
PtCu Nanoparticle Catalyst for Electrocatalytic Glycerol Oxidation: How Does the PtCu Affect to Glycerol Oxidation Reaction Performance by Changing pH Conditions?

Abstract
:1. Introduction
2. Results and Discussion
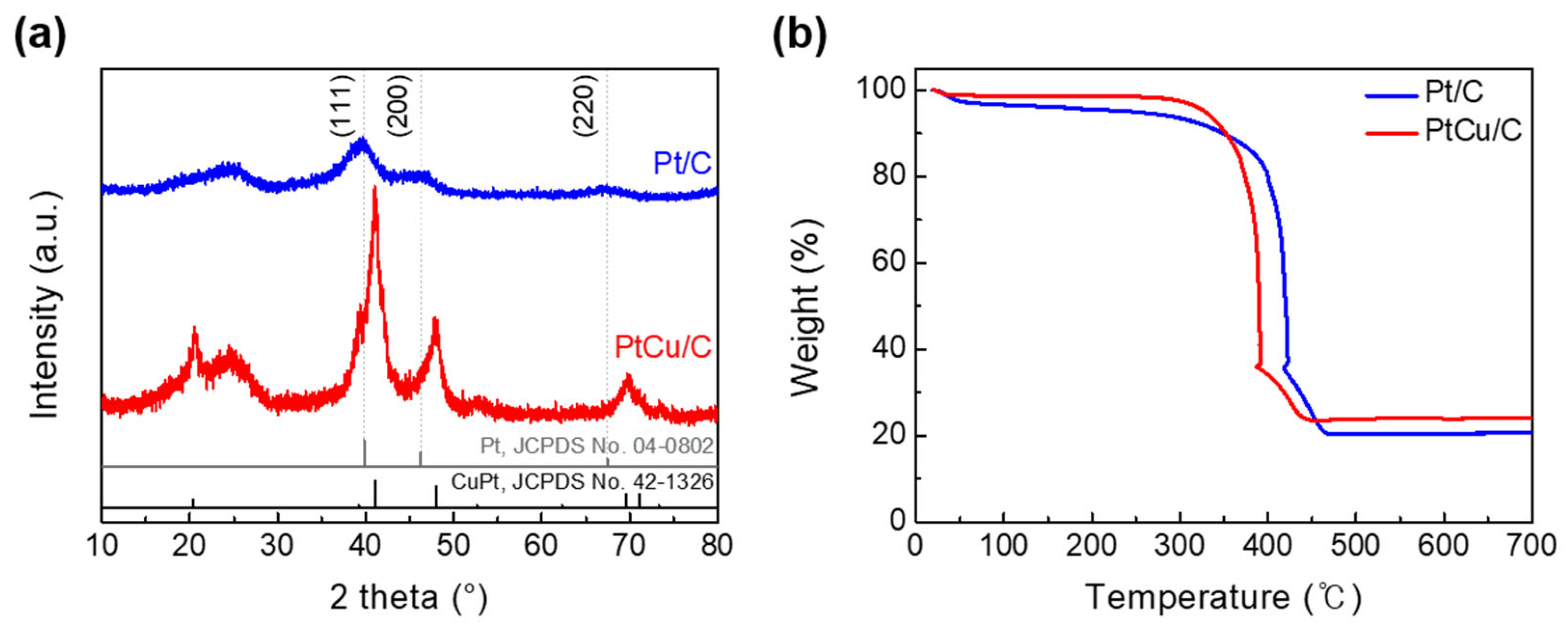
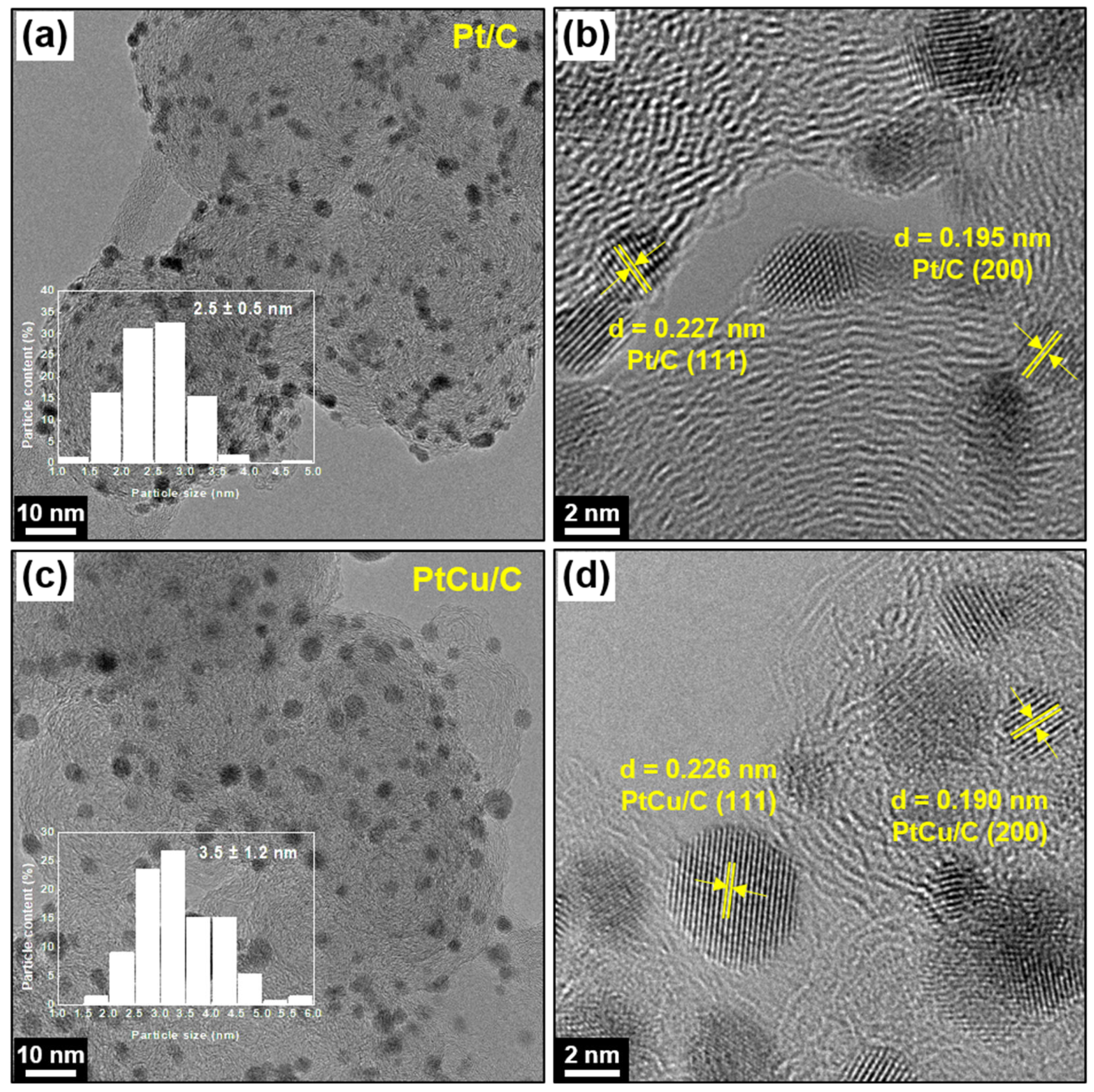
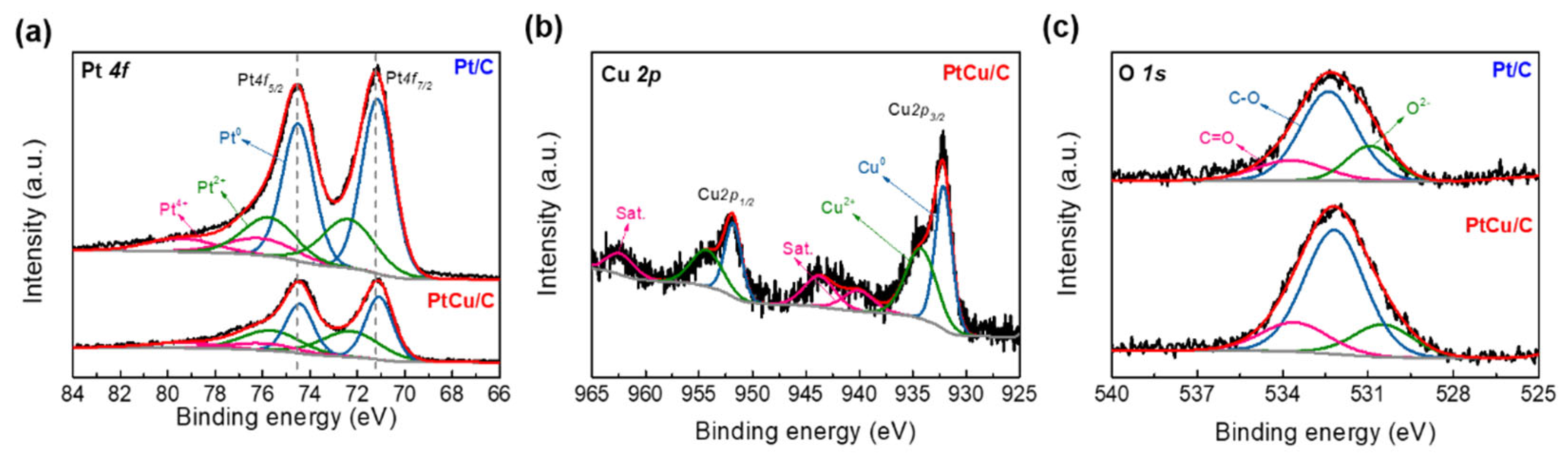
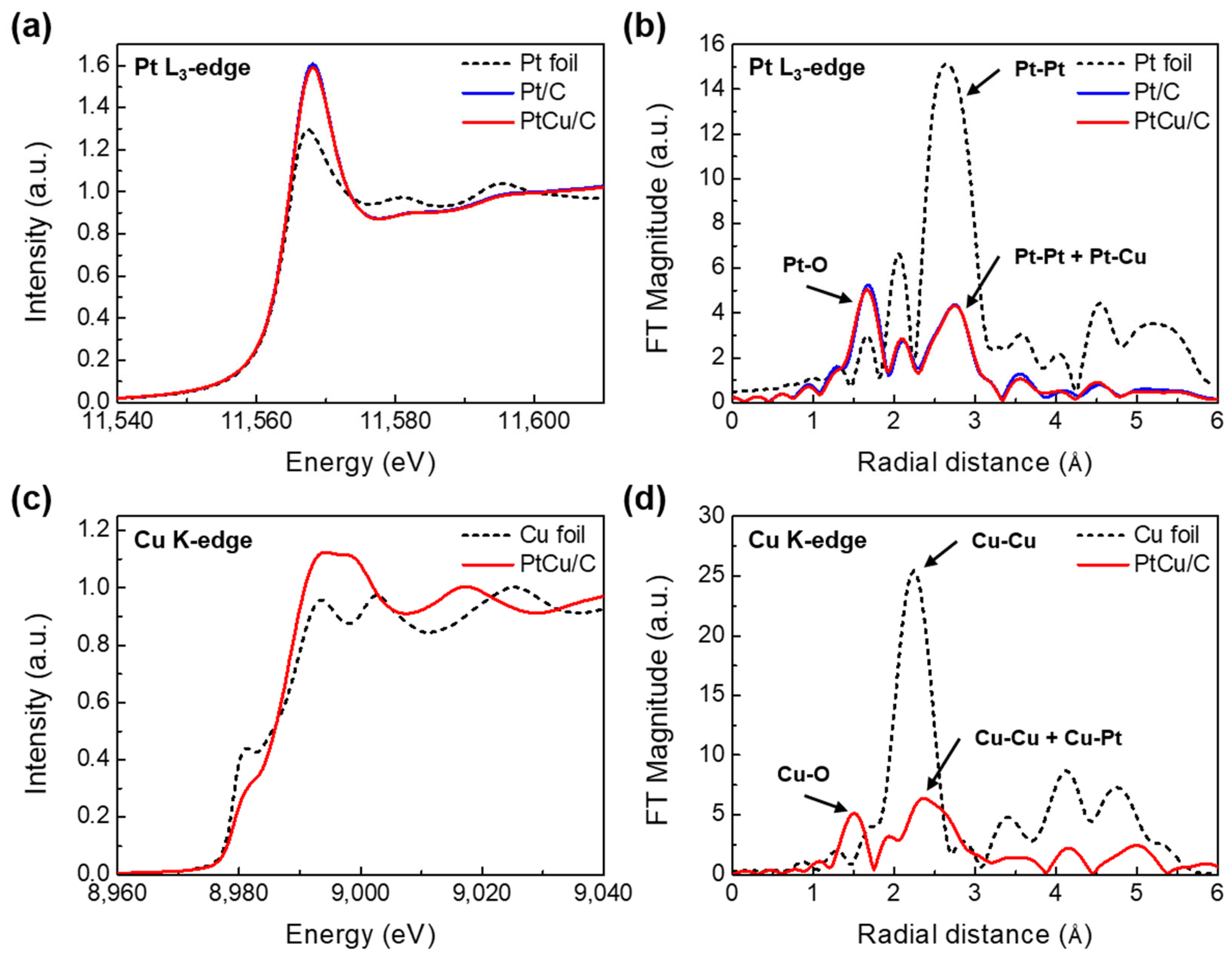
2.1. Physicochemical Characterizations of Pt/C and PtCu/C Catalysts
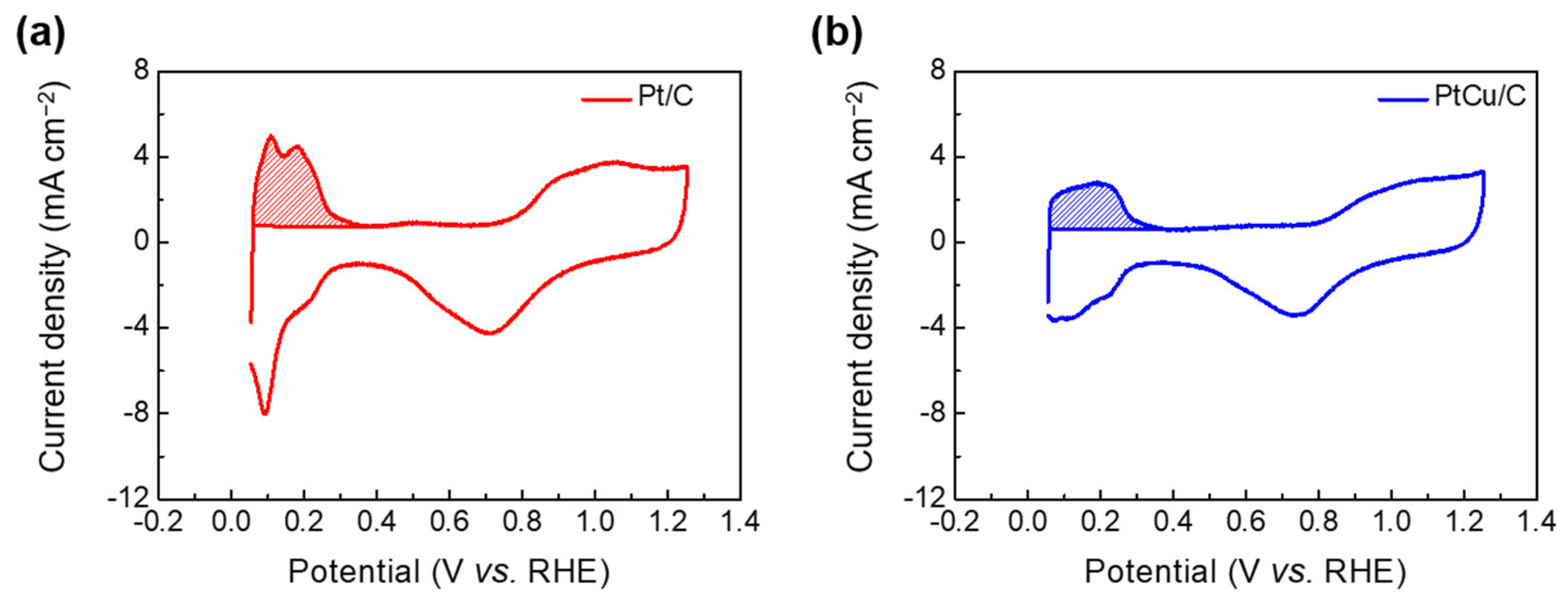
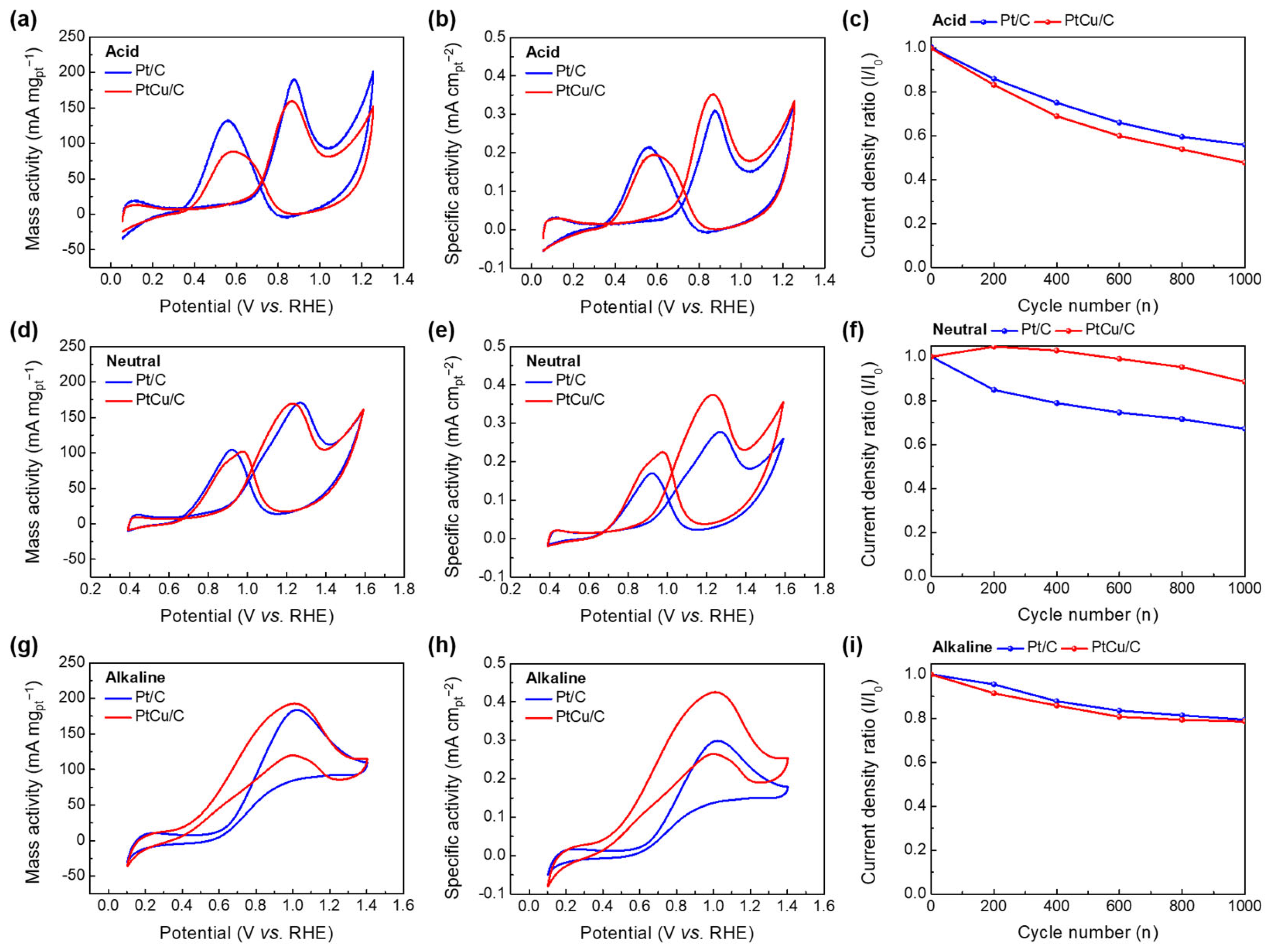
2.2. Electrochemical Performance Measurement of Pt/C and PtCu/C Catalysts
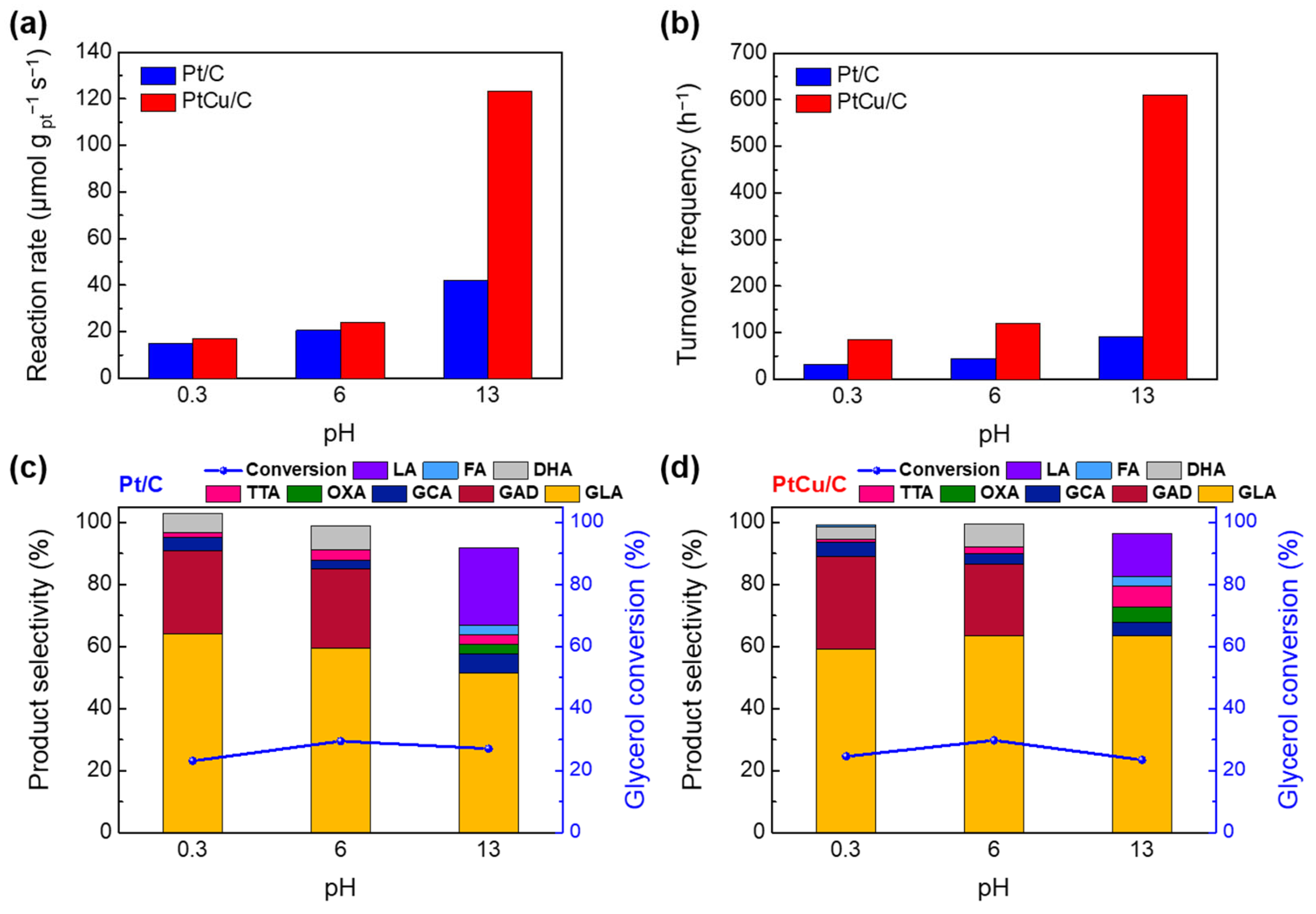
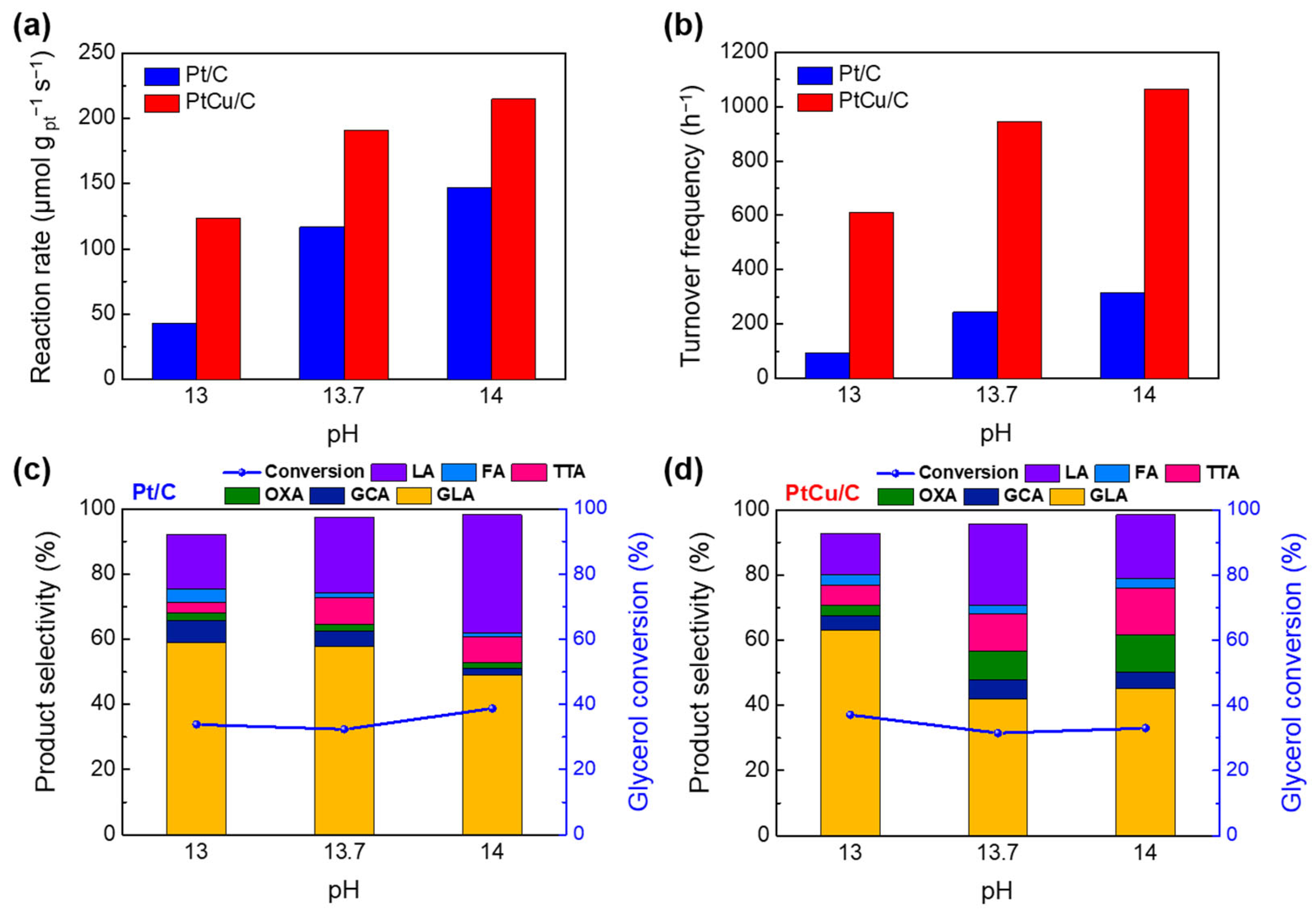
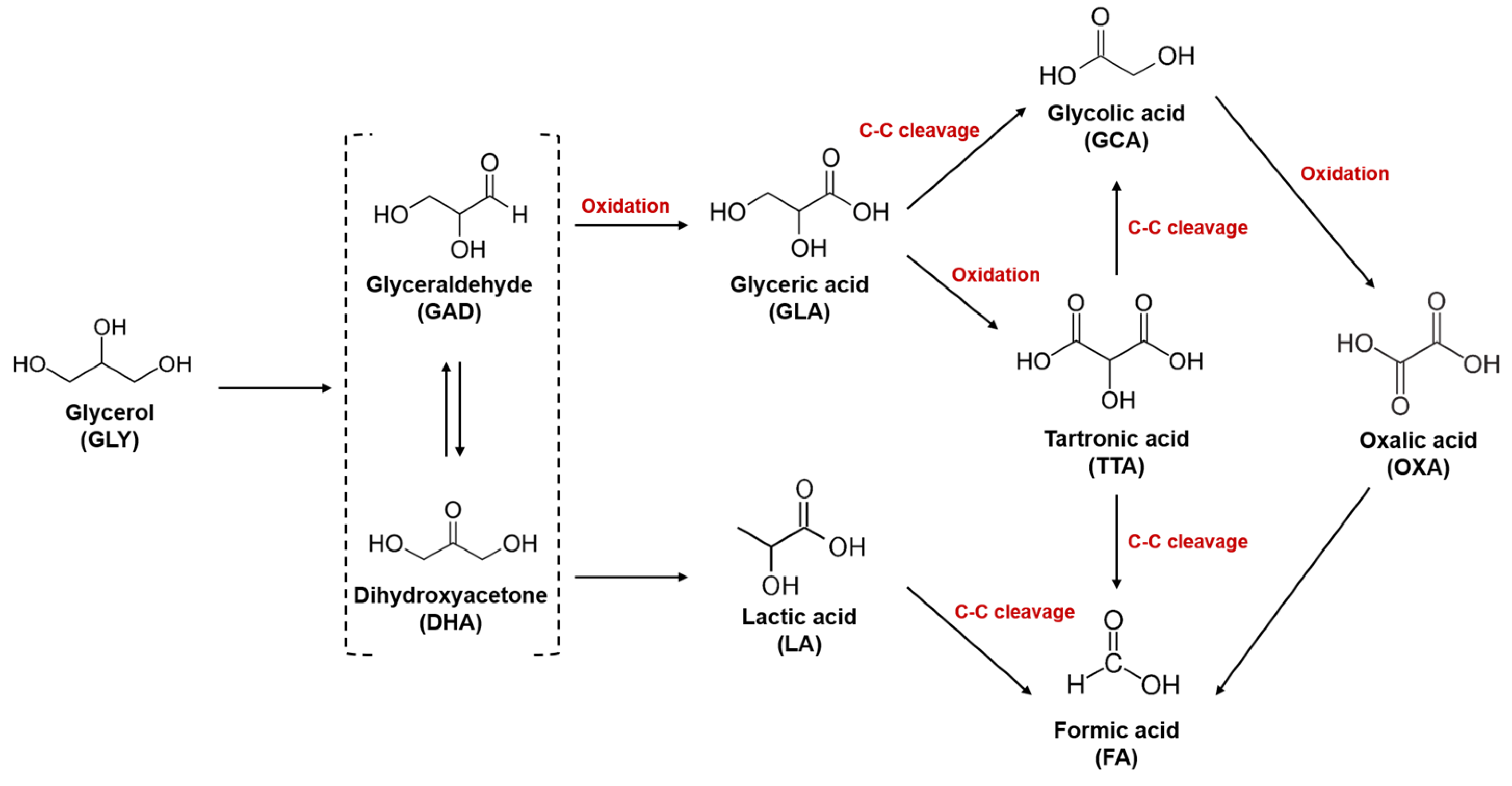
2.3. EGOR Performance Analyses of Pt/C and PtCu/C at Different pH Conditions
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Physicochemical Characterization
3.2.2. Electrochemical Performance Measurement in an Electrochemical Half-Cell System
3.2.3. Electrocatalytic Glycerol Oxidation Reaction Performance in an Electrochemical Batch Reactor System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; He, Q.; Xu, C. Catalytic Conversion of Glycerol into Hydrogen and Value-Added Chemicals: Recent Research Advances. Catalysts 2021, 11, 1455. [Google Scholar] [CrossRef]
- Salaheldeen, M.; Mariod, A.A.; Aroua, M.K.; Rahman, S.M.A.; Soudagar, M.E.M.; Fattah, I.M.R. Current State and Perspectives on Transesterification of Triglycerides for Biodiesel Production. Catalysts 2021, 11, 1121. [Google Scholar] [CrossRef]
- Tabassum, N.; Pothu, R.; Pattnaik, A.; Boddula, R.; Balla, P.; Gundeboyina, R.; Challa, P.; Rajesh, R.; Perugopu, V.; Mameda, N.; et al. Heterogeneous Catalysts for Conversion of Biodiesel-Waste Glycerol into High-Added-Value Chemicals. Catalysts 2022, 12, 767. [Google Scholar] [CrossRef]
- Anitha, M.; Kamarudin, S.K.; Kofli, N.T. The potential of glycerol as a value-added commodity. Chem. Eng. J. 2016, 295, 119–130. [Google Scholar] [CrossRef]
- Abdul Raman, A.A.; Tan, H.W.; Buthiyappan, A. Two-Step Purification of Glycerol as a Value Added by Product from the Biodiesel Production Process. Front. Chem. 2019, 7, 774. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Jiang, Y.; Zhu, Z.; Hou, Z. Selective oxidation of glycerol over different shaped WO3 supported Pt NPs. Mol. Catal. 2022, 523, 111545. [Google Scholar] [CrossRef]
