

An Asymmetric Dinuclear Bis(ansa-Zirconocene) Complex: Synthesis and Performance in Olefin (co-)Polymerization
 and
and
Abstract
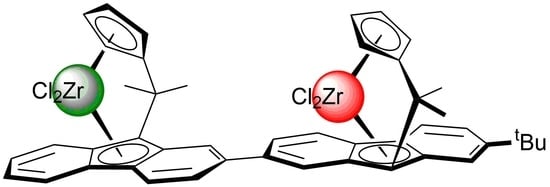
:
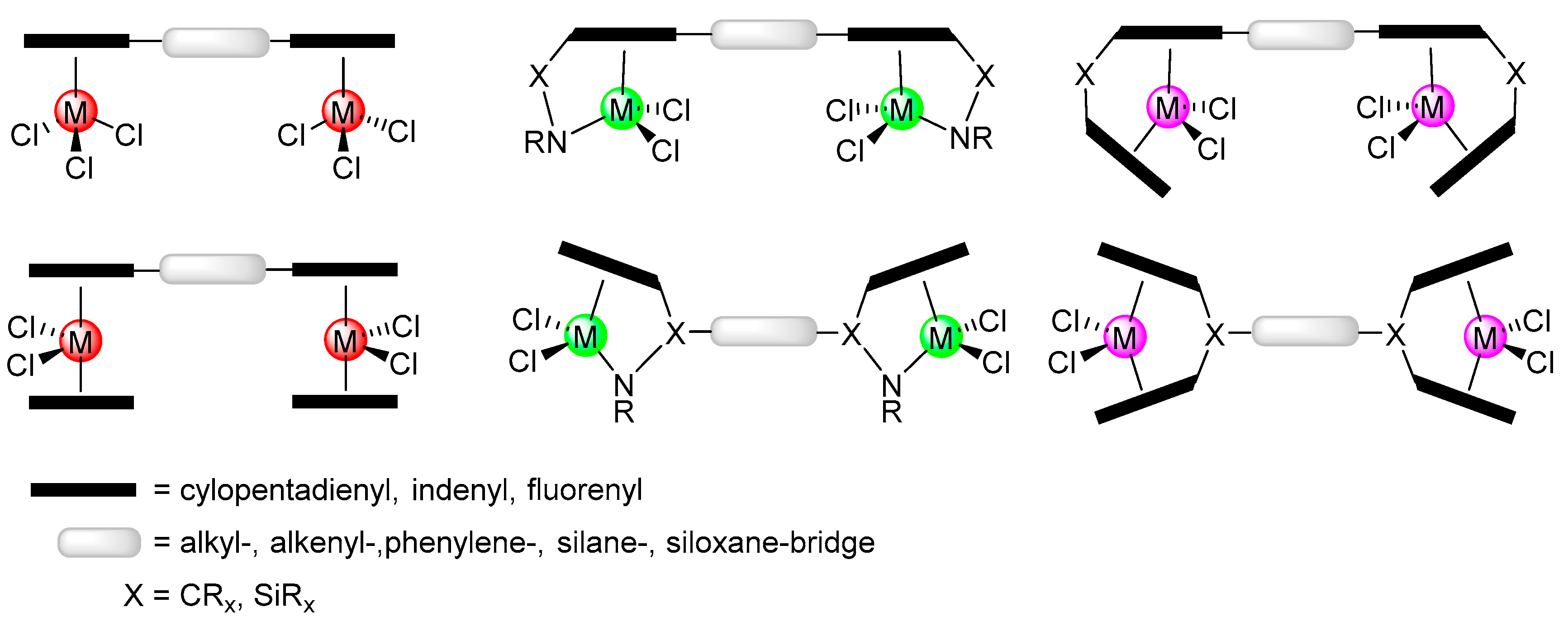
1. Introduction
2. Results and Discussion
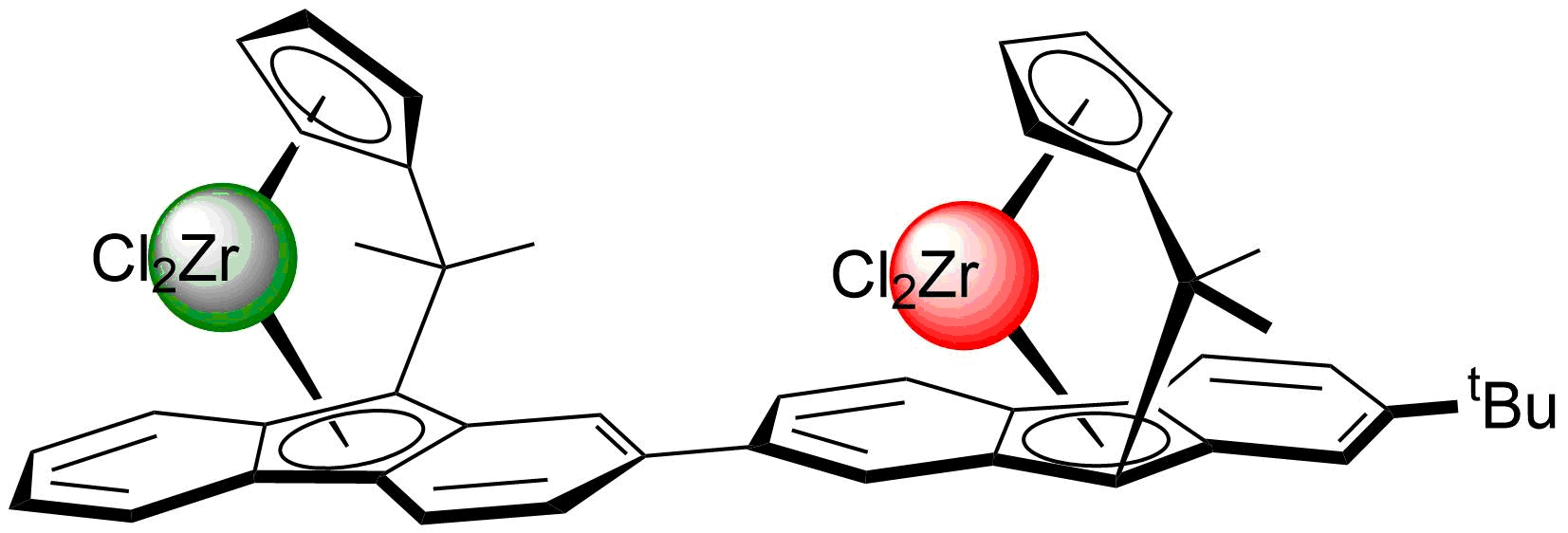
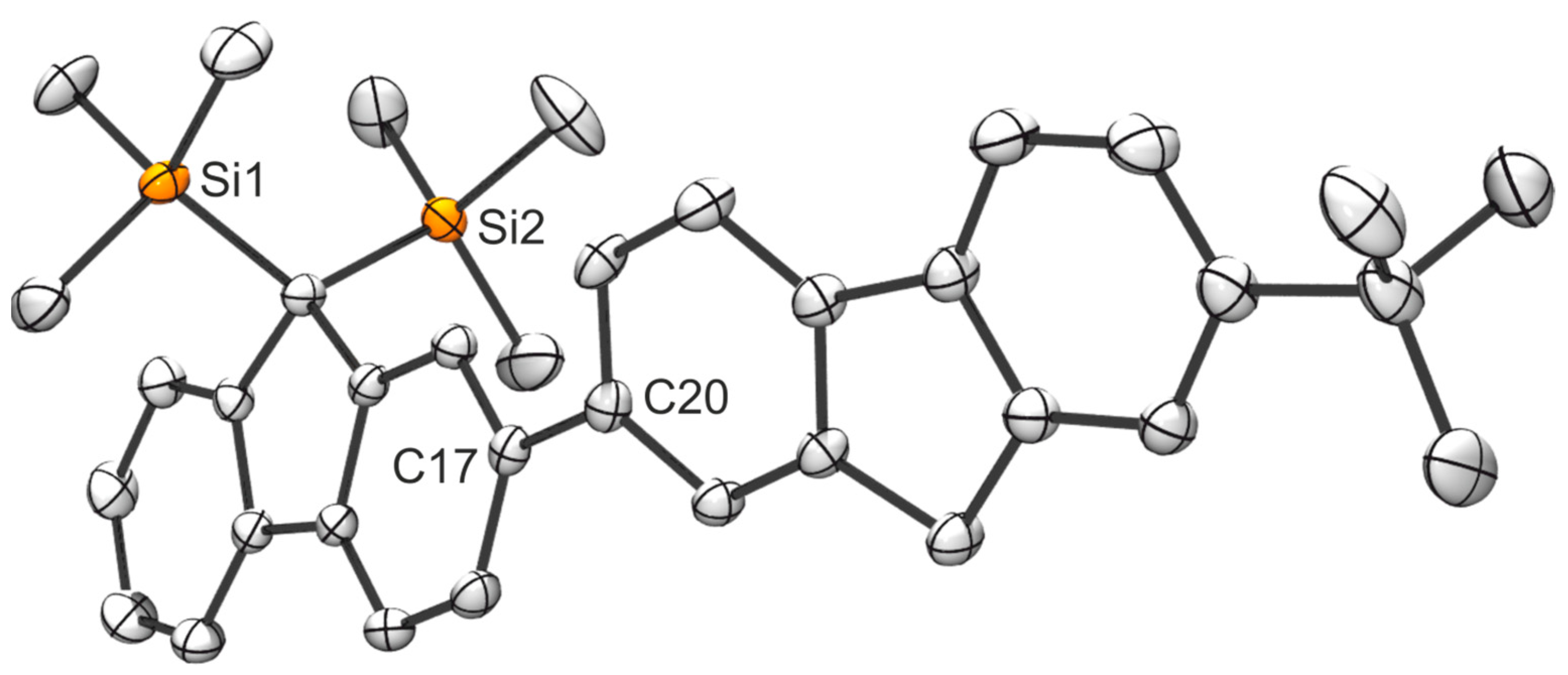
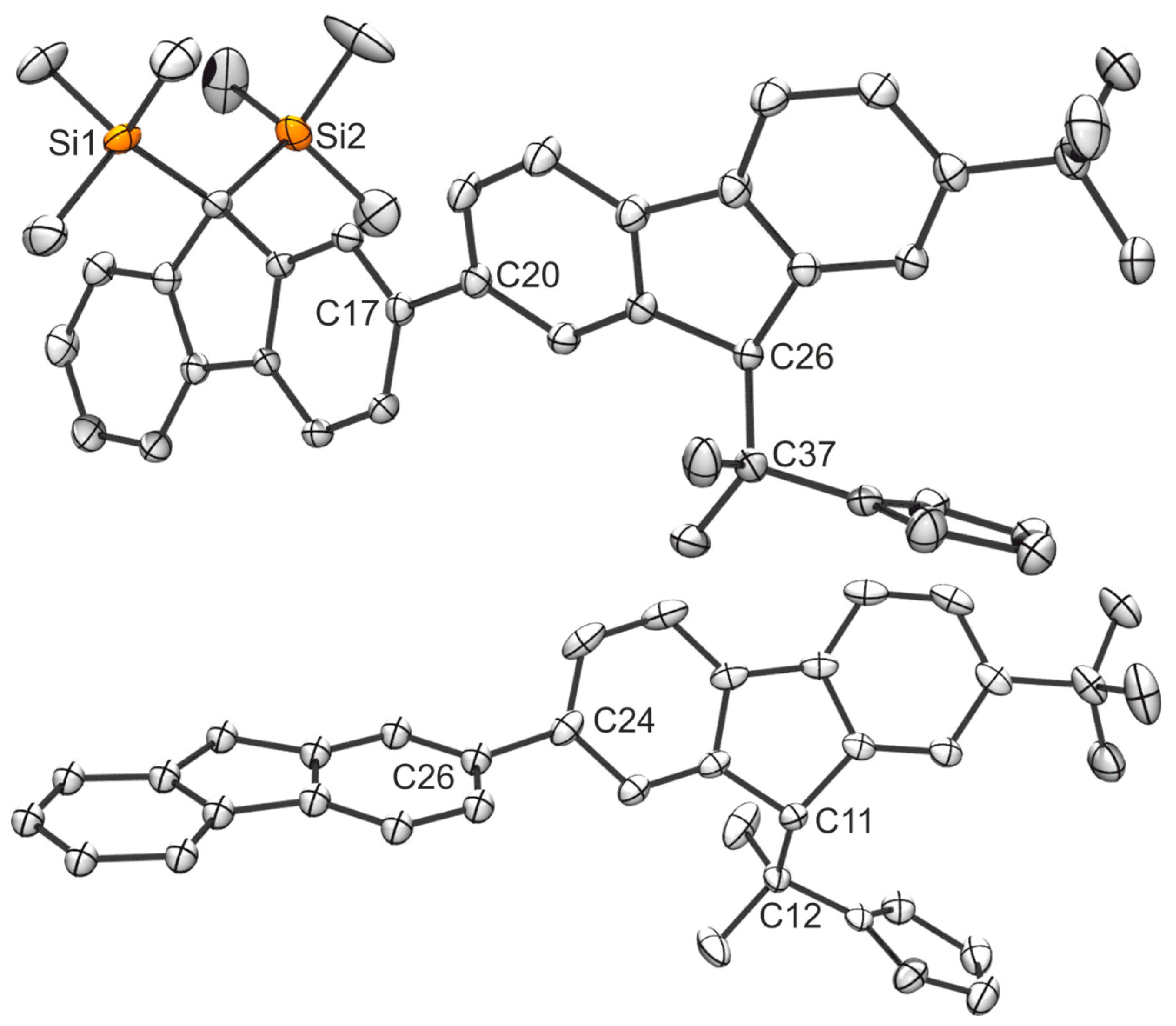
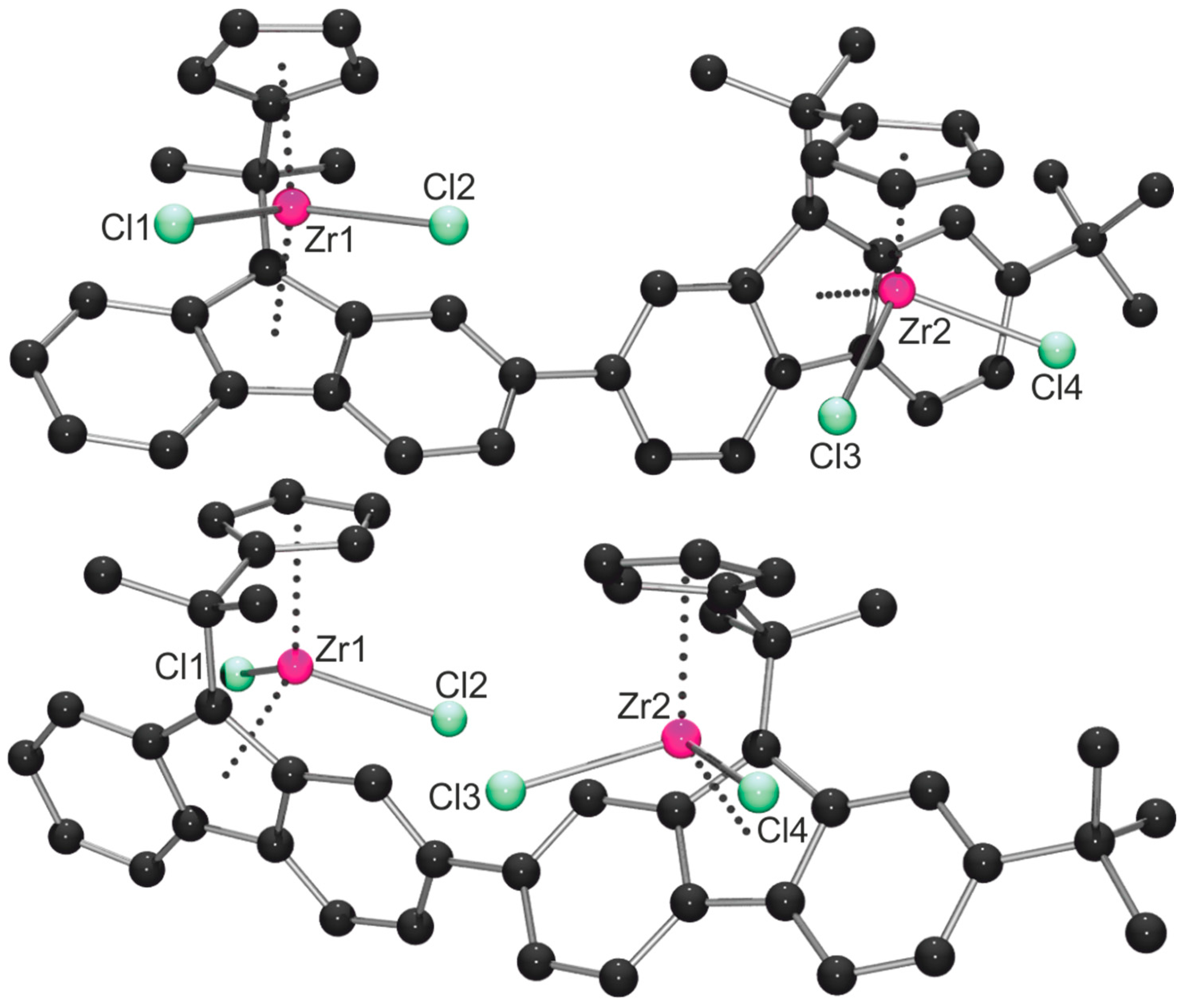
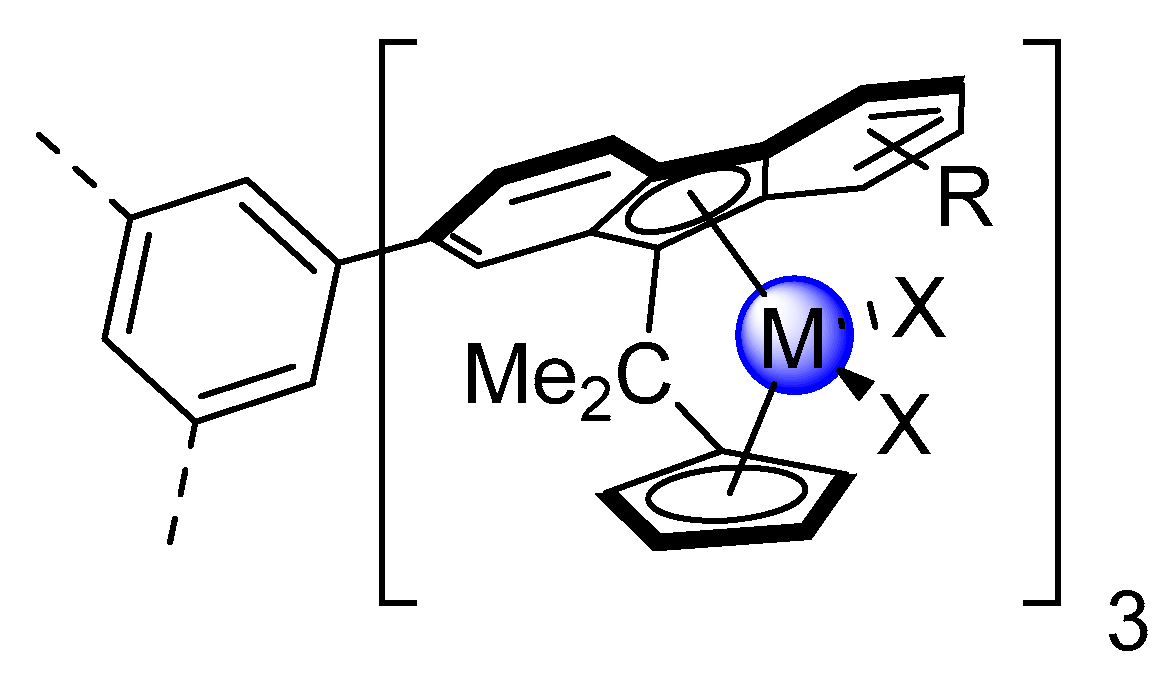
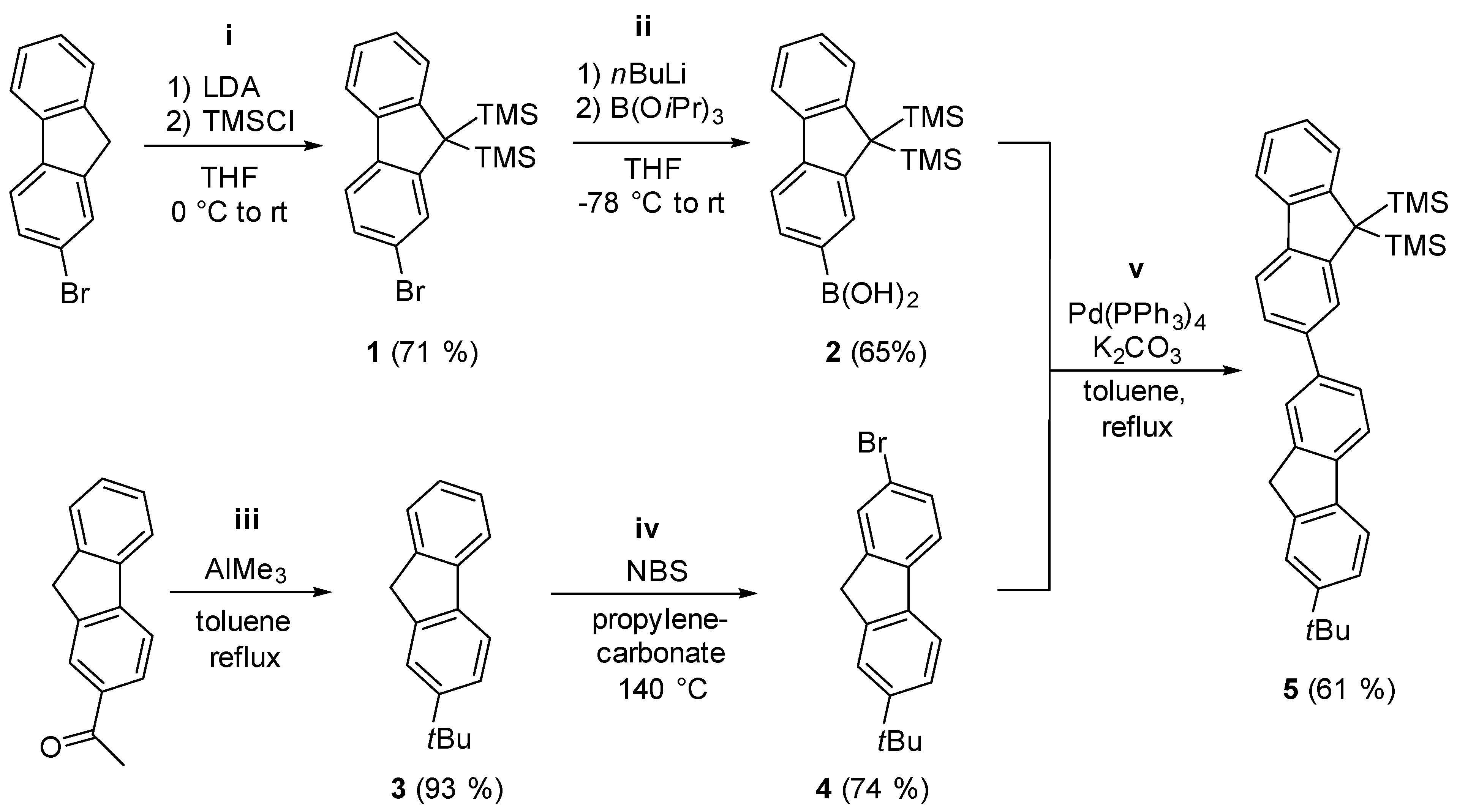
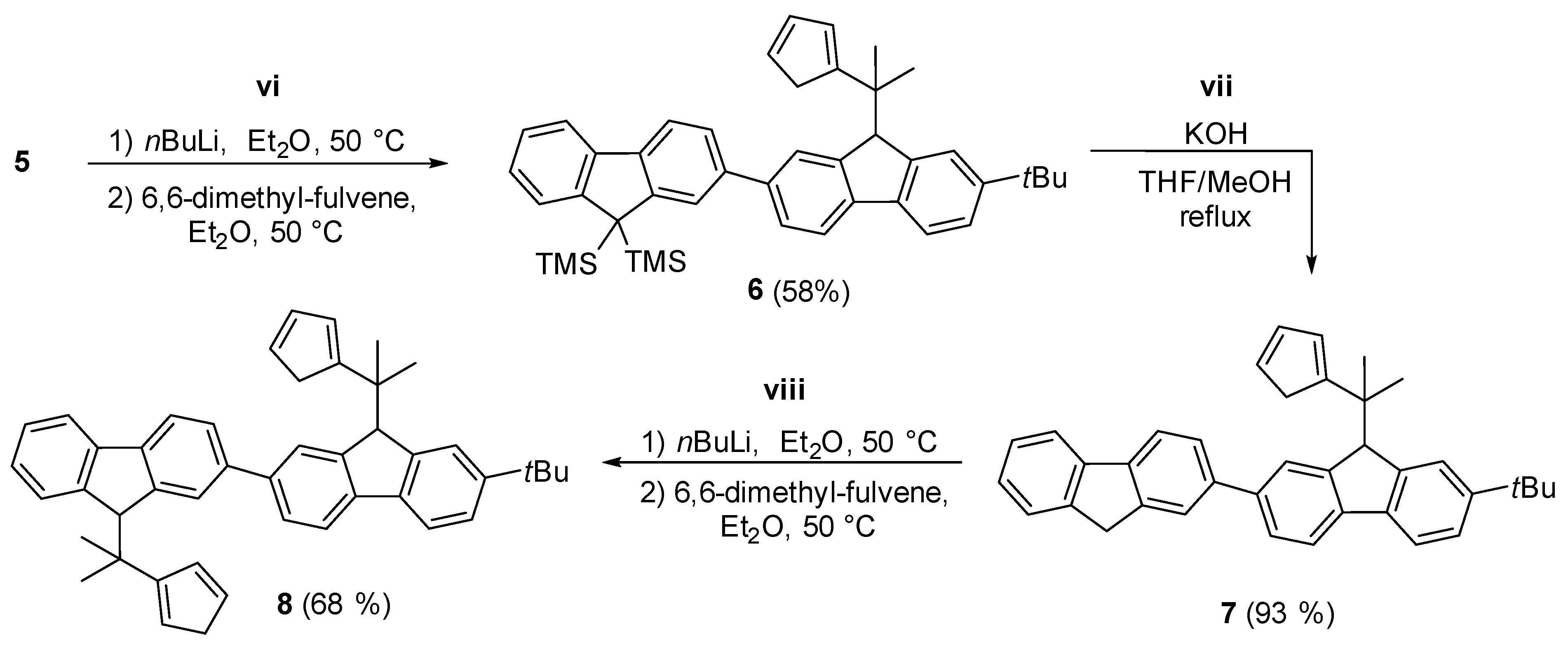
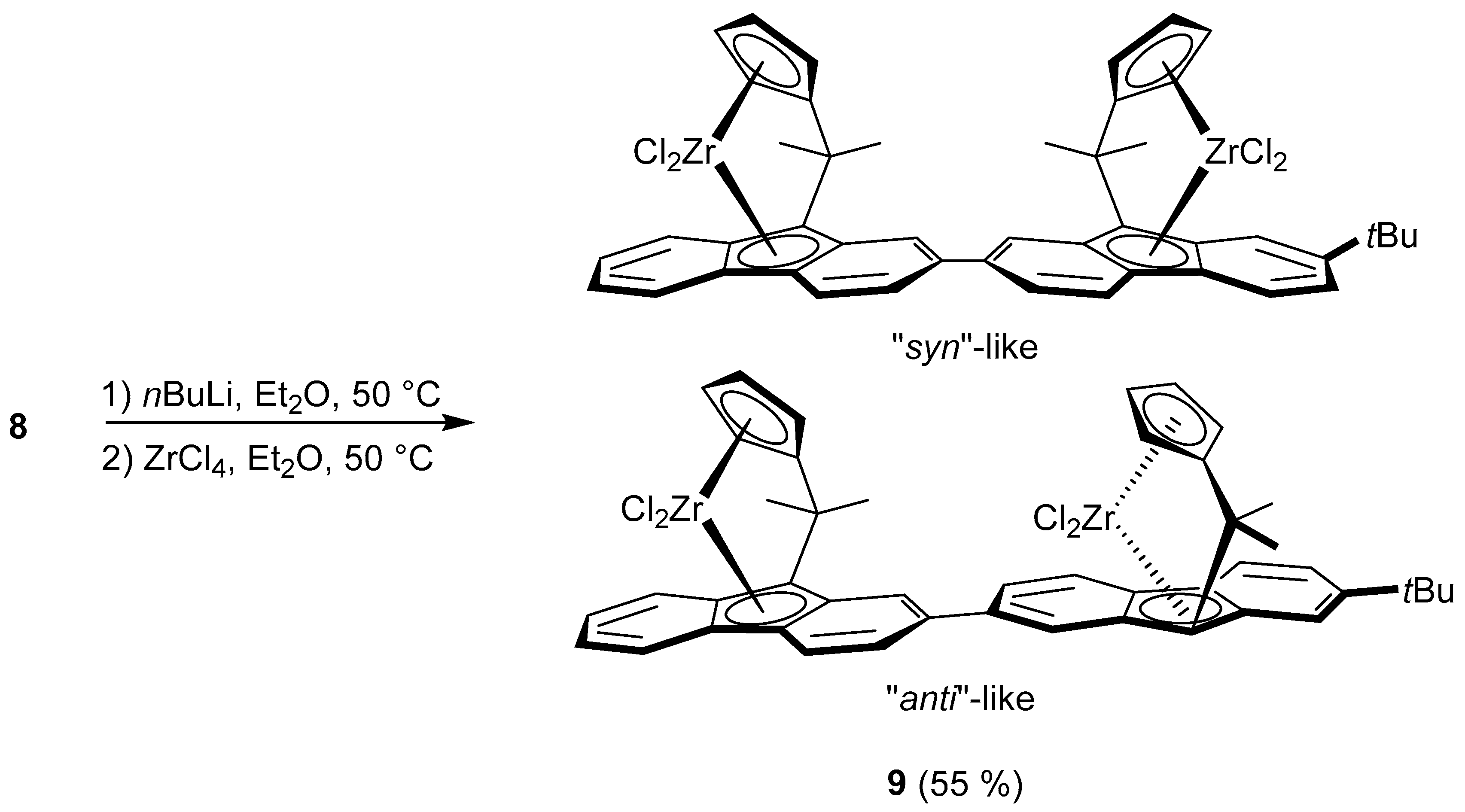
2.1. Synthesis of the Asymmetric Bis(Cp/Flu) Proligand and Corresponding Bis(ansa-Zirconocene)
2.2. Olefin (Co-)Polymerization
3. Materials and Methods
3.1. General Considerations
3.2. Instruments and Measurements
3.3. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ainooson, M.; Meyer, F. Bimetallic Approaches in Olefin Polymerization and Metathesis. In Comprehensive Inorganic Chemistry II, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 433–458. [Google Scholar]
- Delferro, M.; Marks, T.J. Multinuclear olefin polymerization catalysts. Chem. Rev. 2011, 111, 2450–2485. [Google Scholar] [CrossRef] [PubMed]
