
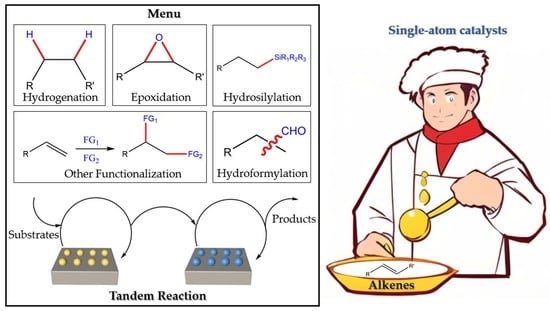
Recent Development of Single-Atom Catalysis for the Functionalization of Alkenes
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
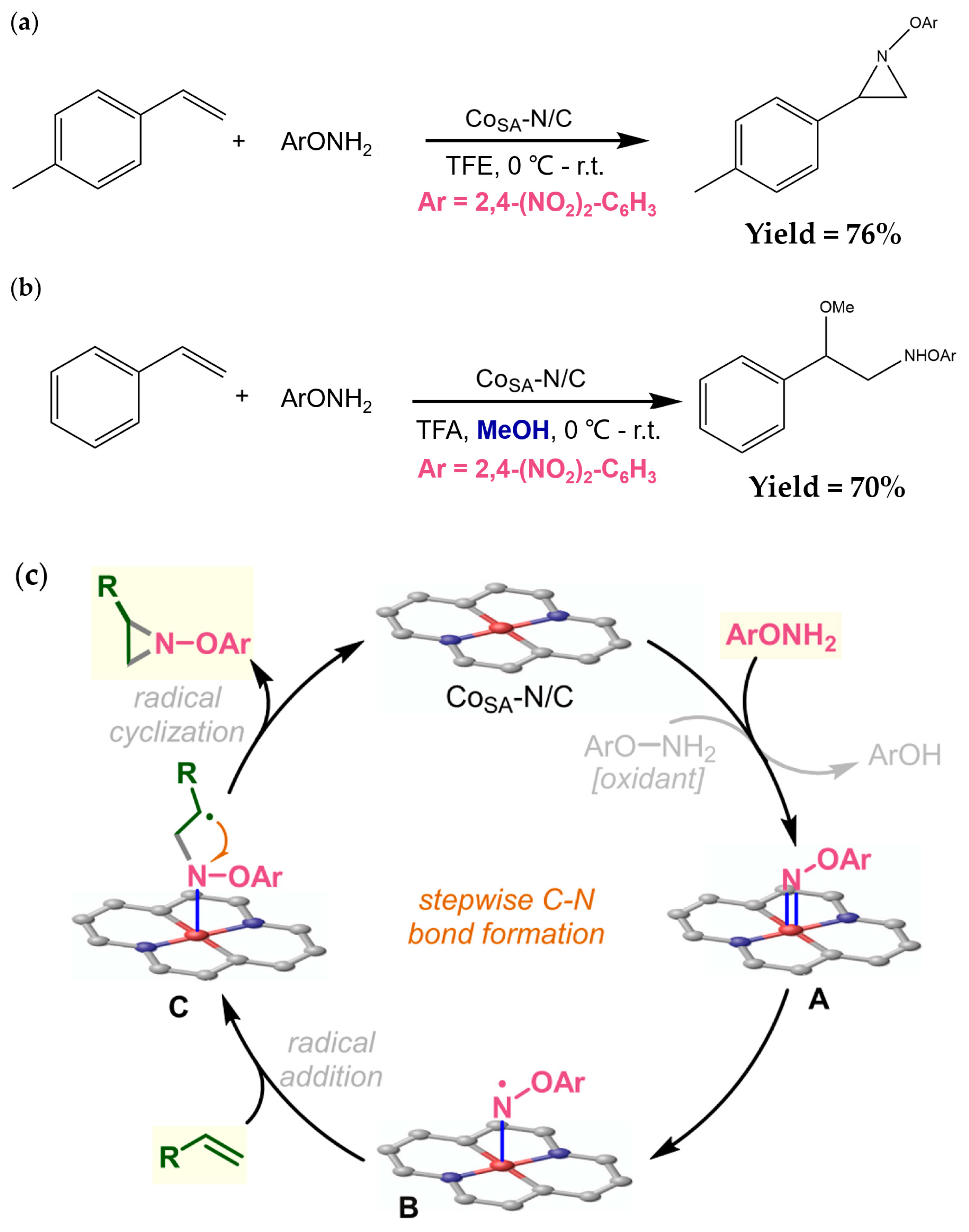{kind=link}
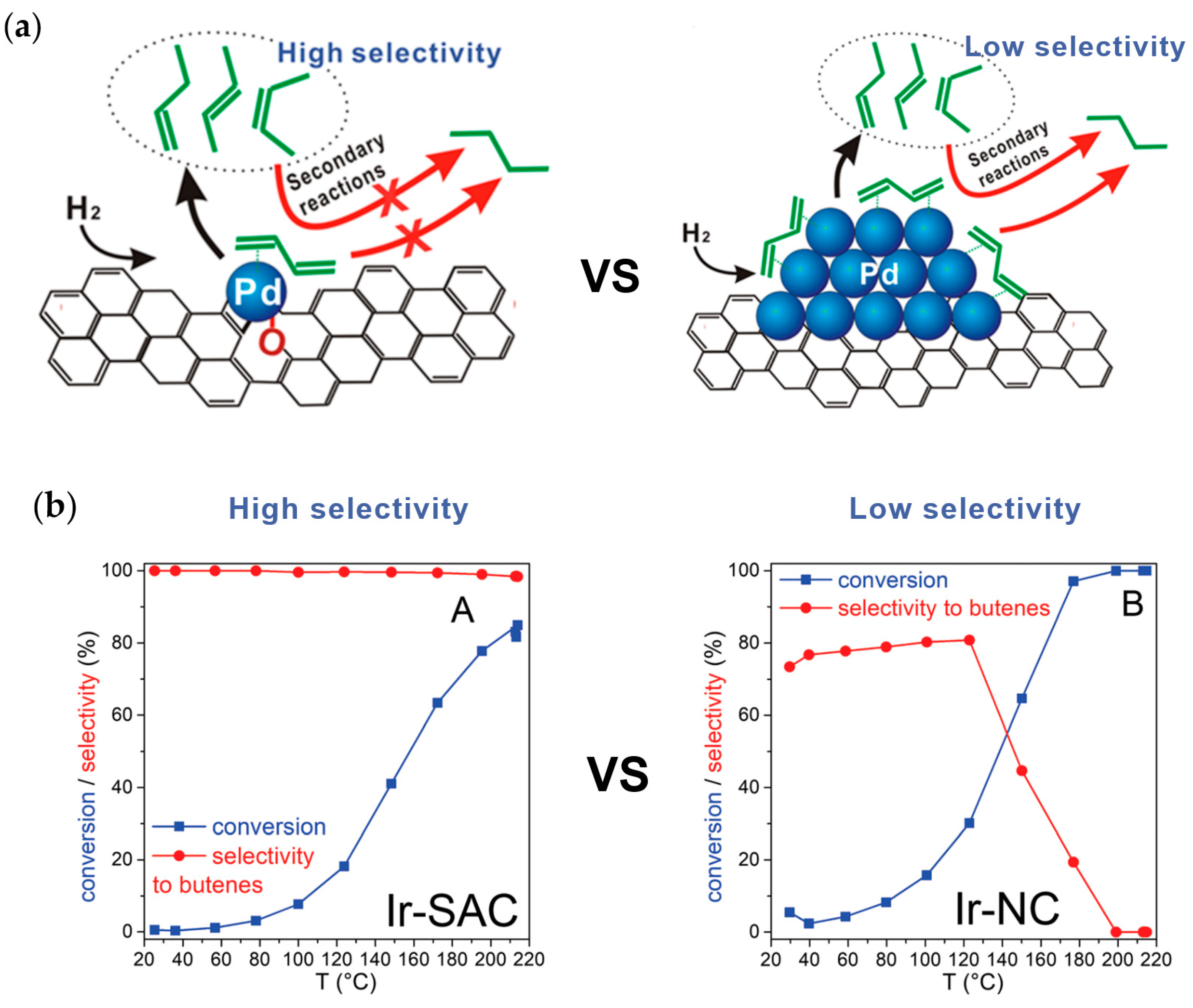{kind=link}
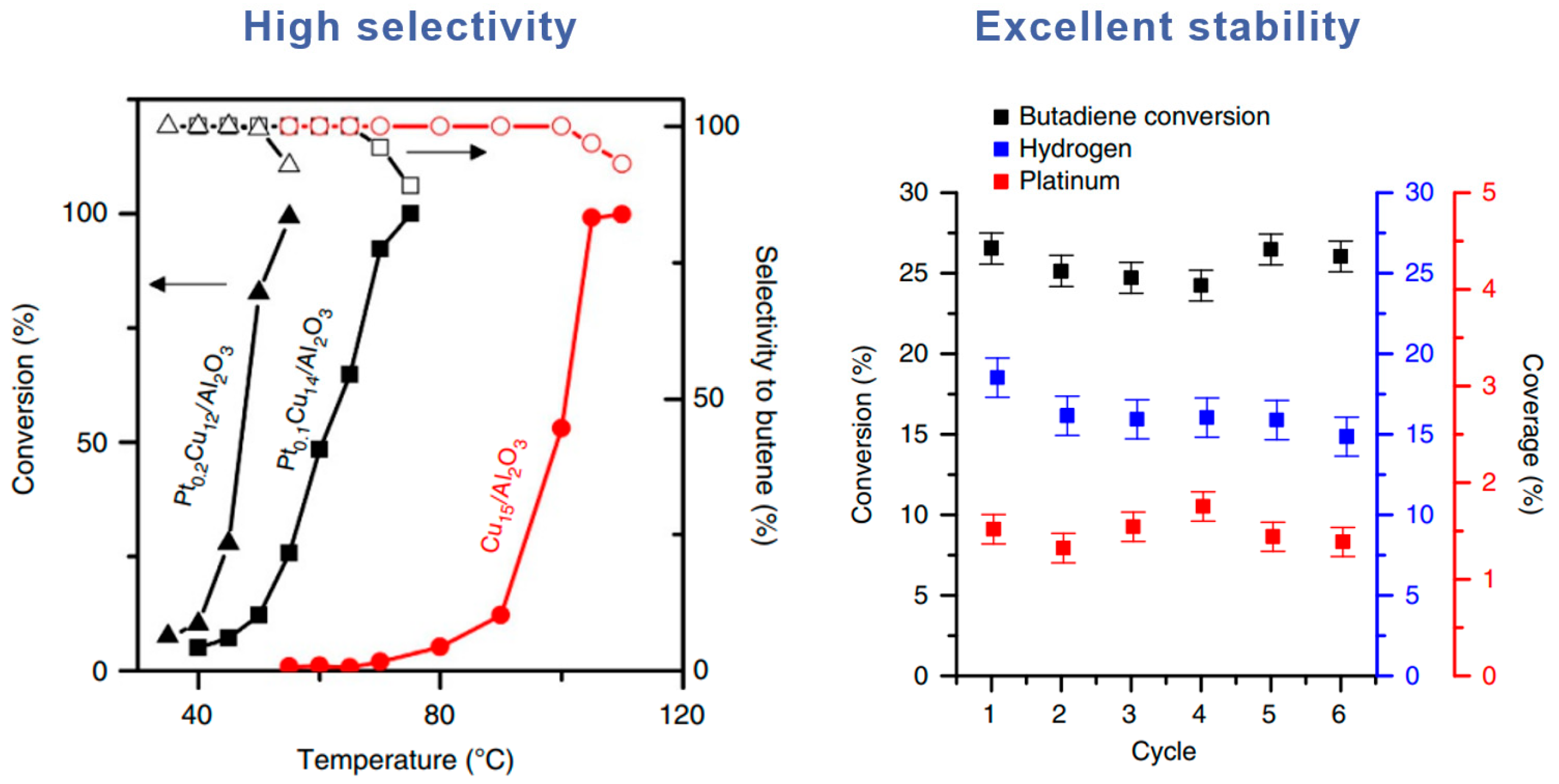{kind=link}
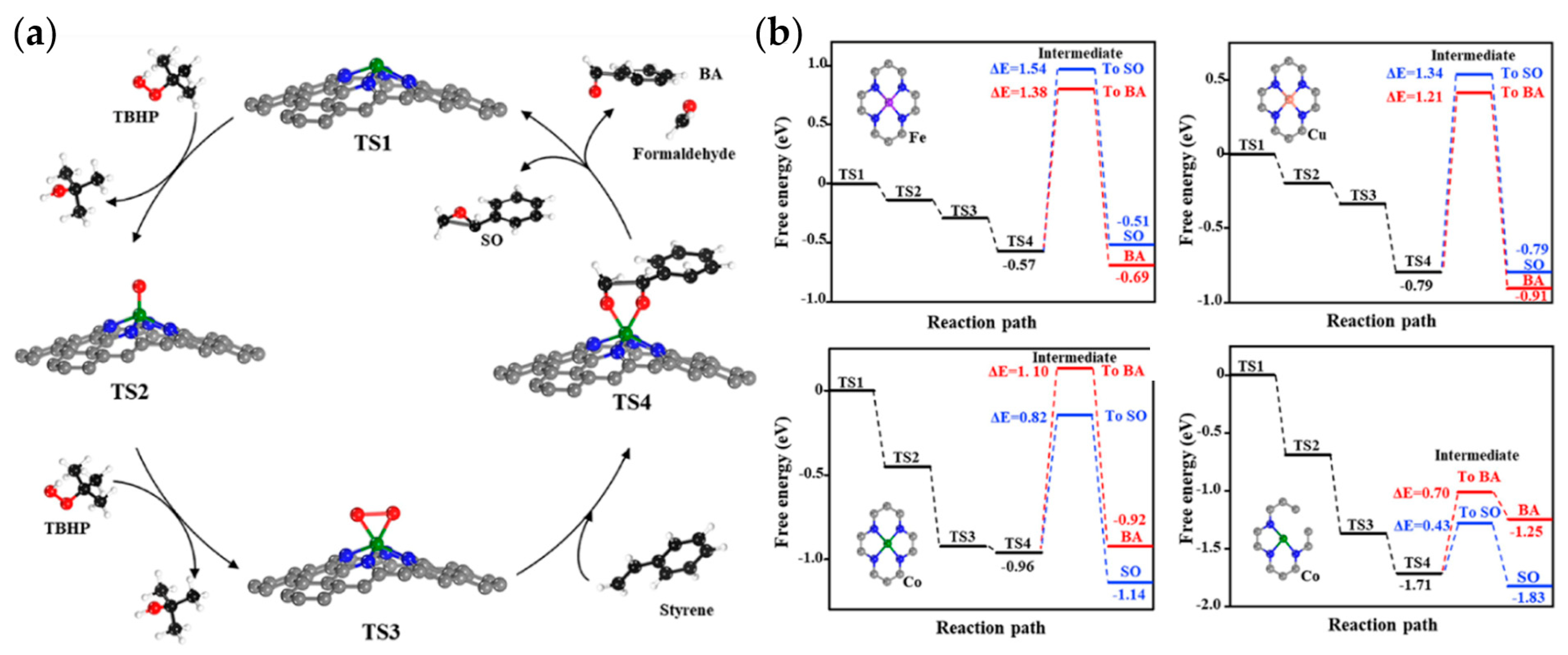{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Functionalization of Alkenes
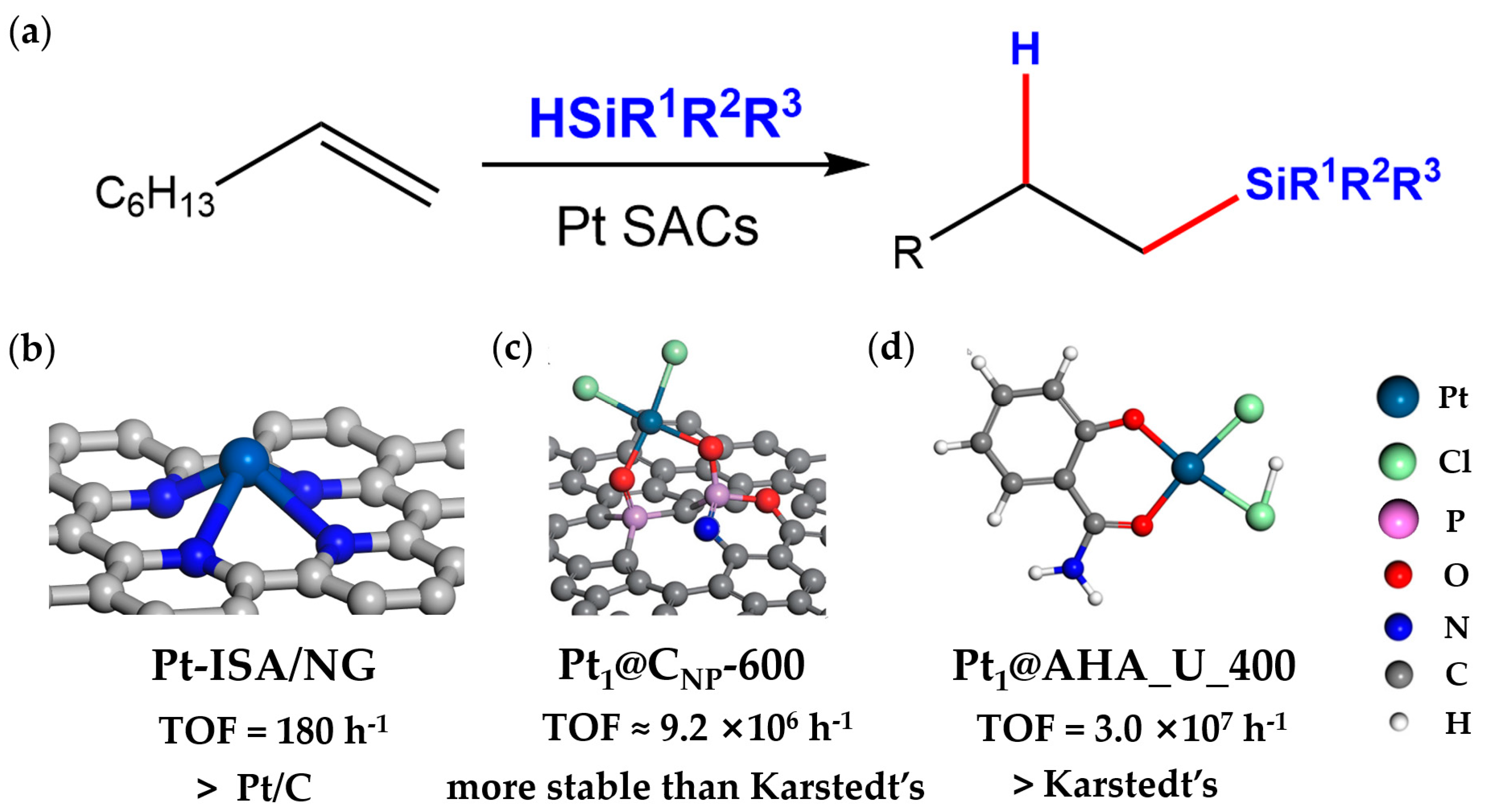
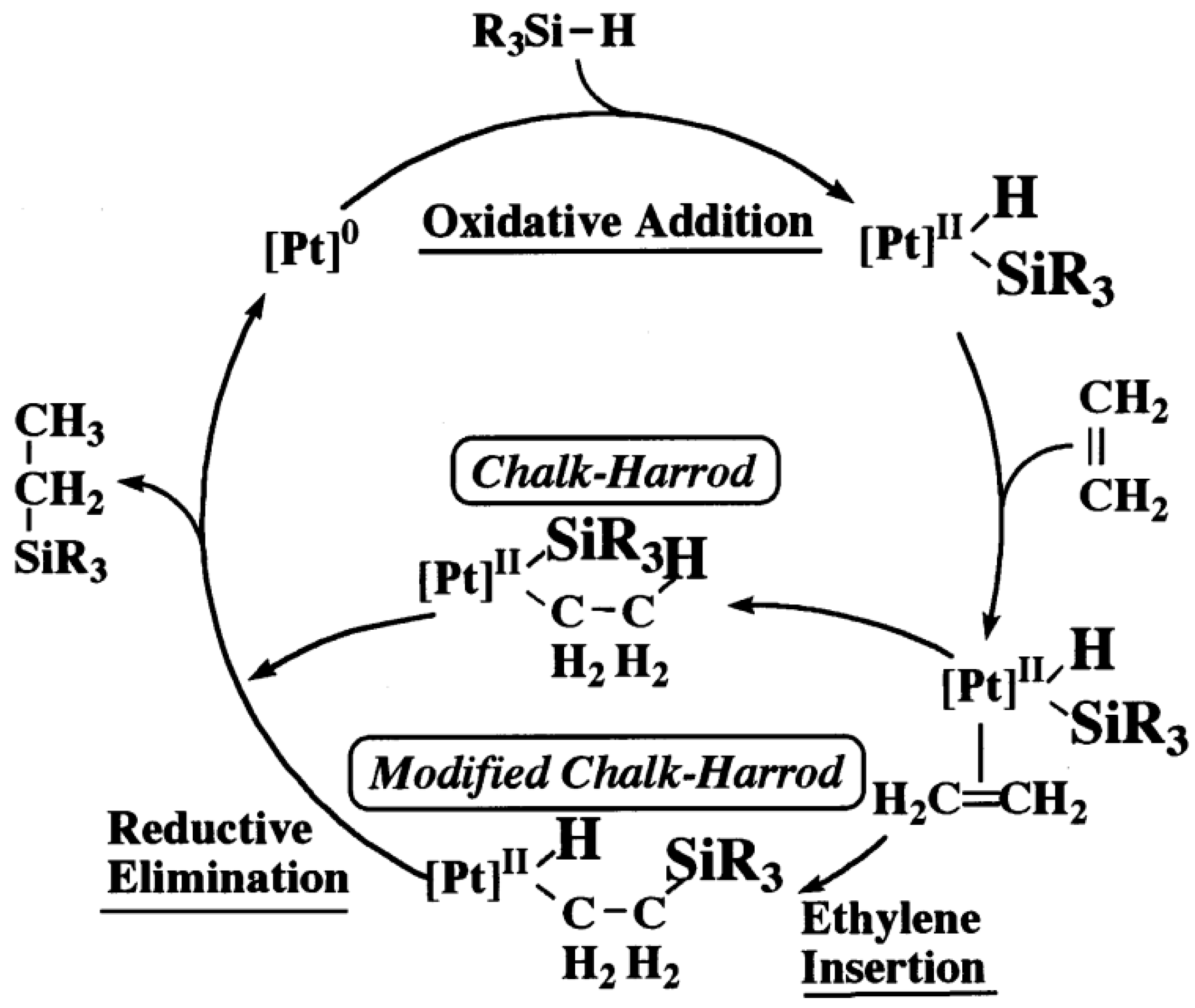
2.1. Hydrosilylation
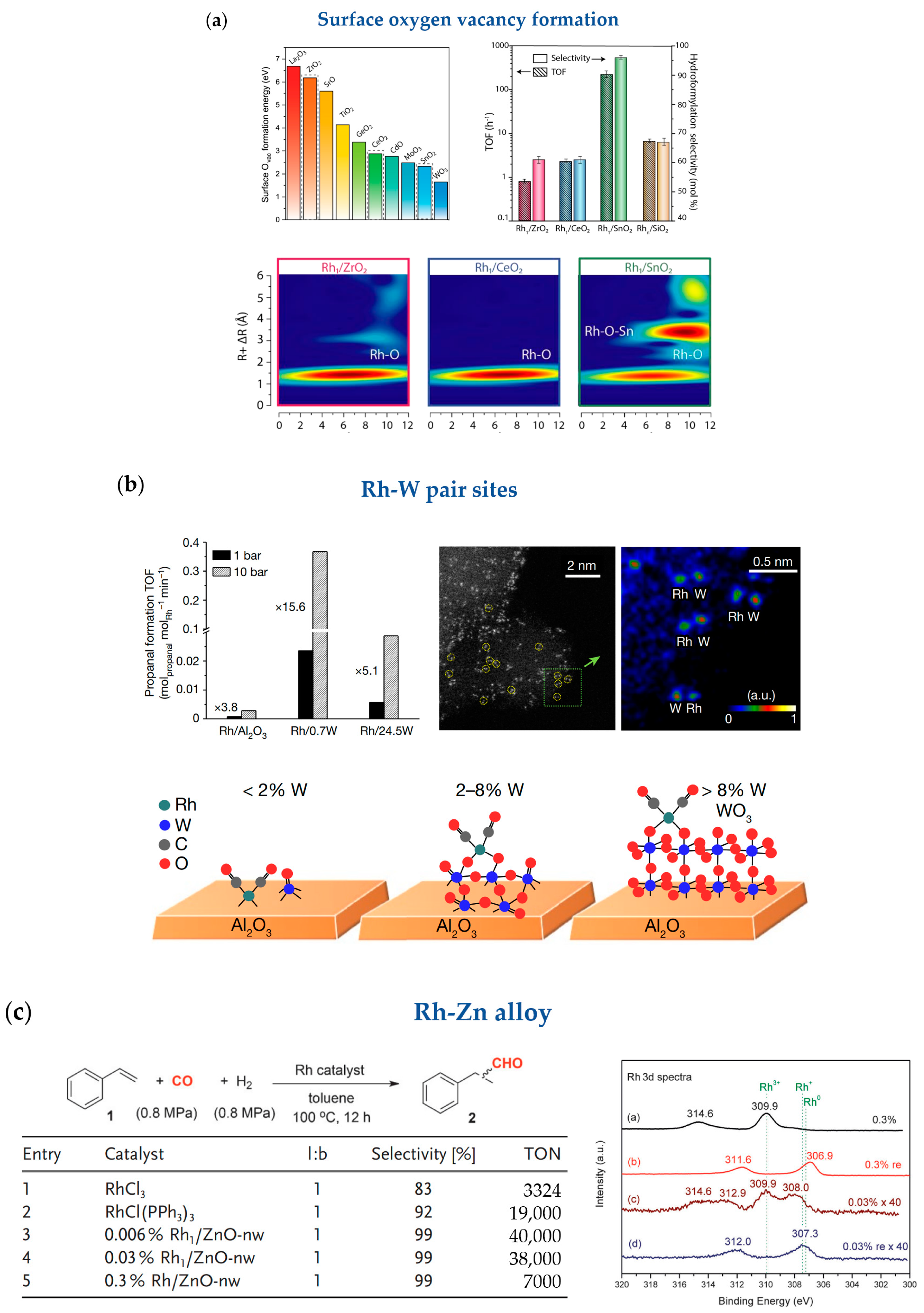
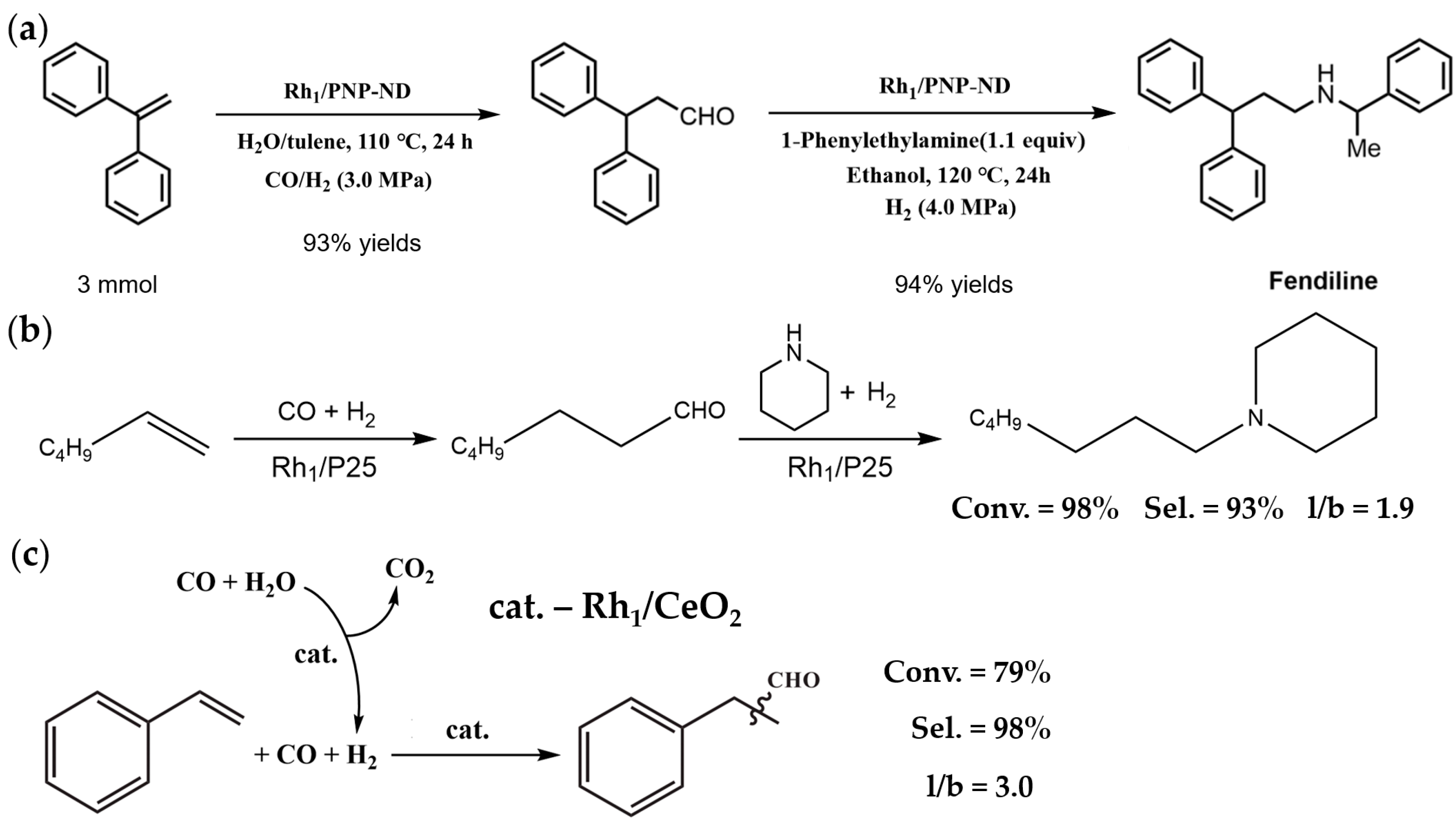
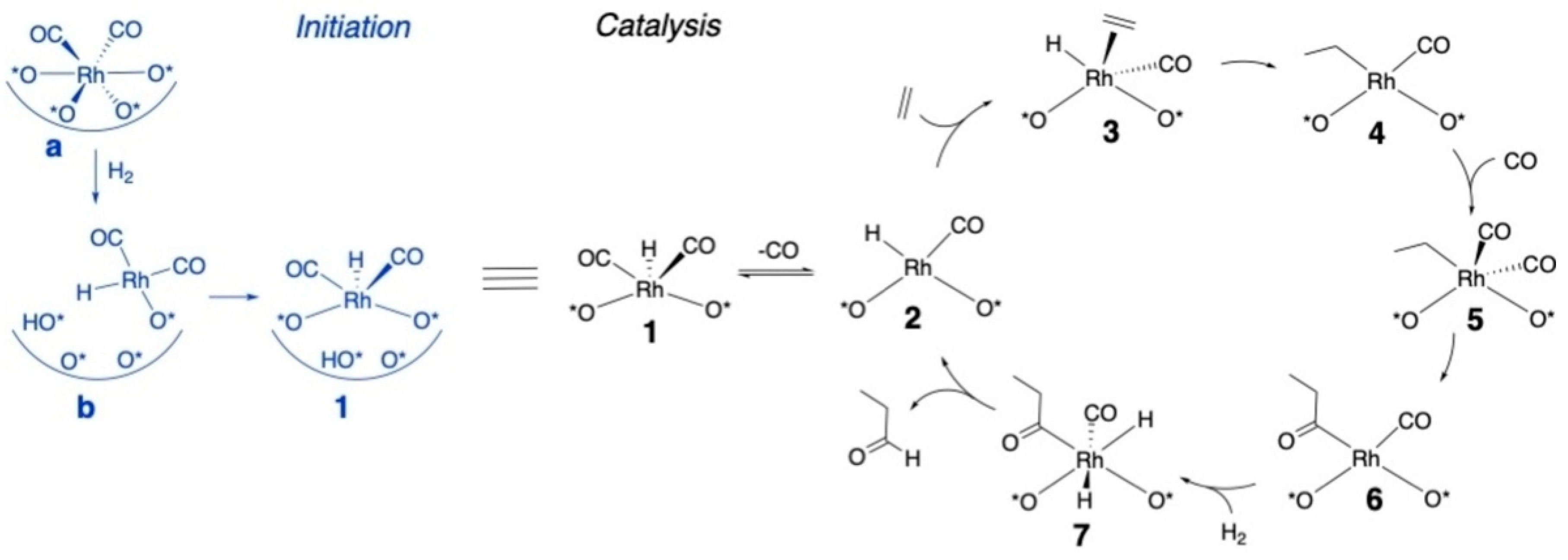
2.2. Hydroformylation
2.3. Constructing C-Y (Y = B, P, S, N) Bonds
2.4. Hydrogenation
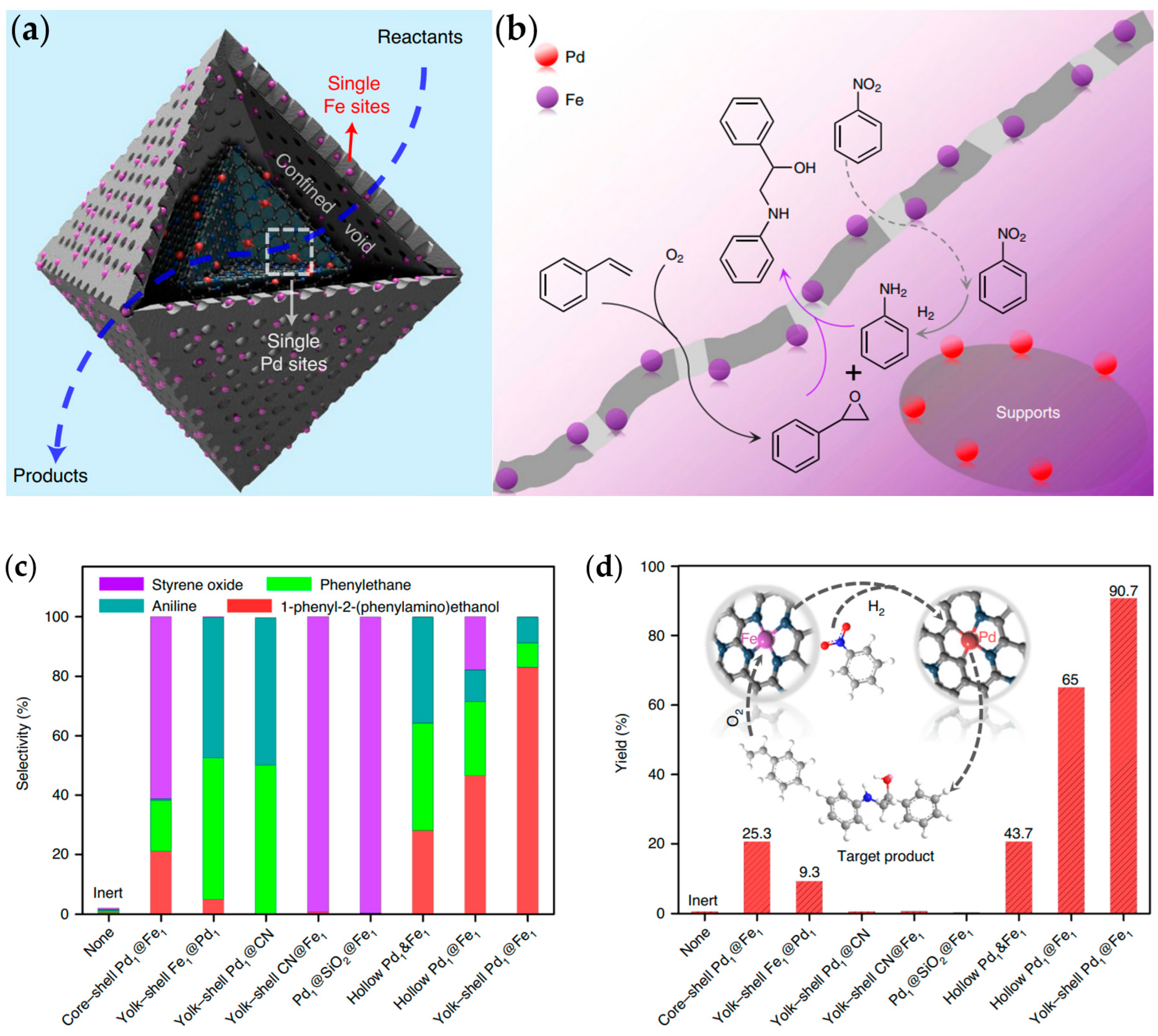
2.5. Epoxidation
3. Tandem Reactions
4. Conclusions and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Komiyama, T.; Minami, Y.; Hiyama, T. Recent Advances in Transition-Metal-Catalyzed Synthetic Transformations of Organosilicon Reagents. ACS Catal. 2016, 7, 631–651. [Google Scholar] [CrossRef]
- Franke, R.; Selent, D.; Borner, A. Applied hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Troegel, D.; Stohrer, J. Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view. Coord. Chem. Rev. 2011, 255, 1440–1459. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, J.; Koh, M.J.; Loh, K.P. Single-Atom Catalysis: From Simple Reactions to the Synthesis of Complex Molecules. Adv. Mater. 2022, 34, e2103882. [Google Scholar] [CrossRef]
- Sakaki, S.; Mizoe, N.; Sugimoto, M.; Musashi, Y. Pt-catalyzed hydrosilylation of ethylene. A theoretical study of the reaction mechanism. Coord. Chem. Rev. 1999, 190–192, 933–960. [Google Scholar] [CrossRef]
