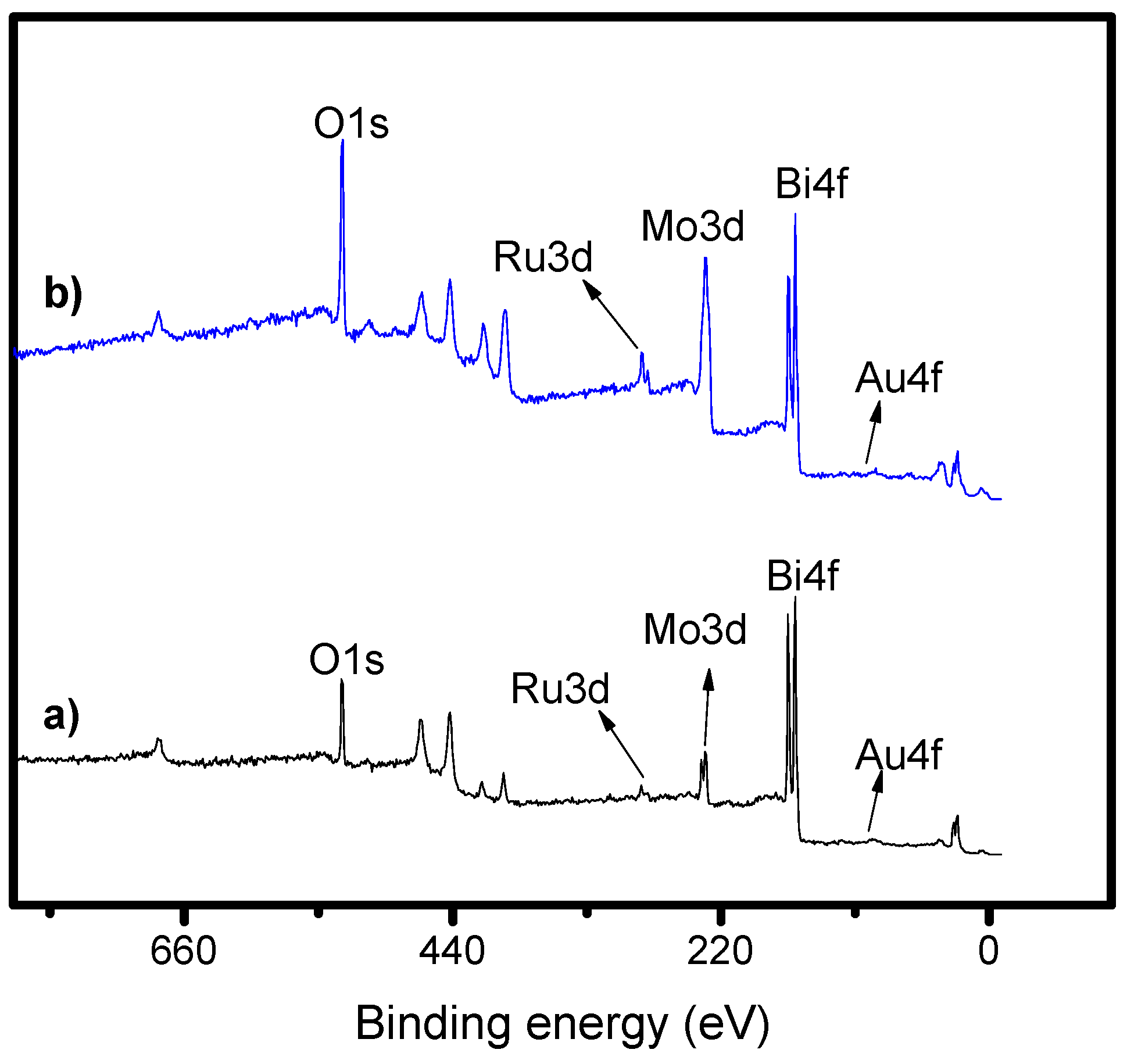
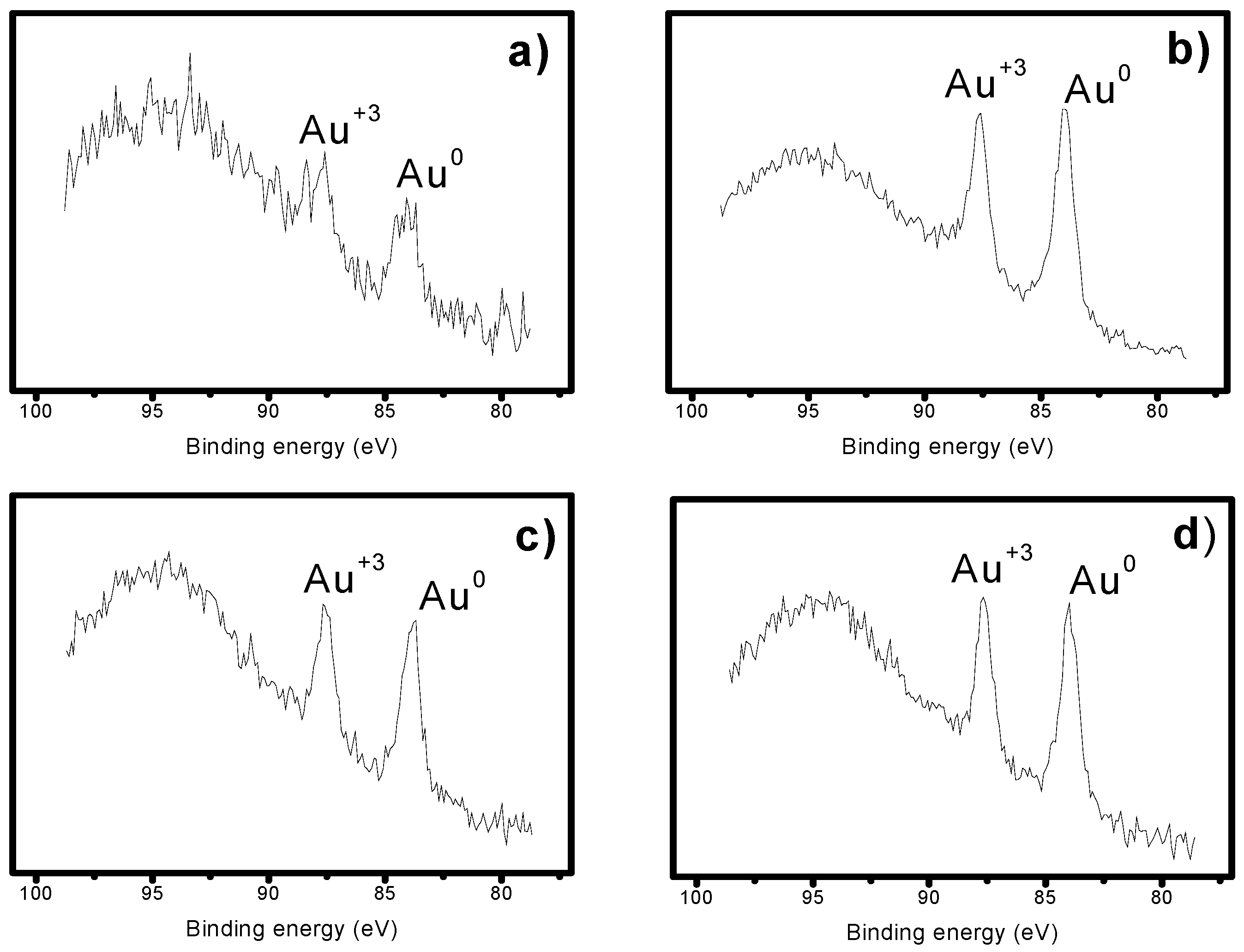
2.3. X-Ray Photoelectron Spectroscopy (XPS)
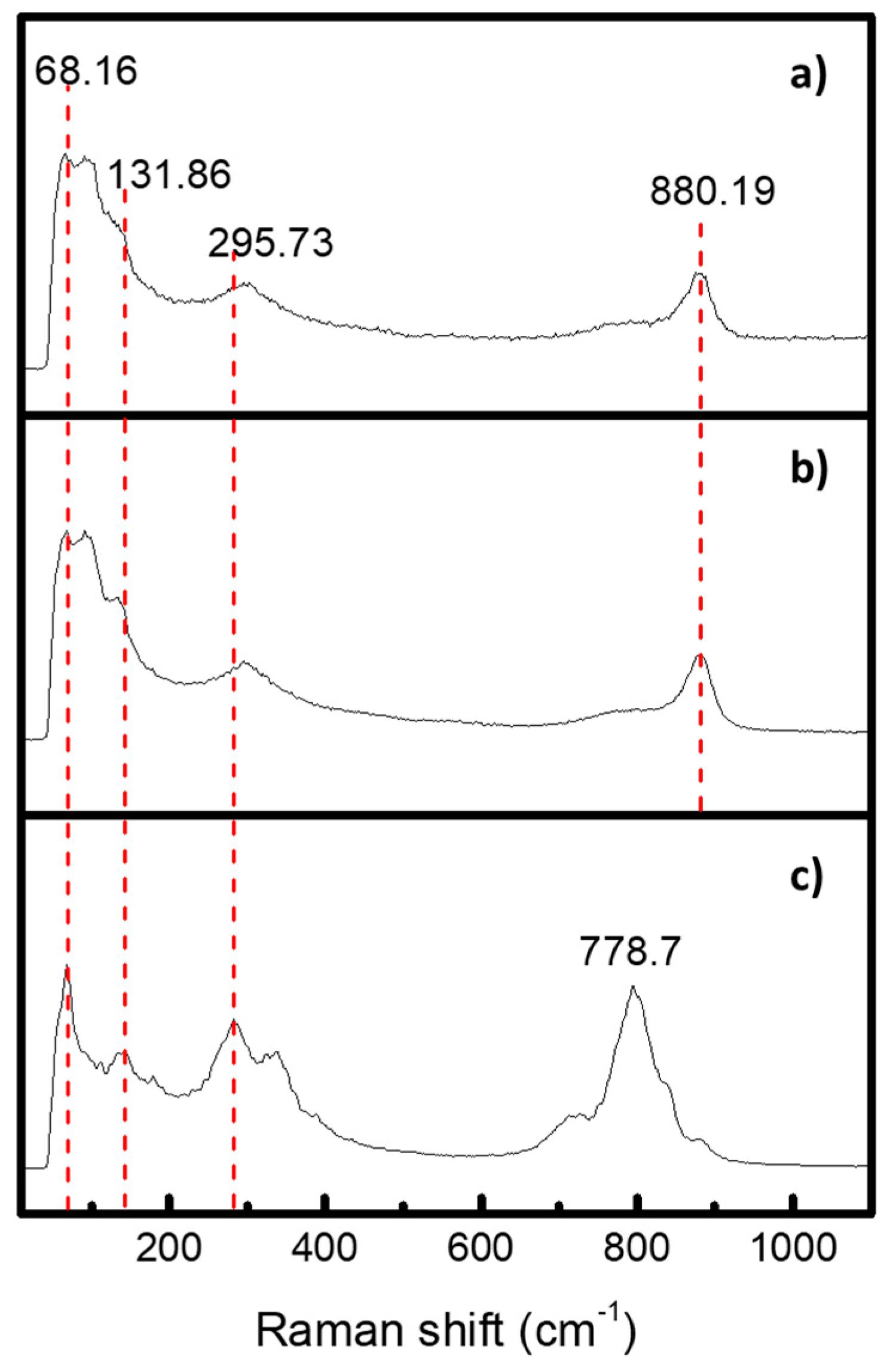
2.4. Raman Spectroscopy
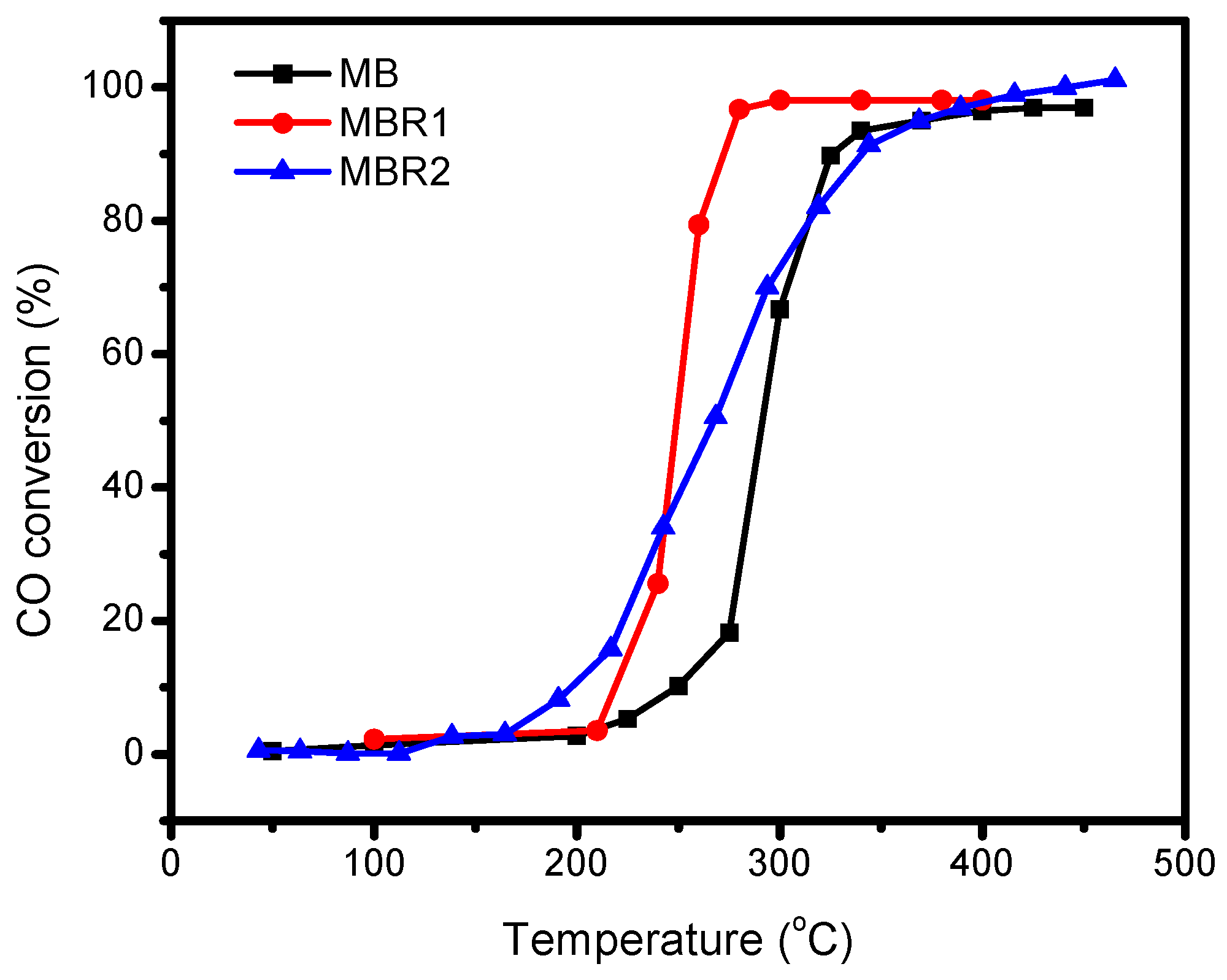
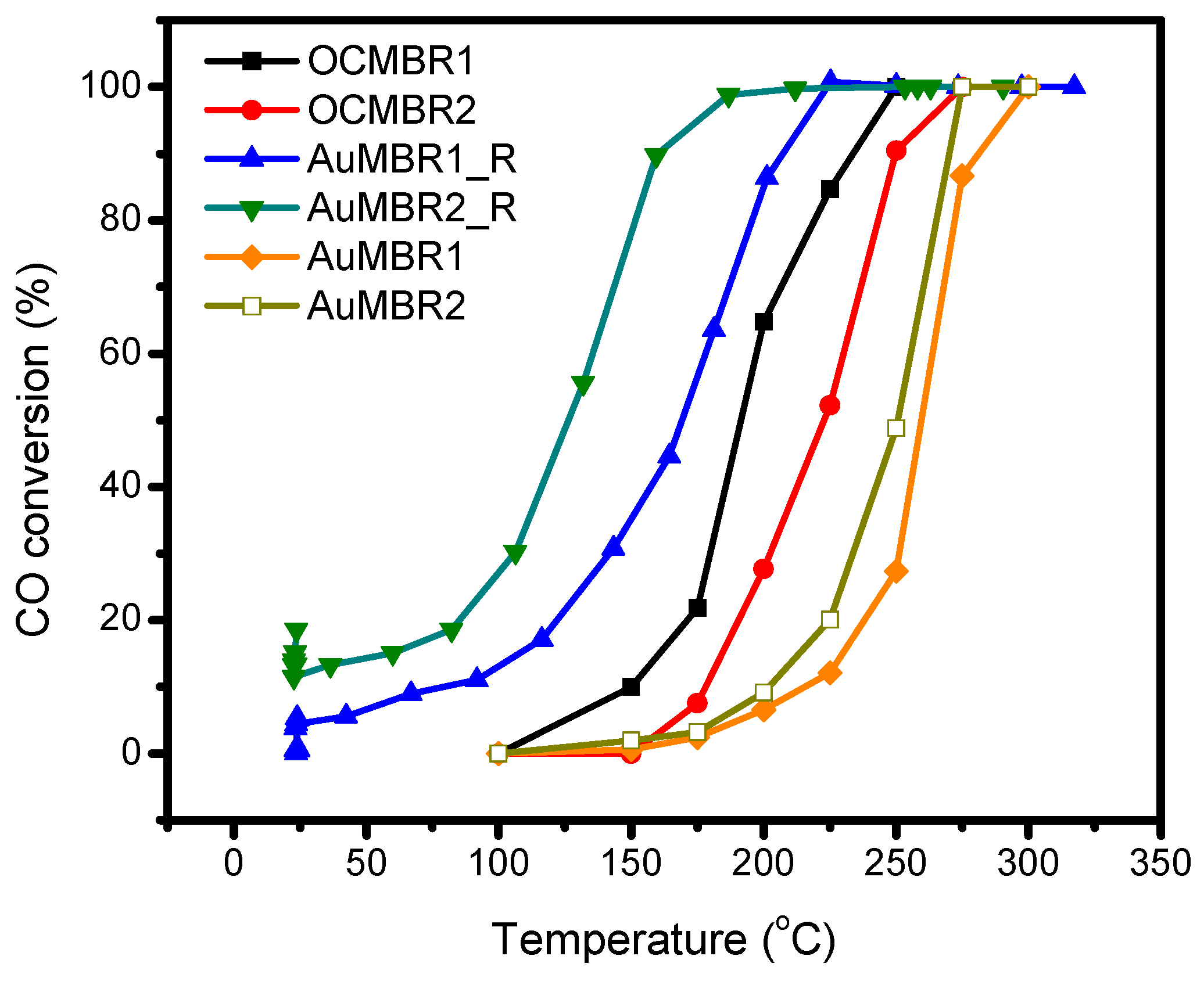
2.5. Catalytic Activity Tests
3. Discussion
4. Materials and Methods
4.1. Catalysts Preparation
4.2. Characterization
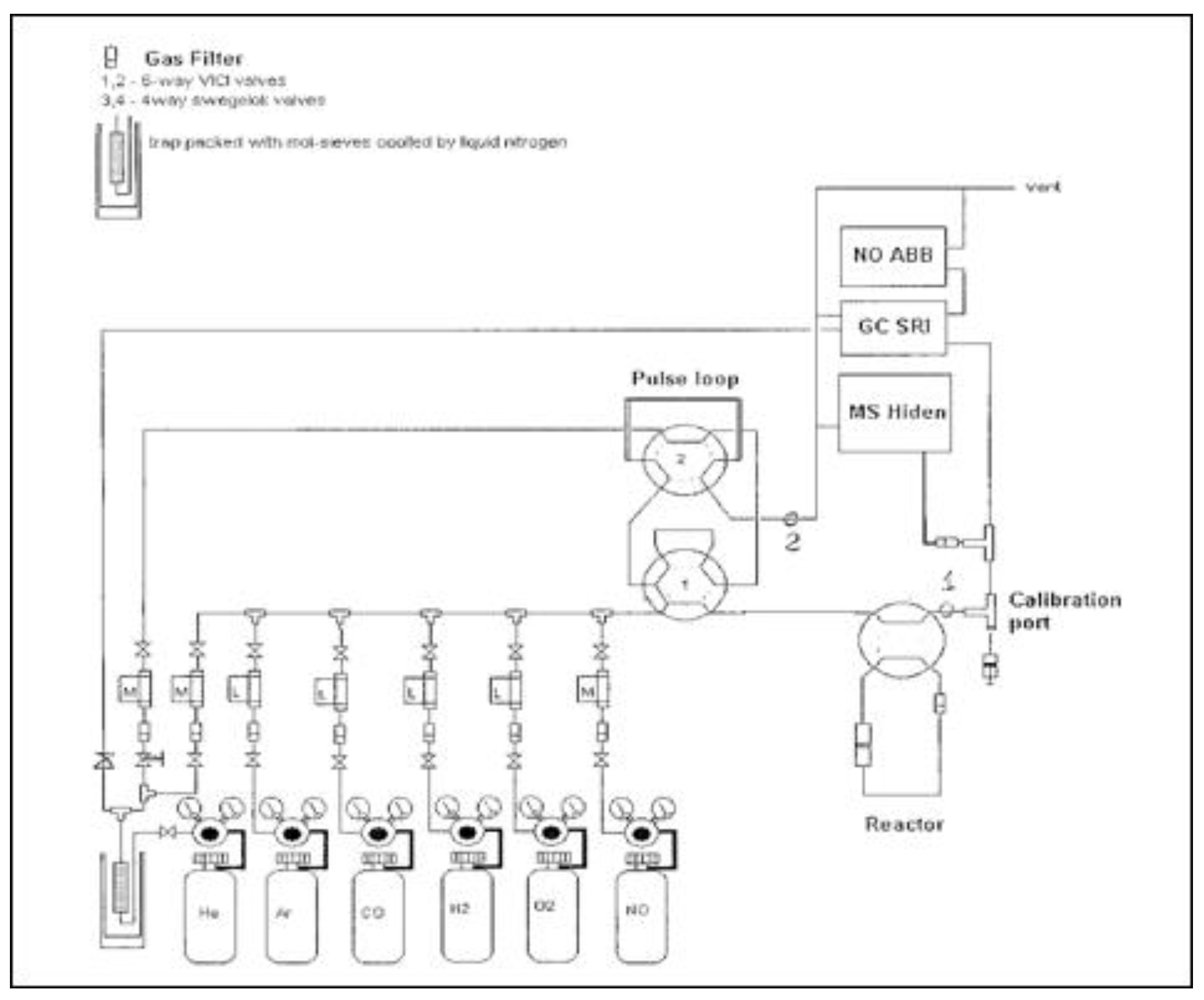
4.3. Catalytic Assessment
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tit, N.; Said, K.; Mahmoud, N.M.; Kouser, S.; Yamani, Z.H. Ab-initio investigation of adsorption of CO and CO2 molecules on graphene: Role of intrinsic defects on gas sensing. Appl. Surf. Sci. 2017, 394, 219–230. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, J.; Tan, Z.; Liu, T.; Weilin, Z.; Li, X.; Huang, C.; Wang, S.; Huang, Z.; Ma, W. Ambient carbon monoxide and increased risk of daily hospital outpatient visits for respiratory diseases in Dougguan, China. Sci. Total Environ. 2019, 668, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.I.; Kolentsova, E.N.; Dimitrov, D.Y.; Avdeev, G.; Tabakova, T. Alumina supported copper-manganese catalysts for combustion of exhaust gases: Catalysts characterization. In Proceedings of the XIII International Conference on Chemical Engineering and Applications, Hong Kong, 27–28 August 2015. [Google Scholar]
- Dey, S.; Dhal, G.C. Materials progress in the control of CO and CO2 emission at ambient conditions: An overview. Mater. Sci. Energy Technol. 2019, 2, 607–623. [Google Scholar] [CrossRef]
- Rangel, R.; Maya-Yescas, R.; García, R. Novel [Ce1−xLaxO2, La2−yCeyO3]/Bi2Mo0.9W0.1O6 catalysts for CO oxidation at low temperature. Catal. Sci. Technol. 2012, 2, 639–642. [Google Scholar] [CrossRef]