- McInnis, J.P.; Delferro, M.; Marks, T.J. Multinuclear Group 4 catalysis: Olefin polymerization pathways modified by strong metal–metal cooperative effects. Acc. Chem. Res. 2014, 47, 2545–2557. [Google Scholar] [CrossRef] [PubMed]
- Jüngling, S.; Müllhaupt, R.; Plenio, H. Cooperative effects in binuclear zirconocenes: Their synthesis and use as catalyst in propene polymerization. J. Organomet. Chem. 1993, 460, 191–195. [Google Scholar] [CrossRef]
- Yan, X.; Chernega, A.; Green, M.L.H.; Sanders, J.; Souter, J.; Ushioda, T. Proximity and cooperativity effects in binuclear d0 olefin polymerization catalysis. Theoretical analysis of structure and reaction mechanism. J. Mol. Catal. A Chem. 1998, 128, 119–141. [Google Scholar] [CrossRef]
- Noh, S.K.; Kim, J.; Jung, J.; Ra, C.S.; Lee, D.-h.; Lee, H.B.; Lee, S.W.; Huh, W.S. Syntheses of polymethylene bridged dinuclear zirconocenes and investigation of their polymerisation activities. J. Organomet. Chem. 1999, 580, 90–97. [Google Scholar] [CrossRef]
- Lee, H.-W.; Park, Y.-H. Polymerization characteristics of in situ supported pentamethylene bridged dinuclear zirconocenes. Catal. Today 2002, 74, 309–320. [Google Scholar] [CrossRef]
- Noh, S.K.; Kim, S.; Yang, Y.; Lyoo, W.S.; Lee, D.-h. Preparation of syndiotactic polystyrene using the doubly bridged dinuclear titanocenes. Eur. Polym. J. 2004, 40, 227–235. [Google Scholar] [CrossRef]