- Cui, X.; Junge, K.; Dai, X.; Kreyenschulte, C.; Pohl, M.M.; Wohlrab, S.; Shi, F.; Bruckner, A.; Beller, M. Synthesis of Single Atom Based Heterogeneous Platinum Catalysts: High Selectivity and Activity for Hydrosilylation Reactions. ACS Cent. Sci. 2017, 3, 580–585. [Google Scholar] [CrossRef]
- Zhu, Y.; Cao, T.; Cao, C.; Luo, J.; Chen, W.; Zheng, L.; Dong, J.; Zhang, J.; Han, Y.; Li, Z.; et al. One-Pot Pyrolysis to N-Doped Graphene with High-Density Pt Single Atomic Sites as Heterogeneous Catalyst for Alkene Hydrosilylation. ACS Catal. 2018, 8, 10004–10011. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Liu, K.; Hou, J.; Badamdorj, B.; Tarakina, N.V.; Wang, M.; Wang, Q.; Wang, X.; Antonietti, M. In-situ Synthesis of -P=N-doped Carbon Nanofibers for Single Atom Catalytic Hydrosilylation. Adv. Mater. 2023, e2209310. [Google Scholar] [CrossRef]
- Liu, K.; Badamdorj, B.; Yang, F.; Janik, M.J.; Antonietti, M. Accelerated Anti-Markovnikov Alkene Hydrosilylation with Humic-Acid-Supported Electron-Deficient Platinum Single Atoms. Angew. Chem., Int. Ed. 2021, 60, 24220–24226. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xiong, Y.; Liu, C.; Zhang, H.; Zhao, M.; Chen, W.; Chen, W.; Huang, W. Electron-rich isolated Pt active sites in ultrafine PtFe3 intermetallic catalyst for efficient alkene hydrosilylation. J. Catal. 2021, 396, 351–359. [Google Scholar] [CrossRef]
- Pan, G.; Hu, C.; Hong, S.; Li, H.; Yu, D.; Cui, C.; Li, Q.; Liang, N.; Jiang, Y.; Zheng, L.; et al. Biomimetic caged platinum catalyst for hydrosilylation reaction with high site selectivity. Nat. Commun. 2021, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, S.; Li, J.; Chen, X.; Ji, Y.; Yu, H.; Bai, D.; Xu, G.; Zhong, Z.; Su, F. Partially charged single-atom Ru supported on ZrO2 nanocrystals for highly efficient ethylene hydrosilylation with triethoxysilane. Nano Res. 2022, 15, 5857–5864. [Google Scholar] [CrossRef]
- Lee, S.; Patra, A.; Christopher, P.; Vlachos, D.G.; Caratzoulas, S. Theoretical Study of Ethylene Hydroformylation on Atomically Dispersed Rh/Al2O3 Catalysts: Reaction Mechanism and Influence of the ReOx Promoter. ACS Catal. 2021, 11, 9506–9518. [Google Scholar] [CrossRef]