- Monteiro, M.R.; Kugelmeier, C.L.; Pinheiro, R.S.; Batalha, M.O.; da Silva César, A. Glycerol from biodiesel production: Technological paths for sustainability. Renew. Sust. Energy Rev. 2018, 88, 109–122. [Google Scholar] [CrossRef]
- D’Angelo, S.C.; Dall’Ara, A.; Mondelli, C.; Pérez-Ramírez, J.; Papadokonstantakis, S. Techno-Economic Analysis of a Glycerol Biorefinery. ACS Sustain. Chem. Eng. 2018, 6, 16563–16572. [Google Scholar] [CrossRef]
- Chun Minh, A.L.; Samudrala, S.P.; Bhattacharya, S. Valorisation of glycerol through catalytic hydrogenolysis routes for sustainable production of value-added C3 chemicals: Current and future trends. Sustain. Energy Fuels 2022, 6, 596–639. [Google Scholar] [CrossRef]
- Dodekatos, G.; Schünemann, S.; Tüysüz, H. Recent Advances in Thermo-, Photo-, and Electrocatalytic Glycerol Oxidation. ACS Catal. 2018, 8, 6301–6333. [Google Scholar] [CrossRef]
- Pagliaro, M.V.; Bellini, M.; Bevilacqua, M.; Filippi, J.; Folliero, M.G.; Marchionni, A.; Miller, H.A.; Oberhauser, W.; Caporali, S.; Innocenti, M.; et al. Carbon supported Rh nanoparticles for the production of hydrogen and chemicals by the electroreforming of biomass-derived alcohols. RSC Adv. 2017, 7, 13971–13978. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Liu, B.; Liu, X.; Senthilkumar, N.; Wang, G.; Wen, Z. Recent Progress in Electrocatalytic Glycerol Oxidation. Energy Technol. 2020, 9, 2000804. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, Y.; Xi, J.; Luo, X. Selective Electro-Oxidation of Glycerol to Dihydroxyacetone by PtAg Skeletons. ACS Appl. Mater. Interfaces 2019, 11, 28953–28959. [Google Scholar] [CrossRef]
- Kim, D.; Oh, L.S.; Tan, Y.C.; Song, H.; Kim, H.J.; Oh, J. Enhancing Glycerol Conversion and Selectivity toward Glycolic Acid via Precise Nanostructuring of Electrocatalysts. ACS Catal. 2021, 11, 14926–14931. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, C.; Zhao, X.; Yan, H.; Li, J.; Lyu, P.; Du, Y.; Xi, S.; Chi, K.; Chi, X.; et al. Promoted Glycerol Oxidation Reaction in an Interface-Confined Hierarchically Structured Catalyst. Adv. Mater. 2019, 31, e1804763. [Google Scholar] [CrossRef]