- Keulks, G.W.; Hall, J.L.; Suzuki, C.D. The catalytic oxidation of propylene: IV. Preparation and characterization of a-bismuth molybdate. J. Catal. 1974, 34, 79–97. [Google Scholar] [CrossRef]
- Trifiro, F.; Hoser, H.; Scarle, R.D. Relationship between structure and activity of mixed oxide as oxidation catalysts. I. Preparation and solid-state reactions of Bi-molybdates. J. Catal. 1972, 25, 12–24. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Singh, A.; Shukla, C.S. Kinetics and mechanism of solid-state reaction between bismuth(III) oxide and molybdenum(VI) oxide. J. Solid State Chem. 1982, 42, 136–148. [Google Scholar] [CrossRef]
- Le, M.T.; Bac, L.H.; van Driessche, I.; Hoste, S.; van Well, W.J.M. The synergy effect between gamma and beta phase of bismuth molybdate catalysts: Is there any relation between conductivity and catalytic activity? Catal. Today 2008, 131, 566–571. [Google Scholar] [CrossRef]
- Le, M.T.; van Craenenbroeck, J.; van Driessche, I.; Hoste, S. Bismuth molybdate catalysts synthesized using spray drying for the selective oxidation of propylene. Appl. Catal. A Gen. 2003, 249, 355–364. [Google Scholar] [CrossRef]
- Carrazán, S.R.G.; Martín, C.; Mateos, R.; Rives, V. Influence of the active phase structure Bi-Mo-Ti-O in the selective oxidation of propene. Catal. Today 2006, 112, 121–125. [Google Scholar] [CrossRef]
- Moro-oka, Y.; Ueda, W. Multicomponent bismuth molybdate catalyst: A highly functionalized catalyst system for the selective oxidation of olefin. Adv. Catal. 1994, 40, 233–273. [Google Scholar]
- Schuh, K.; Kleist, W.; Høj, M.; Trouillet, V.; Beato, P.; Jensen, A.D.; Patzke, G.R.; Grunwaldt, J.D. Selective oxidation of propylene to acrolein by hydrothermally synthesized bismuth molybdates. Appl. Catal. A Gen. 2014, 482, 145–156. [Google Scholar] [CrossRef]