- Amsler, J.; Sarma, B.B.; Agostini, G.; Prieto, G.; Plessow, P.N.; Studt, F. Prospects of Heterogeneous Hydroformylation with Supported Single Atom Catalysts. J. Am. Chem. Soc. 2020, 142, 5087–5096. [Google Scholar] [CrossRef]
- Farpon, M.G.; Henao, W.; Plessow, P.N.; Andres, E.; Arenal, R.; Marini, C.; Agostini, G.; Studt, F.; Prieto, G. Rhodium Single-Atom Catalyst Design through Oxide Support Modulation for Selective Gas-Phase Ethylene Hydroformylation. Angew. Chem. Int. Ed. 2023, 62, e202214048. [Google Scholar] [CrossRef]
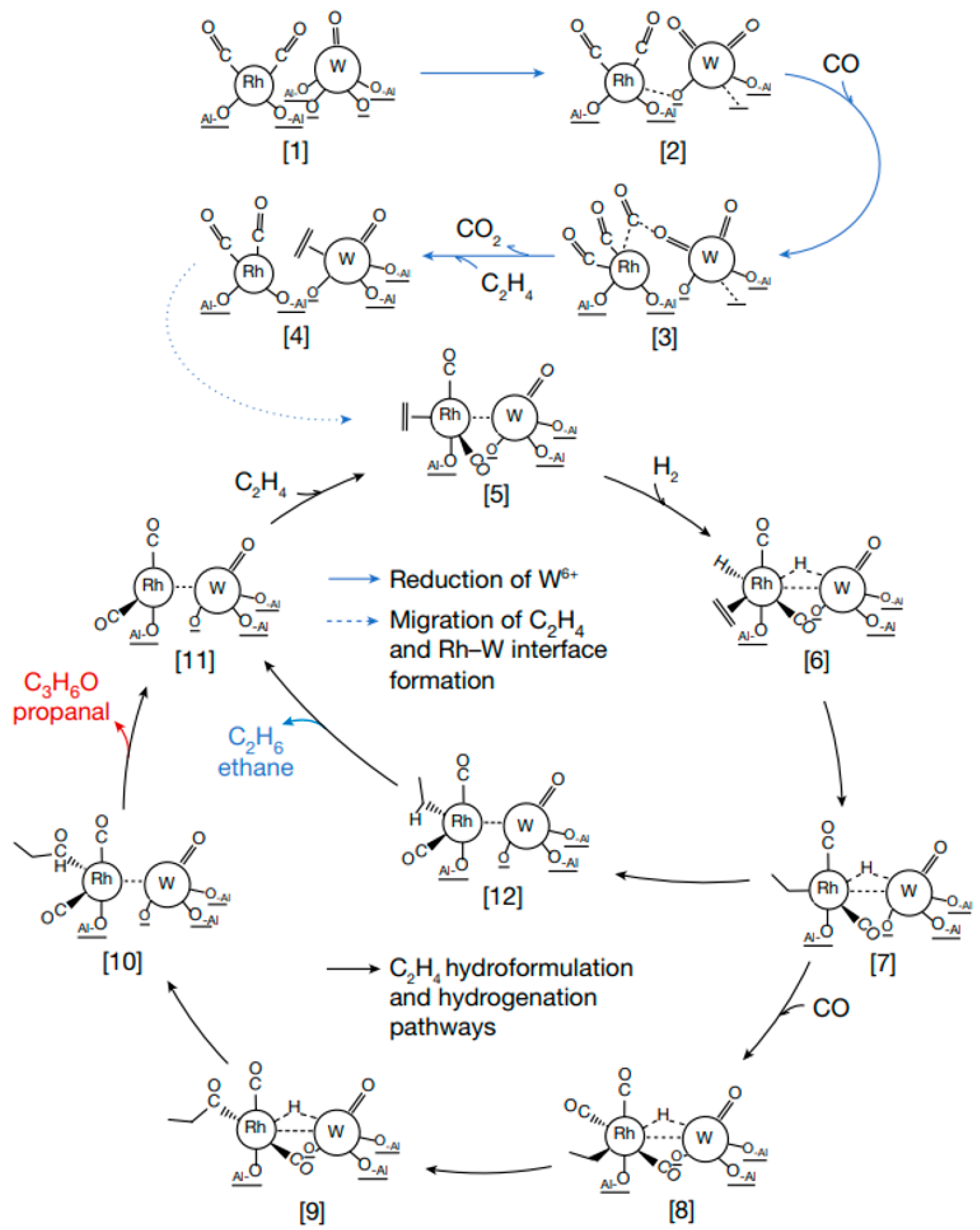
- Ro, I.; Qi, J.; Lee, S.; Xu, M.; Yan, X.; Xie, Z.; Zakem, G.; Morales, A.; Chen, J.G.; Pan, X.; et al. Bifunctional hydroformylation on heterogeneous Rh-WOx pair site catalysts. Nature 2022, 609, 287–292. [Google Scholar] [CrossRef]
- Lang, R.; Li, T.; Matsumura, D.; Miao, S.; Ren, Y.; Cui, Y.T.; Tan, Y.; Qiao, B.; Li, L.; Wang, A.; et al. Hydroformylation of Olefins by a Rhodium Single-Atom Catalyst with Activity Comparable to RhCl(PPh3)3. Angew. Chem. Int. Ed. 2016, 55, 16054–16058. [Google Scholar] [CrossRef]
- Shang, W.; Qin, B.; Gao, M.; Qin, X.; Chai, Y.; Wu, G.; Guan, N.; Ma, D.; Li, L. Efficient Heterogeneous Hydroformylation over Zeolite-Encaged Isolated Rhodium Ions. CCS Chem. 2022, in press. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, H.; Wang, X.; Li, T.; Dai, X.; Zhang, L.; Cui, X.; Shi, F. Confinement of atomically dispersed Rh catalysts within porous monophosphine polymers for regioselective hydroformylation of alkenes. J. Catal. 2021, 401, 321–330. [Google Scholar] [CrossRef]
- Escobar-Bedia, F.J.; Lopez-Haro, M.; Calvino, J.J.; Martin-Diaconescu, V.; Simonelli, L.; Perez-Dieste, V.; Sabater, M.J.; Concepción, P.; Corma, A. Active and Regioselective Ru Single-Site Heterogeneous Catalysts for Alpha-Olefin Hydroformylation. ACS Catal. 2022, 12, 4182–4193. [Google Scholar] [CrossRef]