- Lin, C.; Dong, C.; Kim, S.; Lu, Y.; Wang, Y.; Yu, Z.; Gu, Y.; Gu, Z.; Lee, D.K.; Zhang, K.; et al. Photo-Electrochemical Glycerol Conversion over a Mie Scattering Effect Enhanced Porous BiVO4 Photoanode. Adv. Mater. 2023, 35, e2209955. [Google Scholar] [CrossRef]
- Oh, L.S.; Park, M.; Park, Y.S.; Kim, Y.; Yoon, W.; Hwang, J.; Lim, E.; Park, J.H.; Choi, S.M.; Seo, M.H.; et al. How to Change the Reaction Chemistry on Nonprecious Metal Oxide Nanostructure Materials for Electrocatalytic Oxidation of Biomass-Derived Glycerol to Renewable Chemicals. Adv. Mater. 2023, 35, e2203285. [Google Scholar] [CrossRef]
- Xiong, L.; Qi, H.; Zhang, S.; Zhang, L.; Liu, X.; Wang, A.; Tang, J. Highly Selective Transformation of Biomass Derivatives to Valuable Chemicals by Single-Atom Photocatalyst Ni/TiO2. Adv. Mater. 2023, 35, e2209646. [Google Scholar] [CrossRef]
- Han, J.; Kim, Y.; Jackson, D.H.K.; Chang, H.; Kim, H.W.; Lee, J.; Kim, J.-R.; Noh, Y.; Kim, W.B.; Lee, K.-Y.; et al. Enhanced catalytic performance and changed reaction chemistry for electrochemical glycerol oxidation by atomic-layer-deposited Pt-nanoparticle catalysts. Appl. Catal. B 2020, 273, 119037. [Google Scholar] [CrossRef]
- Lee, D.; Kim, Y.; Kwon, Y.; Lee, J.; Kim, T.-W.; Noh, Y.; Kim, W.B.; Seo, M.H.; Kim, K.; Kim, H.J. Boosting the electrocatalytic glycerol oxidation performance with highly-dispersed Pt nanoclusters loaded on 3D graphene-like microporous carbon. Appl. Catal. B 2019, 245, 555–568. [Google Scholar] [CrossRef]
- Lima, C.C.; Rodrigues, M.V.F.; Neto, A.F.M.; Zanata, C.R.; Pires, C.T.G.V.M.T.; Costa, L.S.; Solla-Gullón, J.; Fernández, P.S. Highly active Ag/C nanoparticles containing ultra-low quantities of sub-surface Pt for the electrooxidation of glycerol in alkaline media. Appl. Catal. B 2020, 279, 119369. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, Y.; Xi, J. Seed-mediated synthesis of PtxAuy@Ag electrocatalysts for the selective oxidation of glycerol. Appl. Catal. B 2019, 245, 604–612. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, J.; Green, S.K.; Huber, G.W.; Kim, W.B. Selective glycerol oxidation by electrocatalytic dehydrogenation. ChemSusChem 2014, 7, 1051–1056. [Google Scholar] [CrossRef]
- Lee, S.; Kim, H.J.; Lim, E.J.; Kim, Y.; Noh, Y.; Huber, G.W.; Kim, W.B. Highly selective transformation of glycerol to dihydroxyacetone without using oxidants by a PtSb/C-catalyzed electrooxidation process. Green Chem. 2016, 18, 2877–2887. [Google Scholar] [CrossRef]