- Brazdil, J.F.; Toft, M.A.; Lin, S.S.Y.; McKenna, S.T.; Zajac, G.; Kaduk, J.A.; Golab, J.T. Characterization of bismuth-cerium-molybdate selective propylene ammoxidation catalysts. Appl. Catal. A Gen. 2015, 495, 115–123. [Google Scholar] [CrossRef]
- Rangel, R.; López, J.L.C.; Espino, J.; Núñez-González, R.; Bartolo-Pérez, P.; Gómez-Cortés, A.; Díaz, G. Estudio del efecto de diferentes soportes mixtos en la actividad catalitica y las caracteristicas estructurales de catalizadores de Bi2MoxW1−xO6. Rev. Int. Contam. Ambient. 2014, 30, 841–848. [Google Scholar]
- Park, J.H.; Shin, C.H. Influence of the catalyst composition in the oxidative dehydrogenation of 1-butene over BiVxMo1−x oxide catalysts. Appl. Catal. A Gen. 2015, 495, 1–7. [Google Scholar] [CrossRef]
- Joo, S.H.; Park, J.Y.; Renzas, J.R.; Butcher, D.R.; Huang, W.; Somorjai, G.A. Size effect of ruthenium nanoparticles in catalytic carbon monoxide oxidation. Nano Lett. 2010, 10, 2709–2713. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Mizuno, N. Supported ruthenium catalyst for the heterogeneous oxidation of alcohols with molecular oxygen. Angew. Chem. Int. Ed. 2002, 114, 4538–4542. [Google Scholar] [CrossRef]
- Satsuma, A.; Yanagihara, M.; Ohyama, J.; Shimizu, K. Oxidation of CO over Ru/Ceria prepared by self-dispersion of Ru metal powder into nano-sized particle. Catal. Today 2013, 201, 62–67. [Google Scholar] [CrossRef]
- Gaálová, J.; Barbier, J.; Rossignol, S. Ruthenium versus platinum on cerium materials in wet air oxidation of acetic acid. J. Hazard. Mater. 2010, 181, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, H.M.; Henkelman, G. CO oxidation mechanism on CeO2-supported Au nanoparticles. J. Am. Chem. Soc. 2011, 134, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Zhang, R.; Lu, K.; Zong, L.; Tong, S.; Wang, X.; Feng, G. Gold supported on ceria nanotubes for CO oxidation. Appl. Surf. Sci. 2017, 416, 183–190. [Google Scholar] [CrossRef]