- Kuwabara, J.; Takeuchi, D.; Osakada, K. Zr/Zr and Zr/Fe dinuclear complexes with flexible bridging ligands. Preparation by olefin metathesis reaction of the mononuclear precursors and properties as polymerization catalysts. Organometallics 2005, 24, 2705–2712. [Google Scholar] [CrossRef]
- Lee, M.H.; Kim, S.K.; Do, Y. Biphenylene-bridged dinuclear group 4 metal complexes: Enhanced polymerization properties in olefin polymerization. Organometallics 2005, 24, 3618–3620. [Google Scholar] [CrossRef]
- Liu, X.; Sun, J.; Zhang, H.; Xiao, X.; Lin, F. Ethylene polymerization by novel phenylenedimethylene-bridged homobinuclear titanocene/MAO systems. Eur. Polym. J. 2005, 41, 1519–1524. [Google Scholar] [CrossRef]
- Noh, S.K.; Jung, W.; Oh, H.; Lee, Y.R.; Lyoo, W.S. Synthesis and styrene polymerization properties of dinuclear half-titanocene complexes with xylene linkage. J. Organomet. Chem. 2006, 691, 5000–5006. [Google Scholar] [CrossRef]
- Xiao, X.; Sun, J.; Li, X.; Li, H.; Wang, Y. Binuclear titanocenes linked by the bridge combination of rigid and flexible segment: Synthesis and their use as catalysts for ethylene polymerization. J. Mol. Catal. A Chem. 2007, 267, 86–91. [Google Scholar] [CrossRef]