- Lee, D.; Kim, Y.; Han, H.; Kim, W.B.; Chang, H.; Chung, T.-M.; Han, J.H.; Kim, H.W.; Kim, H.J. Atomic-layer-deposited SnO2 on Pt/C prevents sintering of Pt nanoparticles and affects the reaction chemistry for the electrocatalytic glycerol oxidation reaction. J. Mater. Chem. A 2020, 8, 15992–16005. [Google Scholar] [CrossRef]
- Benipal, N.; Qi, J.; Liu, Q.; Li, W. Carbon nanotube supported PdAg nanoparticles for electrocatalytic oxidation of glycerol in anion exchange membrane fuel cells. Appl. Catal. B 2017, 210, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Sun, L.; Pan, X.; Zhou, Z.; Zheng, Y.; Yang, X.; Zhao, G. Carbon paper supported gold nanoflowers for tunable glycerol electrooxidation boosting efficient hydrogen evolution. Carbon 2023, 203, 88–96. [Google Scholar] [CrossRef]
- Tran, G.-S.; Vo, T.-G.; Chiang, C.-Y. Earth-abundant manganese oxide nanoneedle as highly efficient electrocatalyst for selective glycerol electro-oxidation to dihydroxyacetone. J. Catal. 2021, 404, 139–148. [Google Scholar] [CrossRef]
- Han, X.; Sheng, H.; Yu, C.; Walker, T.W.; Huber, G.W.; Qiu, J.; Jin, S. Electrocatalytic Oxidation of Glycerol to Formic Acid by CuCo2O4 Spinel Oxide Nanostructure Catalysts. ACS Catal. 2020, 10, 6741–6752. [Google Scholar] [CrossRef]
- Ahmad, M.S.; Ng, K.H.; Chen, C.L.; Kabir, F.; Witoon, T.; Wu, T.Y.; Cheng, C.K. Nitrogen-phosphorous co-doped palladium electrocatalyst for glycerol electro-oxidation reaction (GEOR): An efficient system for mesoxalic acid and dihydroxyacetone production. Fuel 2023, 333, 126471. [Google Scholar] [CrossRef]
- Liu, C.; Hirohara, M.; Maekawa, T.; Chang, R.; Hayashi, T.; Chiang, C.-Y. Selective electro-oxidation of glycerol to dihydroxyacetone by a non-precious electrocatalyst—CuO. Appl. Catal. B 2020, 265, 118543. [Google Scholar] [CrossRef]
- Lee, S.; Kim, H.J.; Choi, S.M.; Seo, M.H.; Kim, W.B. The promotional effect of Ni on bimetallic PtNi/C catalysts for glycerol electrooxidation. Appl. Catal. A Gen. 2012, 429–430, 39–47. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, S.M.; Green, S.; Tompsett, G.A.; Lee, S.H.; Huber, G.W.; Kim, W.B. Highly active and stable PtRuSn/C catalyst for electrooxidations of ethylene glycol and glycerol. Appl. Catal. B 2011, 101, 366–375. [Google Scholar] [CrossRef]