- Acosta, B.; Smolentseva, E.; Beloshapkin, S.; Rangel, R.; Estrada, M.; Fuentes, S.; Simakov, A. Gold supported on ceria nanoparticles and nanotubes. Appl. Catal. A Gen. 2012, 449, 96–104. [Google Scholar] [CrossRef]
- Fonseca, J.; Royer, S.; Bion, N.; Pirault-Roy, L.; do Carmo Rangel, M.; Duprez, D.; Epron, F. Preferential CO oxidation over nanosized gold catalysts supported on ceria and amorphous ceria-alumina. Appl. Catal. B Environ. 2012, 128, 10–20. [Google Scholar] [CrossRef]
- Aneggi, E.; Boaro, M.; Colussi, S.; de Leitenburg, C.; Trovarelli, A. Ceria-based materials in catalysis: Historical perspective and future trends. In Handbook of the Physics and Chemistry of Rare Earths; Bünzli, J.C., Pecharsky, V., Elsevier, B.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 50, pp. 209–242. [Google Scholar] [CrossRef]
- Ansari, S.A.; Khan, M.M.; Ansari, M.O.; Kalathil, S.; Lee, J.; Cho, M.H. Band-gap engineering of CeO2 nanostructure using an electrochemically active biofilm for visible light applications. RSC Adv. 2014, 4, 16782–16791. [Google Scholar] [CrossRef]
- Wu, T.S.; Chen, Y.W.; Weng, S.C.; Lin, C.N.; Lai, C.H.; Huang, Y.J.; Jeng, H.T.; Chang, S.L.; Soo, Y.L. Dramatic band gap reduction incurred by dopant coordination rearrangement in Co-doped nanocrystals of CeO2. Sci. Rep. 2017, 7, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spezzati, G.; Benavidez, A.D.; DeLaRiva, A.T.; Su, Y.; Hofmann, J.P.; Asahina, S.; Olivier, E.J.; Neethling, J.H.; Miller, J.T.; Datye, A.K.; et al. CO oxidation by Pd supported on CeO2 (100) and CeO2 (111) facets. Appl. Catal. B Environ. 2019, 243, 36–46. [Google Scholar] [CrossRef]
