
Identification of Aniline-Degrading Bacteria Using Stable Isotope Probing Technology and Prediction of Functional Genes in Aerobic Microcosms
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
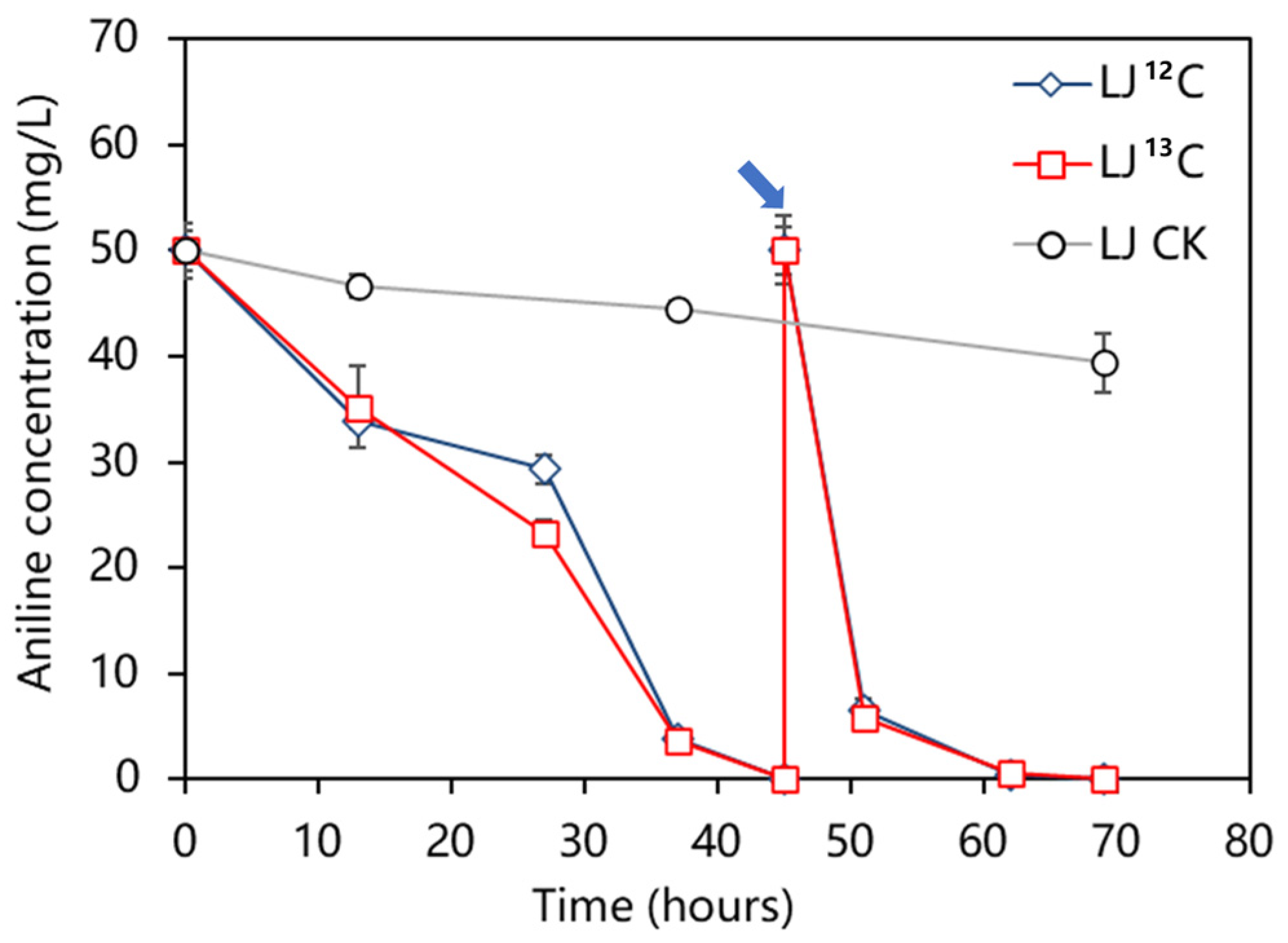
2.1. Variation in Aniline Concentration
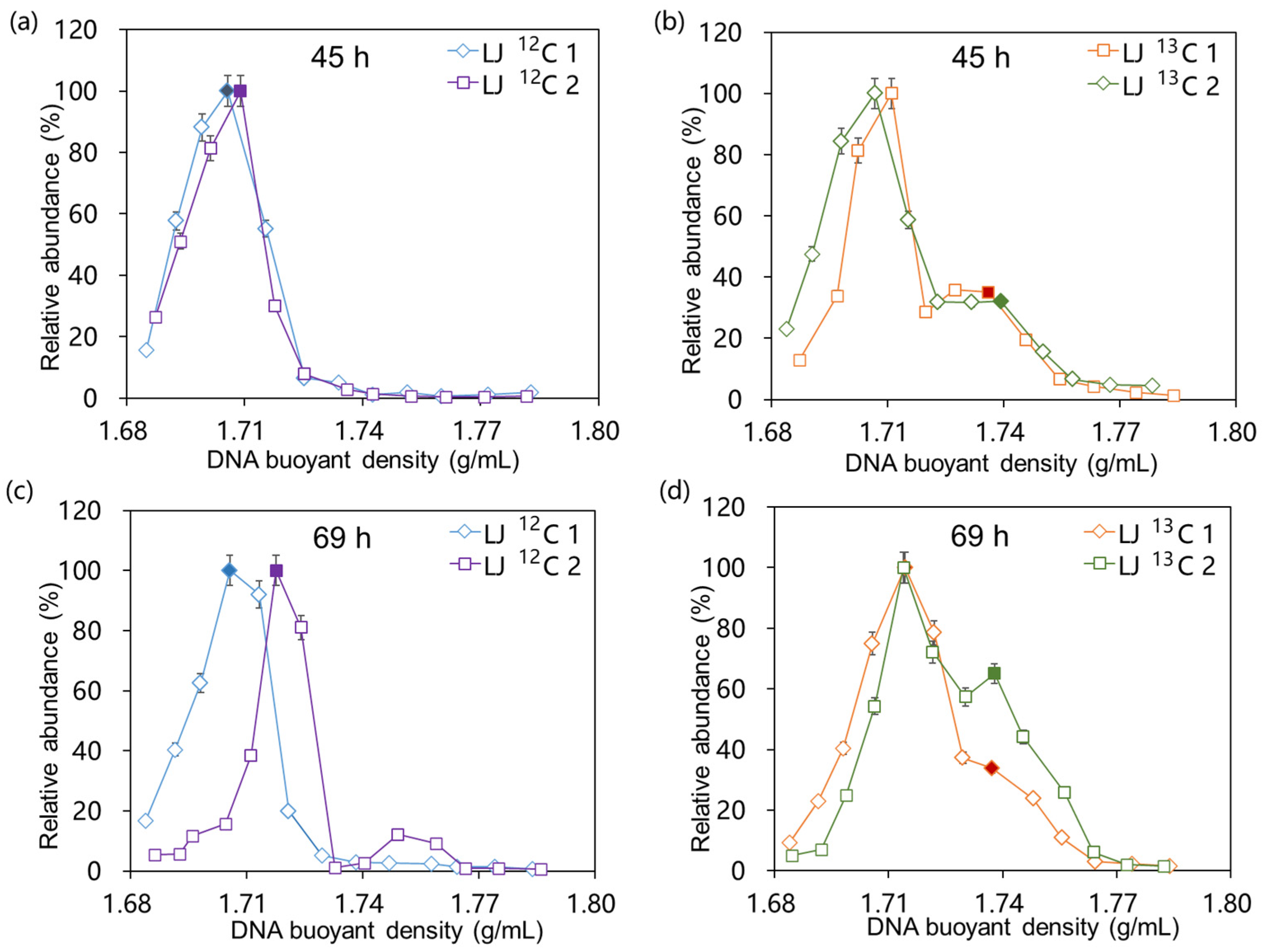
2.2. Isopycnic Density Gradient Centrifugation of DNA
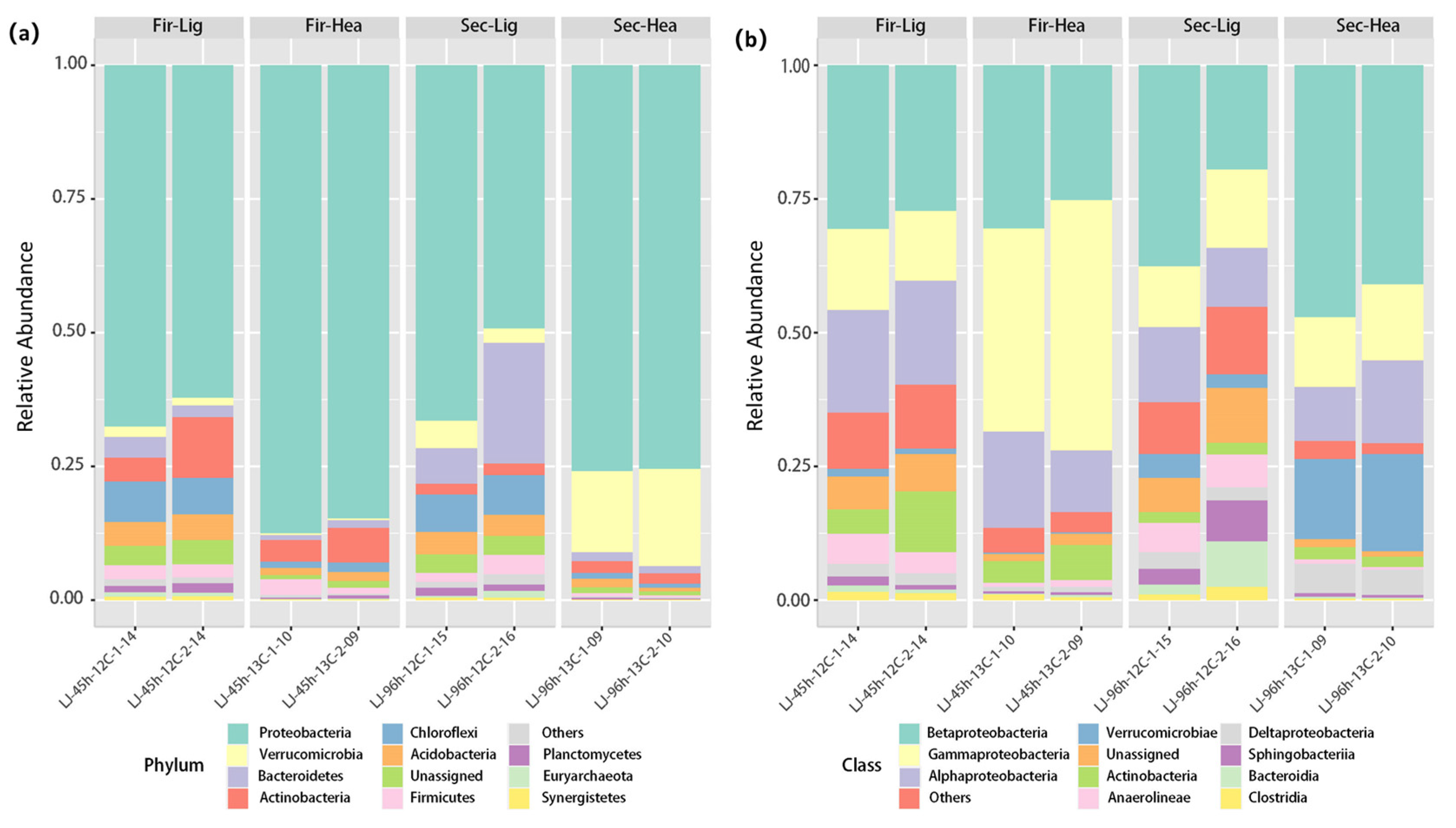
2.3. Distribution of Microorganisms in Different Fractions
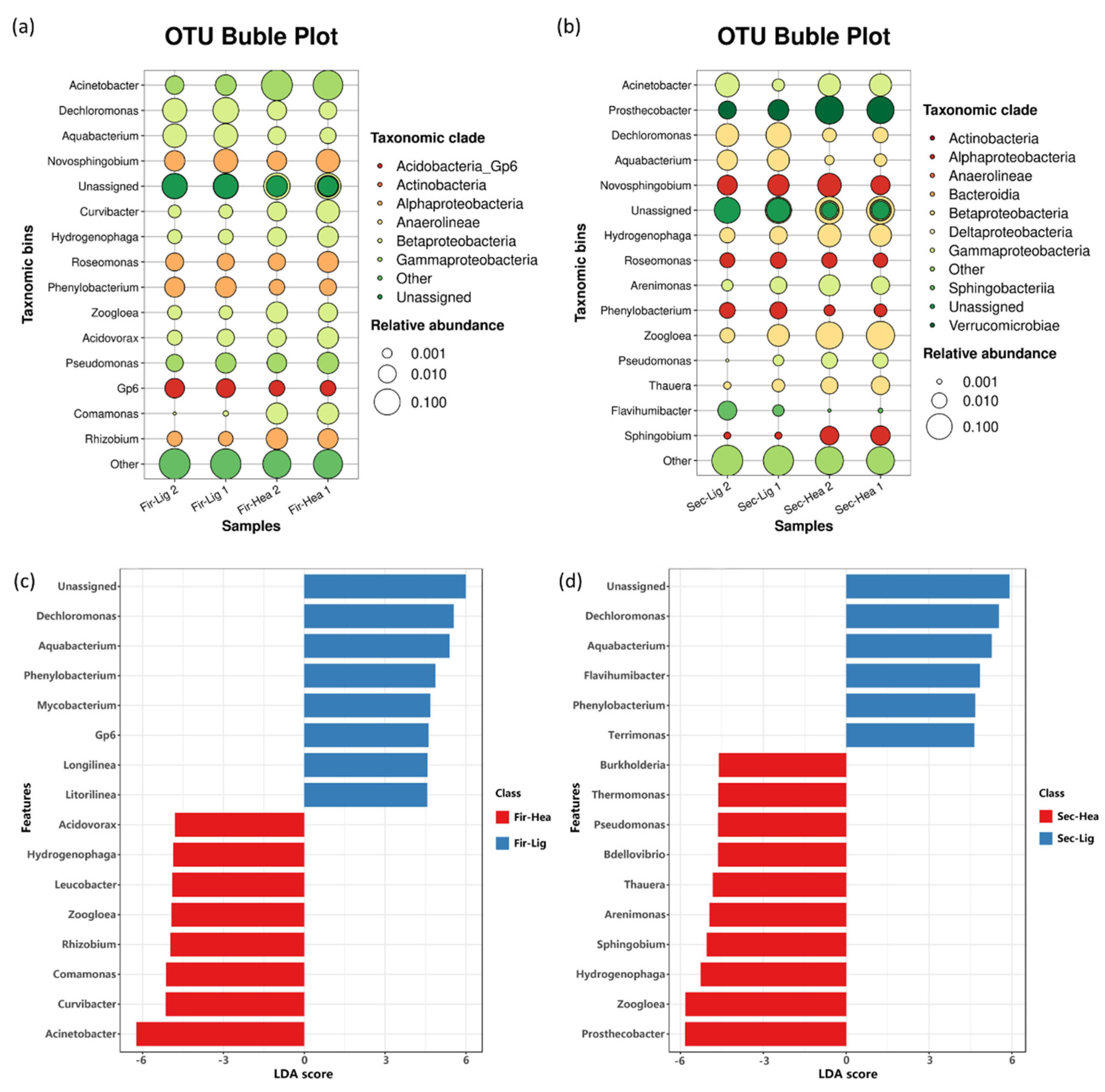
2.4. Enrichment of Functional Bacteria in Heavy Fractions
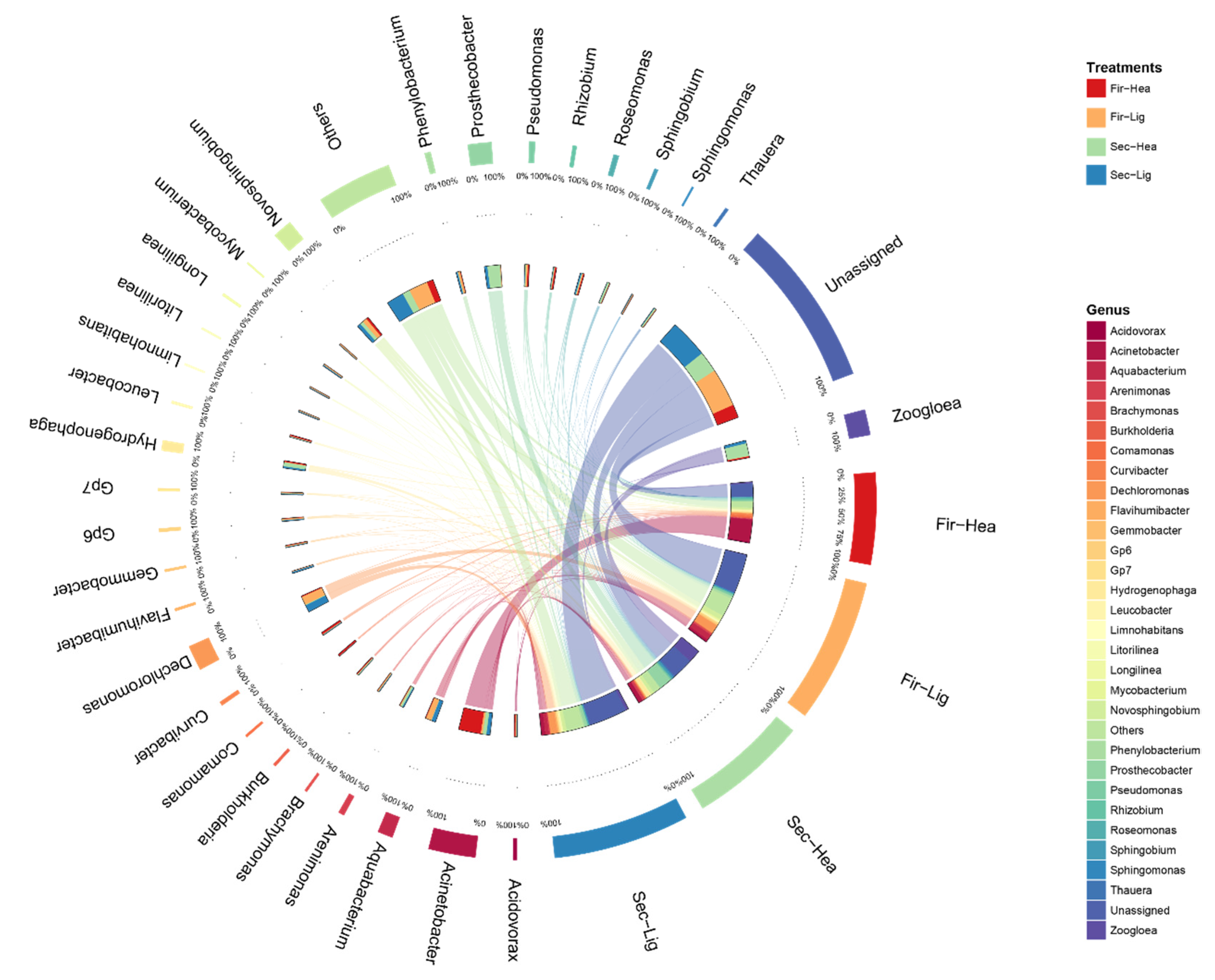
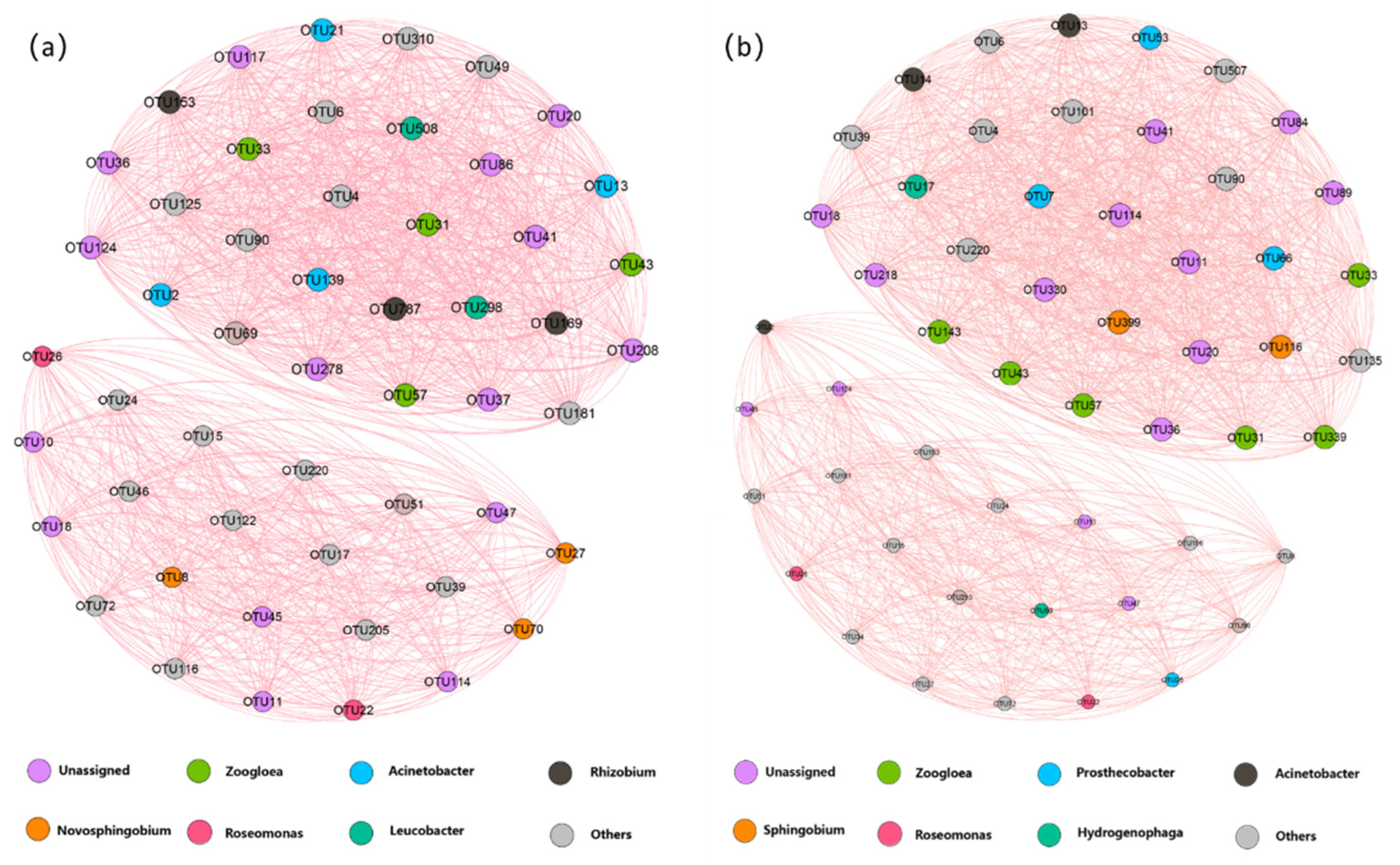
2.5. Co-Occurrence Network of Microbial Community
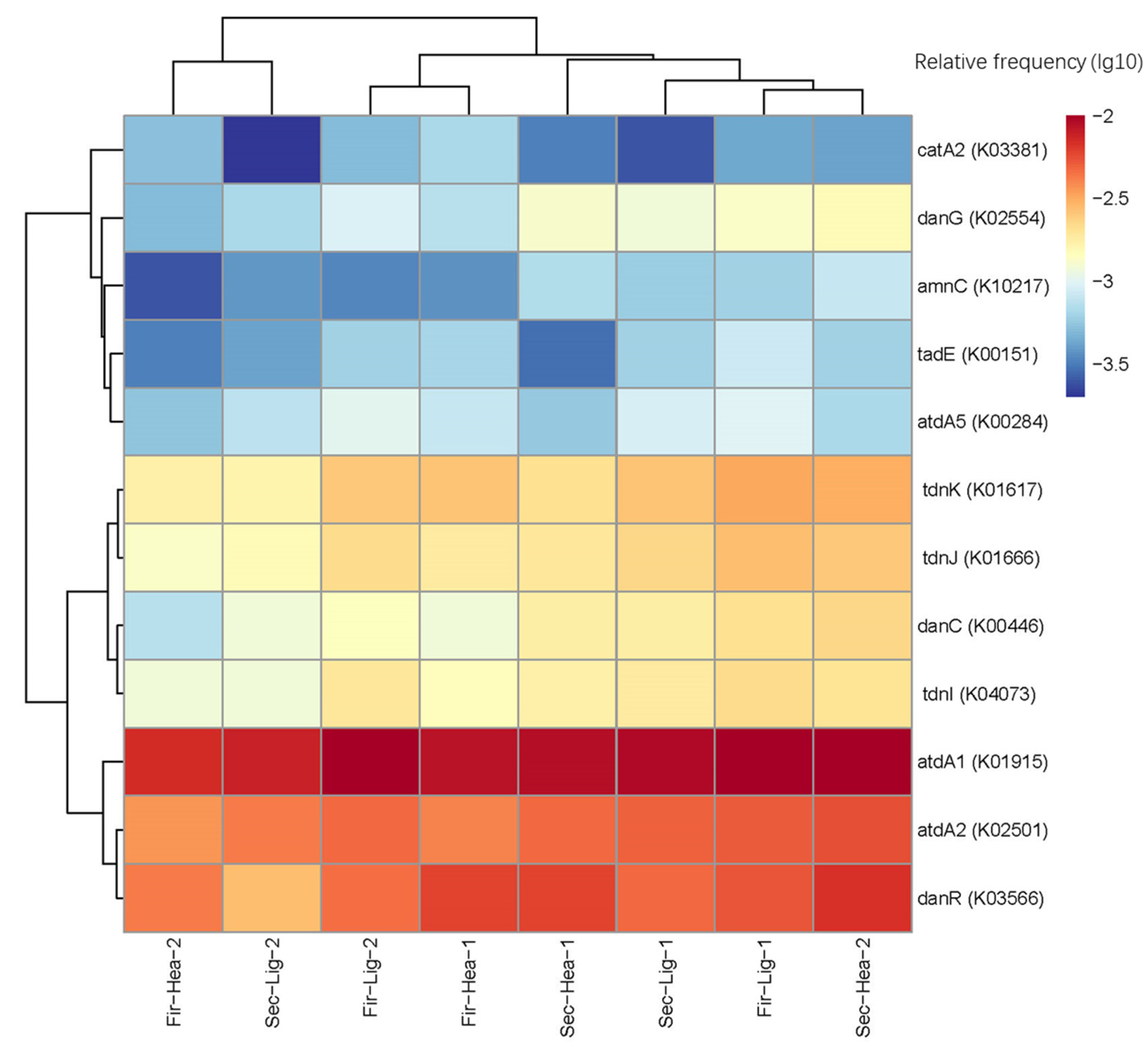
2.6. Prediction of Functional Genes
3. Discussion
3.1. Degradation in Aniline and Uptake of 13C by the Microorganisms
3.2. Aniline-Degrading Bacteria Identified in Aerobic Microcosm
3.3. Aniline-Degradation Genes Possessed by Microorganisms
4. Materials and Methods
4.1. Soil Microcosms Incubations
4.2. DNA Extraction and Quantitative PCR of 16S rRNA Gene
4.3. SIP Gradient Fractionation
4.4. Illumina MiSeq Sequencing and Analysis
4.5. Microbial Functional Prediction Using PICRUSt2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- O’Neill, F.J.; Bromley-Challenor, K.C.A.; Greenwood, R.J.; Knapp, J.S. Bacterial growth on aniline: Implications for the biotreatment of industrial wastewater. Water Res. 2000, 34, 4397–4409. [Google Scholar] [CrossRef]
- Zhu, L.; Lv, M.L.; Dai, X.; Xu, X.Y.; Qi, H.Y.; Yu, Y.W. Reaction kinetics of the degradation of chloroanilines and aniline by aerobic granule. Biochem. Eng. J. 2012, 68, 215–220. [Google Scholar] [CrossRef]