- Linh, N.T.B.; Huyen, N.T.D.; Noh, S.K.; Lyoo, W.S.; Lee, D.-H.; Kim, Y. Preparation of new dinuclear half-titanocene complexes with ortho- and meta-xylene linkages and investigation of styrene polymerization. J. Organomet. Chem. 2009, 694, 3438–3443. [Google Scholar] [CrossRef]
- Reddy, K.P.; Petersen, J.L. Synthesis and characterization of binuclear zirconocene complexes linked by a bridge bis(cyclopentadienyl) ligand. Organometallics 1989, 8, 2107–2113. [Google Scholar] [CrossRef]
- Noh, S.K.; Byun, G.-g.; Lee, C.-s.; Lee, D.; Yoon, K.-b.; Kang, K.S. Synthesis, characterization, and reactivities of the polysiloxane-bridged binuclear metallocenes tetramethyldisiloxanediylbis(cyclopentadienyltitanium trichloride) and hexamethyltrisiloxanediylbis(cyclopentadienyltitanium trichloride). J. Organomet. Chem. 1996, 518, 1–6. [Google Scholar] [CrossRef]
- Xu, S.; Huang, J. Asymmetric binuclear metallocene complexes and their application for olefin polymerization. J. Appl. Polym. Sci. 2013, 130, 2891–2900. [Google Scholar] [CrossRef]
- Murray, R.E.; Jayaratne, K.C.; Yang, Q.; Martin, J.L. To Chevron Phillips Chemical Company. WO Pat. 2009/085129, 2009. [Google Scholar]
- Soga, K.; Ban, H.T.; Uozumi, T. Synthesis of a dinuclear ansa-zirconocene catalyst having a biphenyl bridge and application to ethene polymerization. J. Mol. Catal. A Chem. 1998, 128, 273–278. [Google Scholar] [CrossRef]