- Gong, H.; Zhao, X.; Qin, Y.; Xu, W.; Wei, X.; Peng, Q.; Ma, Y.; Dai, S.; An, P.; Hou, Z. Hydroformylation of olefins catalyzed by single-atom Co(II) sites in zirconium phosphate. J. Catal. 2022, 408, 245–260. [Google Scholar] [CrossRef]
- Wei, B.; Chen, J.; Liu, X.; Hua, K.; Li, L.; Zhang, S.; Luo, H.; Wang, H.; Sun, Y. Achieving rhodium-like activity for olefin hydroformylation by electronic metal-support interaction of single atomic cobalt catalyst. Cell Rep. Phys. Sci. 2022, 3, 101016. [Google Scholar] [CrossRef]
- Xu, Q.; Guo, C.; Tian, S.; Zhang, J.; Chen, W.; Cheong, W.-C.; Gu, L.; Zheng, L.; Xiao, J.; Liu, Q.; et al. Coordination structure dominated performance of single-atomic Pt catalyst for anti-Markovnikov hydroboration of alkenes. Sci. China Mater. 2020, 63, 972–981. [Google Scholar] [CrossRef]
- Xu, Q.; Guo, C.; Li, B.; Zhang, Z.; Qiu, Y.; Tian, S.; Zheng, L.; Gu, L.; Yan, W.; Wang, D.; et al. Al3+ Dopants Induced Mg2+ Vacancies Stabilizing Single-Atom Cu Catalyst for Efficient Free-Radical Hydrophosphinylation of Alkenes. J. Am. Chem. Soc. 2022, 144, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Yang, X.; Sun, Z.; Yang, J.; Han, P.; Liu, Q.; Dong, H.; Gu, M.; Huang, L.; Wang, H. Biomimetic photocatalytic sulfonation of alkenes to access β-ketosulfones with single-atom iron site. Green Chem. 2020, 22, 230–237. [Google Scholar] [CrossRef]
- Yang, J.; Sun, Z.; Yan, K.; Dong, H.; Dong, H.; Cui, J.; Gong, X.; Han, S.; Huang, L.; Wen, J. Single-atom-nickel photocatalytic site-selective sulfonation of enamides to access amidosulfones. Green Chem. 2021, 23, 2756–2762. [Google Scholar] [CrossRef]
- Xue, W.; Zhu, Z.; Chen, S.; You, B.; Tang, C. Atomically Dispersed Co-N/C Catalyst for Divergent Synthesis of Nitrogen-Containing Compounds from Alkenes. J. Am. Chem. Soc. 2023, 145, 4142–4149. [Google Scholar] [CrossRef]