- Simões, M.; Baranton, S.; Coutanceau, C. Enhancement of catalytic properties for glycerol electrooxidation on Pt and Pd nanoparticles induced by Bi surface modification. Appl. Catal. B 2011, 110, 40–49. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.W.; Lee, S.; Han, J.; Lee, D.; Kim, J.R.; Kim, T.W.; Kim, C.U.; Jeong, S.Y.; Chae, H.J.; et al. The Role of Ruthenium on Carbon-Supported PtRu Catalysts for Electrocatalytic Glycerol Oxidation under Acidic Conditions. ChemCatChem 2017, 9, 1683–1690. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shen, Y.; Piao, J. Sustainable Conversion of Glycerol into Value-Added Chemicals by Selective Electro-Oxidation on Pt-Based Catalysts. ChemElectroChem 2018, 5, 1636–1643. [Google Scholar] [CrossRef]
- Sieben, J.M.; Alvarez, A.E.; Sanchez, M.D. Glycerol electrooxidation on carbon-supported Pt-CuO and PtCu-CuO catalysts. Electrochim. Acta 2023, 439, 141672. [Google Scholar] [CrossRef]
- Hu, W.; Knight, D.; Lowry, B.; Varma, A. Selective Oxidation of Glycerol to Dihydroxyacetone over Pt-Bi/C Catalyst: Optimization of Catalyst and Reaction Conditions. Ind. Eng. Chem. Res. 2010, 49, 10876–10882. [Google Scholar] [CrossRef]
- Castagna, R.M.; Sieben, J.M.; Alvarez, A.E.; Duarte, M.M.E. Electrooxidation of ethanol and glycerol on carbon supported PtCu nanoparticles. Int. J. Hydrogen Energy 2019, 44, 5970–5982. [Google Scholar] [CrossRef]
- Dai, C.; Sun, L.; Liao, H.; Khezri, B.; Webster, R.D.; Fisher, A.C.; Xu, Z.J. Electrochemical production of lactic acid from glycerol oxidation catalyzed by AuPt nanoparticles. J. Catal. 2017, 356, 14–21. [Google Scholar] [CrossRef]
- Kwon, Y.; Birdja, Y.; Spanos, I.; Rodriguez, P.; Koper, M.T.M. Highly Selective Electro-Oxidation of Glycerol to Dihydroxyacetone on Platinum in the Presence of Bismuth. ACS Catal. 2012, 2, 759–764. [Google Scholar] [CrossRef]
- Jin, X.; Yan, H.; Zeng, C.; Thapa, P.S.; Subramaniam, B.; Chaudhari, R.V. Phase Transformed PtFe Nanocomposites Show Enhanced Catalytic Performances in Oxidation of Glycerol to Tartronic Acid. Ind. Eng. Chem. Res. 2017, 56, 13157–13164. [Google Scholar] [CrossRef]