- Spaleck, W.; Küber, F.; Bachmann, B.; Fritze, C.; Winter, A. New bridged zirconocenes for olefin polymerization: Binuclear and hybrid structures. J. Mol. Catal. A Chem. 1998, 128, 279–287. [Google Scholar] [CrossRef]
- Alt, H.G.; Ernst, R.; Böhmer, I.K. Dinuclear ansa-zirconocene complexes containing a sandwich and a half-sandwich moiety as catalysts for the polymerization of ethylene. J. Organomet. Chem. 2002, 658, 259–265. [Google Scholar] [CrossRef]
- Jende, L.N.; Vantomme, A.; Welle, A.; Brusson, J.-M.; Carpentier, J.-F.; Kirillov, E. Trinuclear tris(ansa-metallocene) complexes of zirconium and hafnium for olefin polymerization. J. Organomet. Chem. 2018, 878, 19–29. [Google Scholar] [CrossRef]
- Dane, E.L.; Swager, T.M. Carbanionic route to electroactive carbon-centered anion and radical oligomers. Org. Lett. 2010, 12, 4324–4327. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Abboud, K.A.; Miller, S.A. Sterically expanded CGC catalysts: Substituent effects on ethylene and α-olefin polymerization. Dalton Trans. 2013, 42, 9139–9147. [Google Scholar] [CrossRef]
- Tang, W.; Singh, S.P.; Ong, K.H.; Chen, Z.-K. Synthesis of thieno[3,2-b]thiophene derived conjugated oligomers for field-effect transistors applications. J. Mat. Chem. 2010, 20, 1497–1505. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Singer, R.A.; Yang, B.H.; Buchwald, S.L. Highly active palladium catalysts for Suzuki coupling reactions. J. Am. Chem. Soc. 1999, 121, 9550–9561. [Google Scholar] [CrossRef]
- Ewen, J.A.; Jones, R.L.; Razavi, A.; Ferrara, J.D. Syndiospecific propylene polymerizations with Group IVB metallocenes. J. Am. Chem. Soc. 1988, 110, 6255–6256. [Google Scholar] [CrossRef]
- Razavi, A.; Ferrara, J. Preparation and crystal structures of the complexes (η5-C5H4CMe2η5-C13H8)MCl2 (M = Zr, Hf) and their role in the catalytic formation of syndiotactic polypropylene. J. Organomet. Chem. 1992, 435, 299–310. [Google Scholar] [CrossRef]
- Kirillov, E.; Marquet, N.; Razavi, A.; Belia, V.; Hampel, F.; Roisnel, T.; Gladysz, J.A.; Carpentier, J.-F. New C1-symmetric Ph2C-bridged multisubstituted ansa-zirconocenes for highly isospecific propylene polymerization: Synthetic approach via activated fulvenes. Organometallics 2010, 29, 5073–5082. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian Inc.: Pittsburgh, PA, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Precat. | Temp [°C] | [MAO]/[Zr] | mpolym [g] | Prod. [kg(PE)·mol(Zr)−1·h−1] | Tm b [°C] | Mn c [kg·mol−1] | Mw/Mn c |
|---|---|---|---|---|---|---|---|---|
| 1 | 9 | 60 | 5000 | 1.74 | 4640 | 132.2 | 43.7 | 3.3 |
