
Methane Oxidation over the Zeolites-Based Catalysts
Abstract
:1. Introduction
2. Methane Oxidation
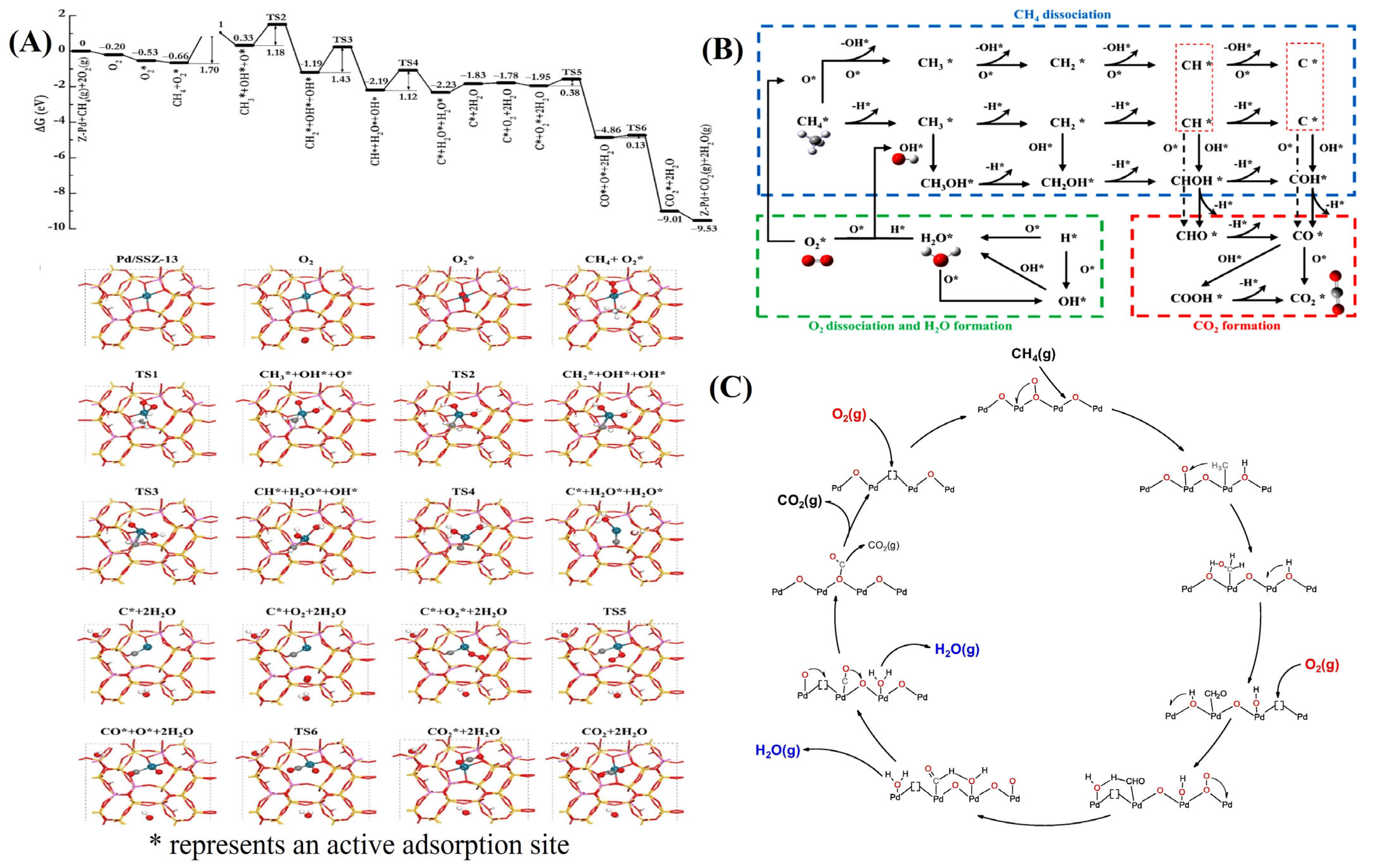
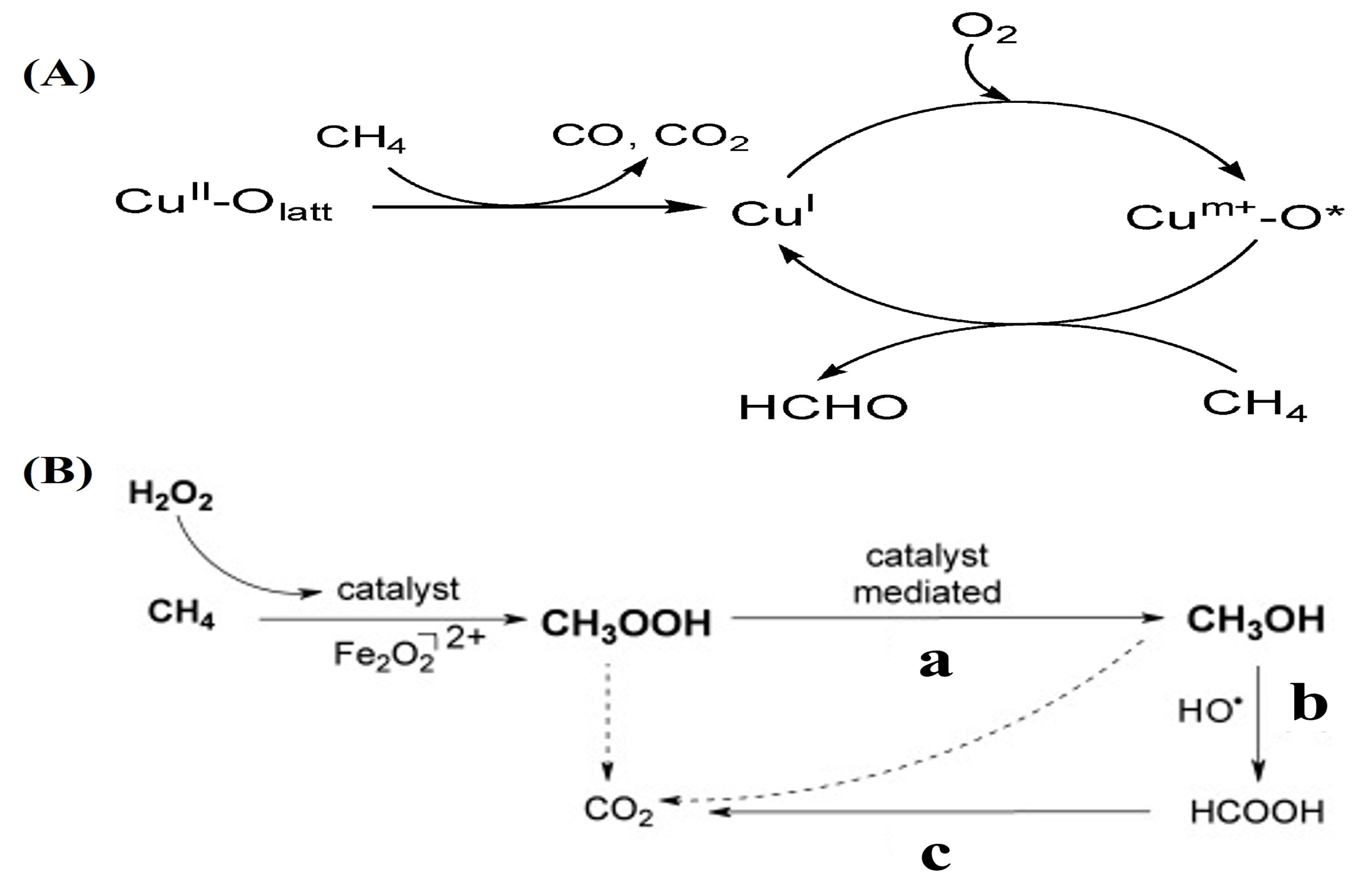