- Nie, R.; Liang, D.; Shen, L.; Gao, J.; Chen, P.; Hou, Z. Selective oxidation of glycerol with oxygen in base-free solution over MWCNTs supported PtSb alloy nanoparticles. Appl. Catal. B 2012, 127, 212–220. [Google Scholar] [CrossRef]
- Peng, X.; Chen, D.; Yang, X.; Wang, D.; Li, M.; Tseng, C.C.; Panneerselvam, R.; Wang, X.; Hu, W.; Tian, J.; et al. Microwave-Assisted Synthesis of Highly Dispersed PtCu Nanoparticles on Three-Dimensional Nitrogen-Doped Graphene Networks with Remarkably Enhanced Methanol Electrooxidation. ACS Appl. Mater. Interfaces 2016, 8, 33673–33680. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Y.; Peng, X.; Wang, X.; Hu, W.; Jing, C.; Tian, S.; Tian, J. Star-like PtCu nanoparticles supported on graphene with superior activity for methanol electro-oxidation. Electrochim. Acta 2015, 177, 86–92. [Google Scholar] [CrossRef]
- Oh, L.S.; Kim, J.Y.; Kim, H.W.; Han, J.; Lim, E.; Kim, W.B.; Park, J.H.; Kim, H.J. Unveiling the enhanced electrocatalytic activity at electrochemically synthesized Pt-WOx hybrid nanostructure interfaces. Chem. Commun. 2021, 57, 11165–11168. [Google Scholar] [CrossRef]
- Lee, D.; Kim, Y.; Kim, H.W.; Choi, M.; Park, N.; Chang, H.; Kwon, Y.; Park, J.H.; Kim, H.J. In situ electrochemically synthesized Pt-MoO3−x nanostructure catalysts for efficient hydrogen evolution reaction. J. Catal. 2020, 381, 1–13. [Google Scholar] [CrossRef]
- Mintsouli, I.; Georgieva, J.; Armyanov, S.; Valova, E.; Avdeev, G.; Hubin, A.; Steenhaut, O.; Dille, J.; Tsiplakides, D.; Balomenou, S.; et al. Pt-Cu electrocatalysts for methanol oxidation prepared by partial galvanic replacement of Cu/carbon powder precursors. Appl. Catal. B 2013, 136–137, 160–167. [Google Scholar] [CrossRef]
- Zou, X.; Fan, H.; Tian, Y.; Zhang, M.; Yan, X. Chemical bath deposition of Cu2O quantum dots onto ZnO nanorod arrays for application in photovoltaic devices. RSC Adv. 2015, 5, 23401–23409. [Google Scholar] [CrossRef]
- Hu, B.; Yuan, J.; Zhang, J.; Shu, Q.; Guan, D.; Yang, G.; Zhou, W.; Shao, Z. High activity and durability of a Pt–Cu–Co ternary alloy electrocatalyst and its large-scale preparation for practical proton exchange membrane fuel cells. Compos. B Eng. 2021, 222, 109082. [Google Scholar] [CrossRef]