| 2 | 9 | 60 | 2000 | 1.71 | 4560 | 131.6 | 48.9 | 3.1 |
| 3 | 9 | 20 | 5000 | 0.14 | 373 | 131.1 | n.d. d | n.d. d |
| 4 | M | 60 | 5000 | 2.79 | 7440 | 132.0 | 39.4 | 3.9 |
| 5 | M | 20 | 5000 | 0.69 | 1840 | 133.1 | 115.5 | 4.1 |
| 6 | M′ | 60 | 5000 | 2.66 | 7090 | 135.1 | 25.7 | 3.5 |
| Entry | Precat. | Temp [°C] | [MAO]/ [Zr] | mpolym [g] | Prod. [kg(PP)·mol(Zr)−1·h−1] | Tm b [°C] | Tcrist b [°C] | Mn c [kg· mol−1] | Mw/Mn c | [rrrr]/[rr]/[r] d [%] | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 9 | 60 | 5000 | 4.31 | 5690 | 102.2 sh118.0 s | 56.6 | 9.9 | 2.3 | 70.0 | 84.1 | 90.4 |
| 8 | 9 | 60 | 2000 | 3.90 | 5150 | 102.1 sh118.4 s | 48.5 | 14.3 | 2.3 | 69.2 | 83.4 | 90.1 |
| 9 | 9 | 20 | 5000 | 5.92 | 7810 | 119.6 sh135.3 s | 75.0 | 21.7 | 2.5 | 78.7 | 88.4 | 92.9 |
| 10 | M | 60 | 5000 | 19.6 | 25,870 | 101.7 sh118.9 s | 51.1 | 16.8 | 2.2 | 74.7 | 86.7 | 92.2 |
| 11 | M | 20 | 5000 | 6.32 | 8340 | 141.5 s150.5 s | 97.9 | 35.5 | 2.2 | 89.0 | 94.8 | 96.8 |
| 12 | M′ | 60 | 5000 | 3.45 | 4550 | 113.1 | 55.5 | 13.2 | 3.6 | 64.2 | 79.3 | 87.7 |
| Run | Precat. | Temp [°C] | [MAO]/ [Zr] | mpolym [g] | Prod. [kg·mol(Zr)−1·h−1] | Tm b [°C] | Mn c [kg·mol−1] | Mw/Mn c | C6 Incorporated d | End-Groups d,e | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mol.% | wt.% | sat. | vinyl | |||||||||