- Li, J.; Liu, X.; Sun, Z.; Sun, Y.; Pan, L. Novel yolk-shell structure bismuth rich bismuth molybdate microsphere for enhanced visible-light photocatalysis. J. Colloid Interface Sci. 2015, 452, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Verykios, X.E. Mechanistic aspects of the low temperature steam reforming of ethanol over supported Pt catalysts. Int. J. Hydrogen Energy 2012, 37, 16333–16345. [Google Scholar] [CrossRef]
- Ferrer, V.; Finol, D.; Ramos, M. Caracterización química y actividad catalítica de óxidos mixtos soportados basados en cerio. Ciencia 2010, 18, 209–219. [Google Scholar]
- Ratova, M.; Kelly, P.J.; West, G.T.; Xia, X.; Gao, Y. Deposition of visible light active photocatalytic bismuth molybdate thin films by reactive magnetron sputtering. Materials 2016, 9, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayame, A.; Uchida, K.; Iwataya, M.; Miyamoto, M. X-ray photoelectron spectroscopic study on α-and γ-bismuth molybdate surfaces exposed to hydrogen, propene, and oxygen. Appl. Catal. A Gen. 2002, 227, 7–17. [Google Scholar] [CrossRef]
- Mun, C.; Ehrhardt, J.J.; Lambert, J.; Madic, C. XPS investigations of ruthenium deposited onto representative inner surfaces of nuclear reactor containment buildings. Appl. Surf. Sci. 2007, 253, 7613–7621. [Google Scholar] [CrossRef] [Green Version]
- Yeung, H.; Chan, H.; Takoudis, C.G.; Weaver, M.J. High-pressure oxidation of ruthenium as probed by surface-enhanced raman and x-ray photoelectron spectroscopies. J. Catal. 1997, 172, 336–345. [Google Scholar] [CrossRef]
- Prusty, D.; Pathak, A.; Mukherjee, M.; Mukherjee, B.; Chowdhury, A. TEM and XPS studies on the faceted nanocrystals of Ce0.8Zr0.2O2. Mater. Charact. 2015, 100, 31–35. [Google Scholar] [CrossRef]
- Aguilar-Guerrero, V.; Lobo-Lapidus, R.J.; Gates, B.C. Genesis of cerium oxide supported gold catalyst for co oxidation: Transformation of mononuclear gold complexes into clusters as characterized by x-ray absorption spectroscopy. J. Phys. Chem. C 2009, 113, 3259–3269. [Google Scholar] [CrossRef]