- Yang, L.H.; Zhang, Y.M.; Bai, Q.; Yan, N.; Xu, H.; Rittmann, B.E. Intimately coupling of photolysis accelerates nitrobenzene biodegradation, but sequential coupling slows biodegradation. J. Hazard. Mater. 2015, 287, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Barrington, S.; Kim, J.W. Biodegradation of pentyl amine and aniline from petrochemical wastewater. J. Environ. Manag. 2007, 83, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Jiang, J.P.; Liu, R.T.; Khan, A.U.; Wang, P. Engineering risk assessment for emergency disposal projects of sudden water pollution incidents. Environ. Sci. Pollut. Res. 2017, 24, 14819–14833. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ding, X. Xiangshui Explosion Accident: Anilines in the Surface Water of Some Rivers in the Park Exceeded the Standard. The Paper, 20:15. Available online: https://www.thepaper.cn/newsDetail_forward_%203182916 (accessed on 22 March 2019).
- Franciscon, E.; Zille, A.; Dias Guirnaro, F.; Ragagnin de Menezes, C.; Durrant, R.L.; Cavaco-Paulo, A. Biodegradation of textile azo dyes by a facultative Staphylococcus arlettae strain VN-11 using a sequential microaerophilic/aerobic process. Int. Biodeterior. Biodegrad. 2009, 63, 280–288. [Google Scholar]
- Vikrant, K.; Giri, B.S.; Raza, N.; Roy, K.; Kim, K.H.; Rai, B.N.; Singh, R.S. Recent advancements in bioremediation of dye: Current status and challenges. Bioresour. Technol. 2018, 253, 355–367. [Google Scholar] [CrossRef]
- Asad, U.K.; Mujaddad, U.R.; Muhammad, Z.; Abdul, B.S.; Ivar, Z. Biodegradation of Brown 706 Dye by Bacterial Strain Pseudomonas aeruginosa. Water 2021, 13, 2959. [Google Scholar]