| 13 | 9 | 60 | 5000 | 3.75 | 10,000 | 100.5 | 27.8 | 2.1 | 4.7 | 12.8 | 5.1 | 2.0 |
| 14 | 9 | 60 | 2000 | 2.50 | 6650 | 104.0 | 31.4 | 2.1 | 3.9 | 10.8 | 5.8 | 2.0 |
| 15 | 9 | 20 | 5000 | 0.32 | 853 | 94.0 113.1 | 44.2 | 3.7 | 4.3 | 11.9 | 5.2 | 0.0 |
| 16 | M | 60 | 5000 | 8.89 | 23,700 | 82.3 | 29.7 | 2.4 | 6.8 | 17.9 | 9.0 | 0.0 |
| 17 | M | 20 | 5000 | 1.96 | 5220 | 95.5 | 75.8 | 2.6 | 4.7 | 12.9 | 4.3 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jende, L.N.; Roisnel, T.; Cirriez, V.; Welle, A.; Kirillov, E.; Carpentier, J.-F. An Asymmetric Dinuclear Bis(ansa-Zirconocene) Complex: Synthesis and Performance in Olefin (co-)Polymerization. Catalysts 2023, 13, 1108. https://doi.org/10.3390/catal13071108
Jende LN, Roisnel T, Cirriez V, Welle A, Kirillov E, Carpentier J-F. An Asymmetric Dinuclear Bis(ansa-Zirconocene) Complex: Synthesis and Performance in Olefin (co-)Polymerization. Catalysts. 2023; 13(7):1108. https://doi.org/10.3390/catal13071108
Chicago/Turabian StyleJende, Lars N., Thierry Roisnel, Virginie Cirriez, Alexandre Welle, Evgueni Kirillov, and Jean-Francois Carpentier. 2023. "An Asymmetric Dinuclear Bis(ansa-Zirconocene) Complex: Synthesis and Performance in Olefin (co-)Polymerization" Catalysts 13, no. 7: 1108. https://doi.org/10.3390/catal13071108