- Kosacki, I.; Petrovsky, V.; Anderson, H.U. Raman spectroscopy of nanocrystalline ceria and zirconia thin films. J. Am. Ceram. Soc. 2002, 85, 2646–2650. [Google Scholar] [CrossRef]
- Bulushev, D.; Yuranov, I.; Suvorova, E.; Buffat, P.; Kiwi-Minsker, L. Highly dispersed gold on activated carbon fibers for low-temperature CO oxidation. J. Catal. 2004, 224, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Santos, V.P.; Carabineiro, S.A.C.; Bakker, J.J.W.; Soares, O.S.G.P.; Chen, X.; Pereira, M.F.R.; Órfão, J.J.M.; Figueiredo, J.L.; Gascon, J.; Kapteijn, F. Stabilized gold on cerium-modified cryptomelane: Highly active in low-temperature CO oxidation. J. Catal. 2014, 309, 58–65. [Google Scholar] [CrossRef]
- Adhikari, R.; Joshie, B.; García, R.N.; de la Rosa, E.; Lee, W.S. Microwave hydrothermal synthesis and infrared to visible upconversion luminescence of Er3+/Yb3+ co-doped bismuth molybdate nanopowder. J. Lumin. 2016, 145, 866–871. [Google Scholar] [CrossRef]
- Sleight, A.; Chen, H. Structure of Bi2Mo2O9: A selective oxidation catalyst. J. Solid State Chem. 1986, 63, 70–75. [Google Scholar]
- Casaletto, M.P.; Longo, A.; Martorana, A.; Prestianni, A.; Venezia, A.M. XPS study of supported gold catalysts: The role of Au0 and Au+g species as active sites. Surf. Interface Anal. 2006, 38, 215–218. [Google Scholar] [CrossRef]
- Piotrowski, T.; Accinno, D.J. Metallography of the precious metals. Metallography 1977, 10, 243–289. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Silva, A.M.T.; Draić, G.; Tavares, P.B.; Figueiredo, J.L. Gold nanoparticles on ceria supports for the oxidation of carbon monoxide. Catal. Today 2010, 154, 21–30. [Google Scholar] [CrossRef]
- Amano, F.; Nogami, K.; Abe, R.; Ohtani, B. Preparation and characterization of bismuth tungstate polycrystalline flake-ball particles for photocatalytic reactions. J. Phys. Chem. C 2008, 112, 9320–9326. [Google Scholar] [CrossRef]