- Chen, H.; Sun, C.R.; Liu, R.H.; Yuan, M.Z.; Mao, Z.H.; Wang, Q.; Zhou, H.B.; Cheng, H.N.; Zhan, W.H.; Wang, Y.G. Enrichment and domestication of a microbial consortium for degrading aniline. J. Water Process Eng. 2021, 42, 102108. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Zhang, Q.; Peng, H.J.; Wei, H.; Feng, J.P.; Su, J.H.; He, J. An attempt to stimulate aniline degrading bioreactor by exogenous auto-inducer: Decontamination performance, sludge characteristics, and microbial community structure response. Bioresour. Technol. 2022, 347, 126675. [Google Scholar] [CrossRef]
- Peng, H.J.; Zhang, Q.; Li, M.; Li, Y.; Zhang, W.L. Identification and characterization of a highly efficient and resistant aniline-degrading strain AD4. Environ. Eng. Sci. 2021, 38, 742–751. [Google Scholar] [CrossRef]
- Qin, X.M.; Hua, Y.D.; Sun, H.; Xie, J.Y.; Zhao, Y.S. Visualization study on aniline-degrading bacteria AN-1 transport in the aquifer with the low-permeability lens. Water Res. 2020, 186, 116329. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.M.; Li, Y.; McGuinness, L.R.; Luo, S.A.; Huang, W.L.; Kerkhof, L.J.; Mack, E.E.; Häggblom, M.M.; Fennell, D.E. Identification of anaerobic aniline-degrading bacteria at a contaminated industrial site. Environ. Sci. Technol. 2015, 49, 11079–11088. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, R.C.; Hanson, B.T.; Chandra, S.; Madsen, E. Community dynamics and functional characteristics of naphthalene-degrading populations in contaminated surface sediments and hypoxic/anoxic groundwater. Environ. Microbiol. 