- Zhou, Z.; Wei, L.; Wang, Y.; Karahan, H.E.; Chen, Z.; Lei, Y.; Chen, X.; Zhai, S.; Liao, X.; Chen, Y. Hydrogen evolution reaction activity of nickel phosphide is highly sensitive to electrolyte pH. J. Mater. Chem. A 2017, 5, 20390–20397. [Google Scholar] [CrossRef]
- Habibi, B.; Ghaderi, S. Synthesis, characterization and electrocatalytic activity of Co@Pt nanoparticles supported on carbon-ceramic substrate for fuel cell applications. Int. J. Hydrogen Energy 2015, 40, 5115–5125. [Google Scholar] [CrossRef]
- Pupo, M.M.S.; López-Suárez, F.E.; Bueno-López, A.; Meneses, C.T.; Eguiluz, K.I.B.; Salazar-Banda, G.R. Sn@Pt and Rh@Pt core–shell nanoparticles synthesis for glycerol oxidation. J. Appl. Electrochem. 2014, 45, 139–150. [Google Scholar] [CrossRef]
- Zuo, Y.; Wu, L.; Cai, K.; Li, T.; Yin, W.; Li, D.; Li, N.; Liu, J.; Han, H. Platinum Dendritic-Flowers Prepared by Tellurium Nanowires Exhibit High Electrocatalytic Activity for Glycerol Oxidation. ACS Appl. Mater. Interfaces 2015, 7, 17725–17730. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Metal Loading 1 (wt%) | Particle Size (nm) | 2θmax 4 (°) | Lattice Parameter 5 (Å) | Pt/Cu (Atomic Ratio) | ||
|---|---|---|---|---|---|---|---|
| TEM 2 | XRD 3 | ICP-OES 6 | XPS 7 | ||||
| Pt/C | 20.9 | 2.5 ± 0.5 | 2.8 | 67.1 | 3.938 | - | - |
| PtCu/C | 23.5 | 3.5 ± 1.2 | 3.9 | 69.8 | 3.806 | 0.81 | 1.38 |
| Catalysts | Binding Energy (eV) /Relative Area (%) | Binding Energy (eV) /Relative Area (%) | Binding Energy (eV) /Relative Area (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Pt/C | Pt 4f7/2 | Pt0 | 71.15/33.40 | - | O2− | 530.93/20.40 | ||
| Pt2+ | 72.38/15.60 | |||||||
| Pt4+ | 76.09/6.90 | C-O | 532.38/62.10 | |||||
| Pt 4f5/2 | Pt0 | 74.50/26.30 | ||||||
| Pt2+ | 75.73/12.30 | C=O | 533.71/17.50 | |||||
| Pt4+ | 79.44/5.50 | |||||||
| PtCu/C | Pt 4f7/2 | Pt0 | 71.08/26.10 | Cu 2p3/2 | Cu0 | 932.13/35.80 | O2− | 530.48/17.10 |
| Pt2+ | 72.29/22.80 | Cu2+ | 934.35/30.10 | |||||
| Pt4+ | 76.00/7.00 | C-O | 532.18/65.60 | |||||
| Pt 4f5/2 | Pt0 | 74.43/20.50 | Cu 2p1/2 | Cu0 | 951.88/18.50 | |||
| Pt2+ | 75.64/18.00 | Cu2+ | 954.10/15.60 | C=O | 533.58/17.20 | |||
| Pt4+ | 79.35/5.50 | |||||||
| Edge | Catalyst | Atom Pair | Coordination Number (N) | R 1 (Å) | ΔE0 2 (eV) | σ 2,3 (10−3 Å2) | R-Factor 4 (%) |
|---|---|---|---|---|---|---|---|
| Pt L3-edge | Pt foil | Pt—Pt | 12.0 | 2.77 ± 0.00 | 8.2 ± 0.7 | 4.8 ± 0.3 | 0.0027 |