- Zhou, X.; Sterbinsky, G.E.; Wasim, E.; Chen, L.; Tait, S.L. Tuning Ligand-Coordinated Single Metal Atoms on TiO2 and their Dynamic Response during Hydrogenation Catalysis. ChemSusChem 2021, 14, 3825–3837. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, M.; Wang, A.; Zhang, T. Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms. Chem. Rev. 2020, 120, 683–733. [Google Scholar] [CrossRef]
- Lang, R.; Du, X.; Huang, Y.; Jiang, X.; Zhang, Q.; Guo, Y.; Liu, K.; Qiao, B.; Wang, A.; Zhang, T. Single-Atom Catalysts Based on the Metal–Oxide Interaction. Chem. Rev. 2020, 120, 11986–12043. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [Green Version]
- Mondelli, C.; Gözaydın, G.; Yan, N.; Pérez-Ramírez, J. Biomass valorisation over metal-based solid catalysts from nanoparticles to single atoms. Chem. Soc. Rev. 2020, 49, 3764–3782. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Wohlrab, S. Review of CO2 Reduction on Supported Metals (Alloys) and Single-Atom Catalysts (SACs) for the Use of Green Hydrogen in Power-to-Gas Concepts. Catalysts 2022, 12, 16. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Mou, X.; Wang, S.; Jiang, X.; Chen, Z.; Jiang, Z.; Lin, R.; Ding, Y. Host-induced alteration of the neighbors of single platinum atoms enables selective and stable hydrogenation of butadiene. Nanoscale 2022, 14, 10506–10513. [Google Scholar] [CrossRef]
- Hu, F.; Leng, L.; Zhang, M.; Chen, W.; Yu, Y.; Wang, J.; Horton, J.H.; Li, Z. Direct Synthesis of Atomically Dispersed Palladium Atoms Supported on Graphitic Carbon Nitride for Efficient Selective Hydrogenation Reactions. ACS Appl. Mater. Interfaces 2020, 12, 54146–54154. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Cheng, H.; Yi, H.; Lin, Y.; Yao, T.; Wang, C.; Li, J.; Wei, S.; Lu, J. Single-Atom Pd1/Graphene Catalyst Achieved by Atomic Layer Deposition: Remarkable Performance in Selective Hydrogenation of 1,3-Butadiene. J. Am. Chem. Soc. 2015, 137, 10484–10487. [Google Scholar] [CrossRef]