- Liao, P.C.; Mar, S.Y.; Ho, W.S.; Huang, Y.S.; Tiong, K.K. Characterization of RuO2 thin films deposited on Si by metal-organic chemical vapor deposition. Thin Solid Film. 1996, 287, 74–79. [Google Scholar] [CrossRef]
- Mukherjee, D.; Rao, B.G.; Reddy, B.M. CO and soot oxidation activity of doped ceria: Influence of dopants. Appl. Catal. B Environ. 2016, 197, 105–115. [Google Scholar] [CrossRef]
- Qadir, K.; Joo, S.H.; Mun, B.S.; Butcher, D.R.; Renzas, J.R.; Aksoy, F.; Liu, Z.; Somorjai, G.A.; Park, J.Y. Intrinsic relation between catalytic activity of CO oxidation on Ru nanoparticles and Ru oxides uncovered with ambient pressure XPS. Nano Lett. 2012, 12, 5761–5768. [Google Scholar] [CrossRef] [PubMed]
- Venkataswamy, P.; Jampaiah, D.; Mukherjee, D.; Aniz, C.U.; Reddy, B.M. Mn-doped ceria solid solutions for CO oxidation at lower temperatures. Catal. Lett. 2016, 146, 2105–2118. [Google Scholar] [CrossRef]
- Mandapaka, R.; Madras, G. Zinc and platinum co-doped ceria for WGS and CO oxidation. Appl. Catal. B Environ. 2017, 211, 137–147. [Google Scholar] [CrossRef]
- Zanella, R. Metodologías para la síntesis de nanopartículas: Controlando forma y tamaño, Mundo Nano. Rev. Interdiscip. Nanocienc. Nanotecnol. 2012, 5, 69–81. [Google Scholar]
- Duan, Y.; Li, Z.; Li, Y.; Zhang, Y.; Li, L.; Li, J. New insight of the Mars-van Krevelen mechanism of the CO oxidation by gold catalyst on the ZnO (101) surface. Comput. Theor. Chem. 2017, 1100, 28–33. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Chen, B.; Zhu, X.; Shi, C.; Zhu, A. CO oxidation activity at room temperature over Au/CeO2 catalysts: Disclosure of induction period and humidity effect. ACS Catal. 2014, 4, 3481–3489. [Google Scholar] [CrossRef]