| Pt/C | Pt—O | 2.6 ± 0.5 | 2.03 ± 0.01 | 14.8 ± 2.0 | 4.2 ± 1.5 | 0.0147 | |
| Pt—Pt | 4.2 ± 1.0 | 2.77 ± 0.01 | 12.4 ± 2.5 | 6.3 ± 1.1 | |||
| PtCu/C | Pt—O | 2.9 ± 0.7 | 2.02 ± 0.02 | 13.6 ± 2.7 | 5.4 ± 2.3 | 0.0124 | |
| Pt—Cu | 0.9 ± 1.5 | 2.68 ± 0.10 | 22.4 ± 15.4 | 9.6 ± 11.2 | |||
| Pt—Pt | 3.0 ± 2.1 | 2.77 ± 0.02 | 9.6 ± 5.2 | 4.7 ± 2.7 | |||
| Cu K-edge | Cu foil | Cu—Cu | 12.0 | 2.54 ± 0.01 | 3.84 ± 1.03 | 8.4 ± 0.6 | 0.0073 |
| PtCu/C | Cu—O | 1.8 ± 1.0 | 1.91 ± 0.03 | −4.30 ± 7.79 | 3.2 ± 4.0 | 0.0102 | |
| Cu—Pt | 2.2 ± 2.3 | 2.66 ± 0.03 | −3.16 ± 8.21 | 4.1 ± 4.1 | |||
| Cu—Cu | 3.1 ± 2.7 | 2.62 ± 0.07 | −8.05 ± 9.38 | 12.5 ± 7.8 |
| Catalyst | Feed Solution (1 M Glycerol in Electrolyte) | ECSA 1 (m2 gPt−1) | Onset Potential 2 (V vs. RHE) | Tafel Slope 2 (mV dec−1) | Peak Potential 3 (V vs. RHE) | Mass Activity 4 (mA mgPt−1) | Specific Activity 4 (mA cmPt−2) |
|---|---|---|---|---|---|---|---|
| Pt/C | 0.5 M H2SO4 | 61.5 | 0.45 | 196.50 | 0.87 | 181.90 ± 6.10 | 0.30 ± 0.01 |
| 0.1 M Na2SO4 | 0.64 | 175.20 | 1.23 | 160.70 ± 13.70 | 0.26 ± 0.02 | ||
| 0.1 M KOH | 0.62 | 160.80 | 1.02 | 190.60 ± 9.74 | 0.31 ± 0.02 | ||
| PtCu/C | 0.5 M H2SO4 | 45.3 | 0.54 | 133.80 | 0.86 | 156.20 ± 3.14 | 0.34 ± 0.01 |
| 0.1 M Na2SO4 | 0.67 | 121.00 | 1.20 | 163.20 ± 5.71 | 0.36 ± 0.01 | ||
| 0.1 M KOH | 0.28 | 107.80 | 1.01 | 199.80 ± 10.00 | 0.44 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, L.S.; Han, J.; Lim, E.; Kim, W.B.; Kim, H.J. PtCu Nanoparticle Catalyst for Electrocatalytic Glycerol Oxidation: How Does the PtCu Affect to Glycerol Oxidation Reaction Performance by Changing pH Conditions? Catalysts 2023, 13, 892. https://doi.org/10.3390/catal13050892
Oh LS, Han J, Lim E, Kim WB, Kim HJ. PtCu Nanoparticle Catalyst for Electrocatalytic Glycerol Oxidation: How Does the PtCu Affect to Glycerol Oxidation Reaction Performance by Changing pH Conditions? Catalysts. 2023; 13(5):892. https://doi.org/10.3390/catal13050892
Chicago/Turabian StyleOh, Lee Seul, Jeonghyun Han, Eunho Lim, Won Bae Kim, and Hyung Ju Kim. 2023. "PtCu Nanoparticle Catalyst for Electrocatalytic Glycerol Oxidation: How Does the PtCu Affect to Glycerol Oxidation Reaction Performance by Changing pH Conditions?" Catalysts 13, no. 5: 892. https://doi.org/10.3390/catal13050892