2018, 20, 3543–3559. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.K. Bacterial degradation of monocyclic aromatic amines. Front. Microbiol. 2015, 6, 820. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, J.L.; Liu, S.J.; Liu, Z.P. A novel and complete gene cluster involved in the degradation of aniline by Delftia sp. AN3. J. Environ. Sci. 2018, 20, 717–724. [Google Scholar] [CrossRef]
- Liu, H.Q.; Lin, H.Z.; Song, B.R.; Sun, X.X.; Xu, R.; Kong, T.L.; Xu, F.Q.; Li, B.Q.; Sun, W.M. Stable-isotope probing coupled with high-throughput sequencing reveals bacterial taxa capable of degrading aniline at three contaminated sites with contrasting pH. Sci. Total Environ. 2021, 771, 144807. [Google Scholar] [CrossRef]
- Kim, S.I.; Leem, S.H.; Choi, J.S.; Chung, Y.H.; Kim, S.; Park, Y.M.; Park, Y.K.; Lee, Y.N.; Ha, K.S. Cloning and characterization of two catA genes in Acinetobacter lwoffii K24. J. Bacteriol. 1997, 179, 5226–5231. [Google Scholar] [CrossRef]
- Liang, Q.F.; Takeo, M.; Chen, M.; Zhang, W.; Xu, Y.Q.; Lin, M. Chromosome-encoded gene cluster for the metabolic pathway that converts aniline to TCA-cycle intermediates in Delftia tsuruhatensis AD9. Microbiology 2005, 151, 3435–3446. [Google Scholar] [CrossRef]
- Fujii, T.; Takeo, M.; Maeda, Y. Plasmid-encoded genes specifying aniline oxidation from Acinetobacter sp. strain YAA. Microbiology 1997, 143, 93–99. [Google Scholar] [CrossRef]