- Zhao, E.; Li, M.; Xu, B.; Wang, X.L.; Jing, Y.; Ma, D.; Mitchell, S.; Pérez-Ramírez, J.; Chen, Z. Transfer Hydrogenation with a Carbon-Nitride-Supported Palladium Single-Atom Photocatalyst and Water as a Proton Source. Angew. Chem. Int. Ed. 2022, 61, e202207410. [Google Scholar] [CrossRef]
- Liu, W.; Morfin, F.; Provost, K.; Bahri, M.; Baaziz, W.; Ersen, O.; Piccolo, L.; Zlotea, C. Unveiling the Ir single atoms as selective active species for the partial hydrogenation of butadiene by operando XAS. Nanoscale 2022, 14, 7641–7649. [Google Scholar] [CrossRef] [PubMed]
- Lucci, F.R.; Liu, J.; Marcinkowski, M.D.; Yang, M.; Allard, L.F.; Flytzani-Stephanopoulos, M.; Sykes, E.C. Selective hydrogenation of 1,3-butadiene on platinum-copper alloys at the single-atom limit. Nat. Commun. 2015, 6, 8550. [Google Scholar] [CrossRef] [Green Version]
- Talib, S.H.; Yu, X.; Yu, Q.; Baskaran, S.; Li, J. Non-noble metal single-atom catalysts with phosphotungstic acid (PTA) support: A theoretical study of ethylene epoxidation. Sci. China Mater. 2020, 63, 1003–1014. [Google Scholar] [CrossRef]
- Cao, R.; Thapa, R.; Kim, H.; Xu, X.; Gyu Kim, M.; Li, Q.; Park, N.; Liu, M.; Cho, J. Promotion of oxygen reduction by a bio-inspired tethered iron phthalocyanine carbon nanotube-based catalyst. Nat. Commun. 2013, 4, 2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, H.; Ye, R.; Jian, P.; Wang, L. Promotional effect of Mn-doping on the catalytic performance of NiO sheets for the selective oxidation of styrene. J. Colloid Interface Sci. 2021, 585, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.-X.; Liu, L.; Han, Z.-B. Palladium nanoparticles supported on UiO-66-NH2 as heterogeneous catalyst for epoxidation of styrene. Inorg. Chem. Commun. 2019, 100, 51–55. [Google Scholar] [CrossRef]