- Kimling, J.; Maier, M.; Okenve, B.; Kotaidis, V.; Ballot, H.; Plech, A. Turkevich method for gold nanoparticle synthesis revisited. J. Phys. Chem. B 2006, 110, 15700–15707. [Google Scholar] [CrossRef] [PubMed]
- Bueno-López, A.; Krishna, K.; Makkee, M.; Moulijn, J.A. Active oxygen from CeO2 and its role in catalyzed soot oxidation. Catal. Lett. 2005, 99, 203–205. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Chemical Composition (Atomic Percent) | |||||
|---|---|---|---|---|---|---|
| Bi | Mo | Ru | O | Ce | Au | |
| MBR1 | 22.05 | 13.62 | 1.56 | 64.34 | - | - |
| MBR2 | 29.81 | 12.56 | 1.49 | 57.63 | - | - |
| OCMBR1 | 16.1 | 13.65 | 2.12 | 64.67 | 3.46 | - |
| OCMBR2 | 16.72 | 13.47 | 1.89 | 64.95 | 3.21 | - |
| AuMBR1 | 16.04 | 19.34 | 1.69 | 61.73 | - | 1.2 |
| AuMBR2 | 14.49 | 22.29 | 1.35 | 60.54 | - | 1.33 |
| Catalysts | Au Species (%) | ||
|---|---|---|---|
| Au+3 | Au0 | ||
| AuMBR1 | Before reduction | 61.34 | 38.66 |
| After reduction | 39.24 | 60.76 | |
| AuMBR2 | Before reduction | 45.03 | 54.97 |
| After reduction | 38.56 | 61.44 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, E.E.; Rangel, R.; Lara, J.; Bartolo-Pérez, P.; Alvarado-Gil, J.J.; Galván, D.H.; García, R. {CeO2/Bi2Mo1−xRuxO6} and {Au/Bi2Mo1−xRuxO6} Catalysts for Low-Temperature CO Oxidation. Catalysts 2019, 9, 947. https://doi.org/10.3390/catal9110947
González EE, Rangel R, Lara J, Bartolo-Pérez P, Alvarado-Gil JJ, Galván DH, García R. {CeO2/Bi2Mo1−xRuxO6} and {Au/Bi2Mo1−xRuxO6} Catalysts for Low-Temperature CO Oxidation. Catalysts. 2019; 9(11):947. https://doi.org/10.3390/catal9110947
Chicago/Turabian StyleGonzález, Edson Edain, Ricardo Rangel, Javier Lara, Pascual Bartolo-Pérez, Juan José Alvarado-Gil, Donald Homero Galván, and Rafael García. 2019. "{CeO2/Bi2Mo1−xRuxO6} and {Au/Bi2Mo1−xRuxO6} Catalysts for Low-Temperature CO Oxidation" Catalysts 9, no. 11: 947. https://doi.org/10.3390/catal9110947