- Boon, N.; Goris, J.; De Vos, P.; Verstraete, W.; Top, E.M. Genetic diversity among 3-chloroaniline- and aniline-degrading strains of the Comamonadaceae. Appl. Environ. Microbiol. 2001, 67, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Hao, Z.Y.; Sun, R.; Bartlam, M.; Wang, Y.Y. Genome sequence of a typical ultramicro bacterium, Curvibacter sp. Strain PAE-UM, capable of phthalate ester degradation. Genome Announc. 2016, 4, e01510–15. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.F.; Wu, Q.P.; Gu, Q.H.; Zhou, Q.; Zhang, J.M. Community structure analysis and biodegradation potential of aniline-degrading bacteria in biofilters. Curr. Microbiol. 2018, 75, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Urata, M.; Uchida, E.; Nojiri, H.; Omori, T.; Obo, R.; Miyaura, N.; Ouchiyama, N. Genes involved in aniline degradation by Delftia acidovorans strain 7N and its distribution in the natural environment. Biosci. Biotechnol. Biochem. 2004, 68, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Kim, J. Sphingobium aromaticivastans sp. nov. a novel aniline- and benzene-degrading, and antimicrobial compound producing bacterium. Arch. Microbiol. 2019, 201, 155–161. [Google Scholar] [CrossRef]
- Chen, H.; Gao, X.Y.; Wang, C.Q.; Shao, J.J.; Xu, X.Y.; Zhu, L. Efficient 2,4-dichloronitrobenzene removal in the coupled BES-UASB reactor: Effect of external voltage mode. Bioresour. Technol. 2017, 241, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Wan, S.G.; An, T.C. Efficient bio-deodorization of aniline vapor in a biotrickling filter: Metabolic mineralization and bacterial community analysis. Chemosphere 2012, 87, 253–258. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Yin, J.; Wang, J.H.; Tian, W.D. Performance and microbial community in hybrid anaerobic baffled reactor-constructed wetland for nitrobenzene wastewater. Bioresour. Technol. 2012, 118, 128–135. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, Y.; Zhang, Q.; Zhang, W.; Li, M.; Feng, J.; Su, J.; He, J.; Zhong, M. Control of aeration time in the aniline degrading-bioreactor with the analysis of metagenomic: Aniline degradation and nitrogen metabolism. Bioresour. Technol. 2022, 344, 126281. [Google Scholar] [CrossRef]
- Takeo, M.; Ohara, A.; Sakae, S.; Okamoto, Y.; Kitamura, C.; Kato, D.; Negoro, S. Mechanism of bacterial aniline oxidation via γ-glutamylanilide: Function of a glutamine synthetase-like protein. J. Bacteriol. 2013, 195, 4406–4414. [Google Scholar] [CrossRef]
- Fukumori, F.; Saint, C.P. Nucleotide sequences and regulational analysis involved in conversion of aniline to catechol in Pseudomonas putida UCC22 (pTDN1). J. Bacteriol. 1997, 179, 399–408. [Google Scholar] [CrossRef]
- Król, J.E.; Penrod, J.T.; McCaslin, H.; Rogers, L.M.; Yano, H.; Stancik, A.D.; Dejonghe, W.; Brown, C.J.; Parales, R.E.; Wuertz, S.; et al. Role of IncP-1 β plasmids pWDL7::rfp and pNB8c in chloroaniline catabolism as determined by genomic and functional analyses. Appl. Environ. Microbiol. 2012, 78, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, W.L.; He, Q.L.; Li, M.; Li, Y.; Huang, W.S. Effects of dissolved oxygen concentrations on a bioaugmented sequencing batch rector treating aniline-laden wastewater: Reactor performance, microbial dynamics and functional genes. Bioresour. Technol. 2020, 313, 123598. [Google Scholar] [CrossRef]