- Jia, B.; Bai, L.; Han, Z.; Li, R.; Huangfu, J.; Li, C.; Zheng, J.; Qu, Y.; Leng, K.; Wang, Y.; et al. High-Performance Styrene Epoxidation with Vacancy-Defect Cobalt Single-Atom Catalysts. ACS Appl. Mater. Interfaces 2022, 14, 10337–10343. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zou, Y.; Zhang, M.; Gou, W.; Zhang, S.; Qu, Y. Multiscale porous single-atom Co catalysts for epoxidation with O2. J. Mater. Chem. A 2022, 10, 6016–6022. [Google Scholar] [CrossRef]
- Li, Z.; Li, H.; Yuan, D.; Leng, L.; Zhang, M.; Di, M.; Horton, J.H.; Wang, J.; Sun, L.; Sun, W. Photoinduction of palladium single atoms supported on defect-containing γ-AlOOH nanoleaf for efficient trans-stilbene epoxidation. Chem. Eng. J. 2022, 429, 132149. [Google Scholar] [CrossRef]
- Dong, X.; Wang, C.; Zhang, M.; Ji, S.; Leng, L.; Hugh Horton, J.; Dong, H.; Qiao, M.; Wang, Y.; Zhang, J.; et al. Atomically dispersed single ruthenium sites anchored on bismuth tungstate with synergistic geometric and electronic effects for epoxidation of trans-stilbene. Chem. Eng. J. 2023, 454, 139940. [Google Scholar] [CrossRef]
- Yang, H.; Wang, X.; Liu, Q.; Huang, A.; Zhang, X.; Yu, Y.; Zhuang, Z.; Li, G.; Li, Y.; Peng, Q.; et al. Heterogeneous Iridium Single-Atom Molecular-like Catalysis for Epoxidation of Ethylene. J. Am. Chem. Soc. 2023, 145, 6658–6670. [Google Scholar] [CrossRef]
- Chung, M.; Jin, K.; Zeng, J.S.; Ton, T.N.; Manthiram, K. Tuning Single-Atom Dopants on Manganese Oxide for Selective Electrocatalytic Cyclooctene Epoxidation. J. Am. Chem. Soc. 2022, 144, 17416–17422. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Liang, G.; Ru, T.; Liu, X.; Qi, H.; Wang, A.; Chen, F.E. Phosphorus coordinated Rh single-atom sites on nanodiamond as highly regioselective catalyst for hydroformylation of olefins. Nat. Commun. 2021, 12, 4698. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, Y.; Tao, F.F. C-N Coupling through Hydroaminoalkylation on a Single-Atom Rh Heterogeneous Catalyst. Angew. Chem. Int. Ed. 2023, 62, e202214332. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, F.; Lang, R.; Wang, H.; Su, Y.; Qiao, B.; Wang, A.; Zhang, T. Styrene Hydroformylation with In Situ Hydrogen: Regioselectivity Control by Coupling with the Low-Temperature Water-Gas Shift Reaction. Angew. Chem. Int. Ed. 2020, 59, 7430–7434. [Google Scholar] [CrossRef]
- Sarma, B.B.; Kim, J.; Amsler, J.; Agostini, G.; Weidenthaler, C.; Pfander, N.; Arenal, R.; Concepcion, P.; Plessow, P.; Studt, F.; et al. One-Pot Cooperation of Single-Atom Rh and Ru Solid Catalysts for a Selective Tandem Olefin Isomerization-Hydrosilylation Process. Angew. Chem. Int. Ed. 2020, 59, 5806–5815. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, H.; Zhu, X.; Qu, Y.; Xiong, C.; Xue, Z.; Zhang, Q.; Liu, X.; Zhou, F.; Mou, X.; et al. Simultaneous oxidative and reductive reactions in one system by atomic design. Nat. Catal. 2021, 4, 134–143. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, Y.; Ji, Y.; Lang, R.; Fang, Y.; Wu, C.D. The Opportunities and Challenges in Single-Atom Catalysis. ChemCatChem 2023, 15, e202201176. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Ji, Y.; Jiang, Y.; Lang, R.; Fang, Y.; Qiao, B. Recent Development of Single-Atom Catalysis for the Functionalization of Alkenes. Catalysts 2023, 13, 730. https://doi.org/10.3390/catal13040730
Yu X, Ji Y, Jiang Y, Lang R, Fang Y, Qiao B. Recent Development of Single-Atom Catalysis for the Functionalization of Alkenes. Catalysts. 2023; 13(4):730. https://doi.org/10.3390/catal13040730
Chicago/Turabian StyleYu, Xuetong, Yuxia Ji, Yan Jiang, Rui Lang, Yanxiong Fang, and Botao Qiao. 2023. "Recent Development of Single-Atom Catalysis for the Functionalization of Alkenes" Catalysts 13, no. 4: 730. https://doi.org/10.3390/catal13040730