- Shin, K.A.; Spain, J.C. Pathway and evolutionary implications of diphenylamine biodegradation by Burkholderia sp. Strain JS667. Appl. Environ. Microb. 2009, 75, 2694–2704. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, W.X.; Liu, B.B.; He, J.; Shen, Q.R.; Zhao, F.J. Anaerobic arsenite oxidation by an autotrophic arsenite-oxidizing bacterium from an arsenic-contaminated paddy soil. Environ. Sci. Technol. 2015, 49, 5956–5964. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, Z.; Azmat, U.; Sultan, A.; Mian, M.; Roy, H.S.; Ivar, Z.; Amir, S. Novel magnetite nanocomposites (Fe3O4/C) for efficient immobilization of ciprofloxacin from aqueous solutions through adsorption pretreatment and membrane processes. Water 2022, 14, 724. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Alves, R.J.E.; Zhang, D.D.; Han, L.L.; He, J.Z.; Zhang, L.M. Time-dependent shifts in populations and activity of bacterial and archaeal ammonia oxidizers in response to liming in acidic soils. Soil Boil. Biochem. 2017, 112, 77–89. [Google Scholar] [CrossRef]
- Li, Y.B.; Guo, L.F.; Häggblom, M.M.; Yang, R.; Li, M.Y.; Sun, X.X.; Chen, Z.; Li, F.B.; Su, X.F.; Yan, G.; et al. Serratia spp. are responsible for nitrogen fixation fueled by As(III) oxidation, a novel biogeochemical process identified in mine tailings. Environ. Sci. Technol. 2022, 56, 2033–2043. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Zhang, M.M.; Li, Z.; Häggblom, M.M.; Young, L.L.; Li, F.B.; He, Z.J.; Lu, G.M.; Xu, R.; Sun, X.X.; Qiu, L.; et al. Bacteria responsible for nitrate-dependent antimonite oxidation in antimony-contaminated paddy soil revealed by the combination of DNA-SIP and metagenomics. Soil Boil. Biochem. 2021, 156, 108194. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Guo, X.X.; Song, G.S.; Li, Y.Y.; Zhao, L.; Wang, J. Switch of bacteria community under oxygen depletion in sediment of Bohai Sea. Front. Mar. Sci. 2022, 9, 2296–7745. [Google Scholar] [CrossRef]
- Sun, X.X.; Song, B.R.; Xu, R.; Zhang, M.M.; Gao, P.; Lin, H.Z.; Sun, W.M. Root-associated (rhizosphere and endosphere) microbiomes of the Miscanthus sinensis and their response to the heavy metal contamination. J. Environ. Sci. 2021, 104, 387–398. [Google Scholar] [CrossRef]
- Toole, D.R.; Zhao, J.; Martens-Habbena, W.; Strauss, S.L. Bacterial functional prediction tools detect but underestimate metabolic diversity compared to shotgun metagenomics in southwest Florida soils. Appl. Soil Ecol. 2021, 168, 104129. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Ghani, M.U.; Sun, W.; Sun, X.; Liu, H.; Yan, G.; Yang, R.; Huang, Y.; Ren, Y.; Song, B. Identification of Aniline-Degrading Bacteria Using Stable Isotope Probing Technology and Prediction of Functional Genes in Aerobic Microcosms. Catalysts 2024, 14, 64. https://doi.org/10.3390/catal14010064
Li B, Ghani MU, Sun W, Sun X, Liu H, Yan G, Yang R, Huang Y, Ren Y, Song B. Identification of Aniline-Degrading Bacteria Using Stable Isotope Probing Technology and Prediction of Functional Genes in Aerobic Microcosms. Catalysts. 2024; 14(1):64. https://doi.org/10.3390/catal14010064
Chicago/Turabian StyleLi, Baoqin, Muhammad Usman Ghani, Weimin Sun, Xiaoxu Sun, Huaqing Liu, Geng Yan, Rui Yang, Ying Huang, Youhua Ren, and Benru Song. 2024. "Identification of Aniline-Degrading Bacteria Using Stable Isotope Probing Technology and Prediction of Functional Genes in Aerobic Microcosms" Catalysts 14, no. 1: 64. https://doi.org/10.3390/catal14010064