2.1. Complete Oxidation of Methane
2.2. Reaction Mechanism of Complete Oxidation of Methane
- (1)
- O2 → O*
- (2)
- CH4 → CH4*
- (3)
- O* + CH4* → CH3* + OH* + O*
- (4)
- CH2* + OH* + OH* → CH* + H2O* + OH*
- (5)
- C* + H2O* + OH* → C* + 2H2O
- (6)
- C* + O2 + 2H2O → C* + O2* + 2H2O
- (7)
- CO* + O* + 2H2O → CO2* + 2H2O

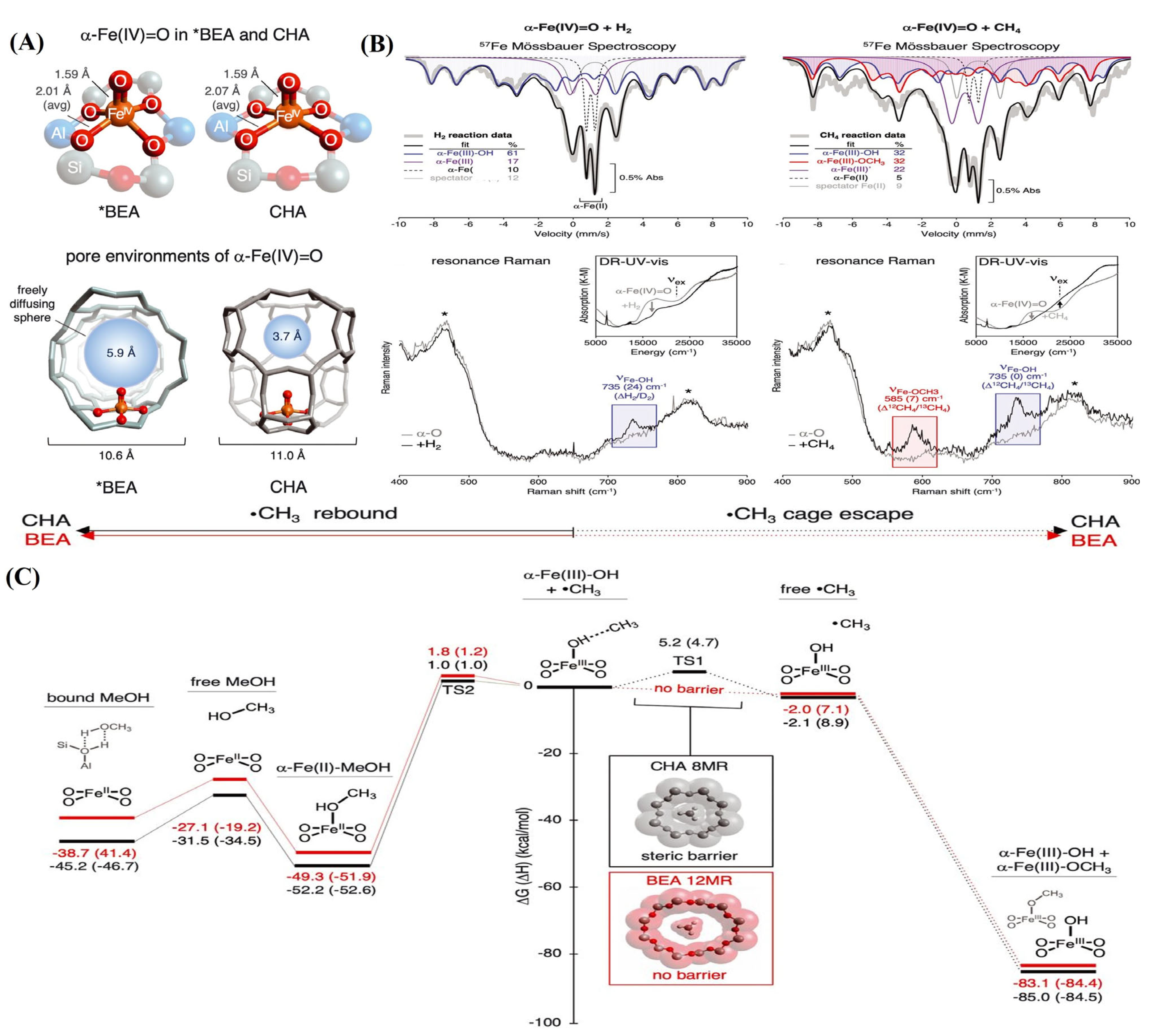
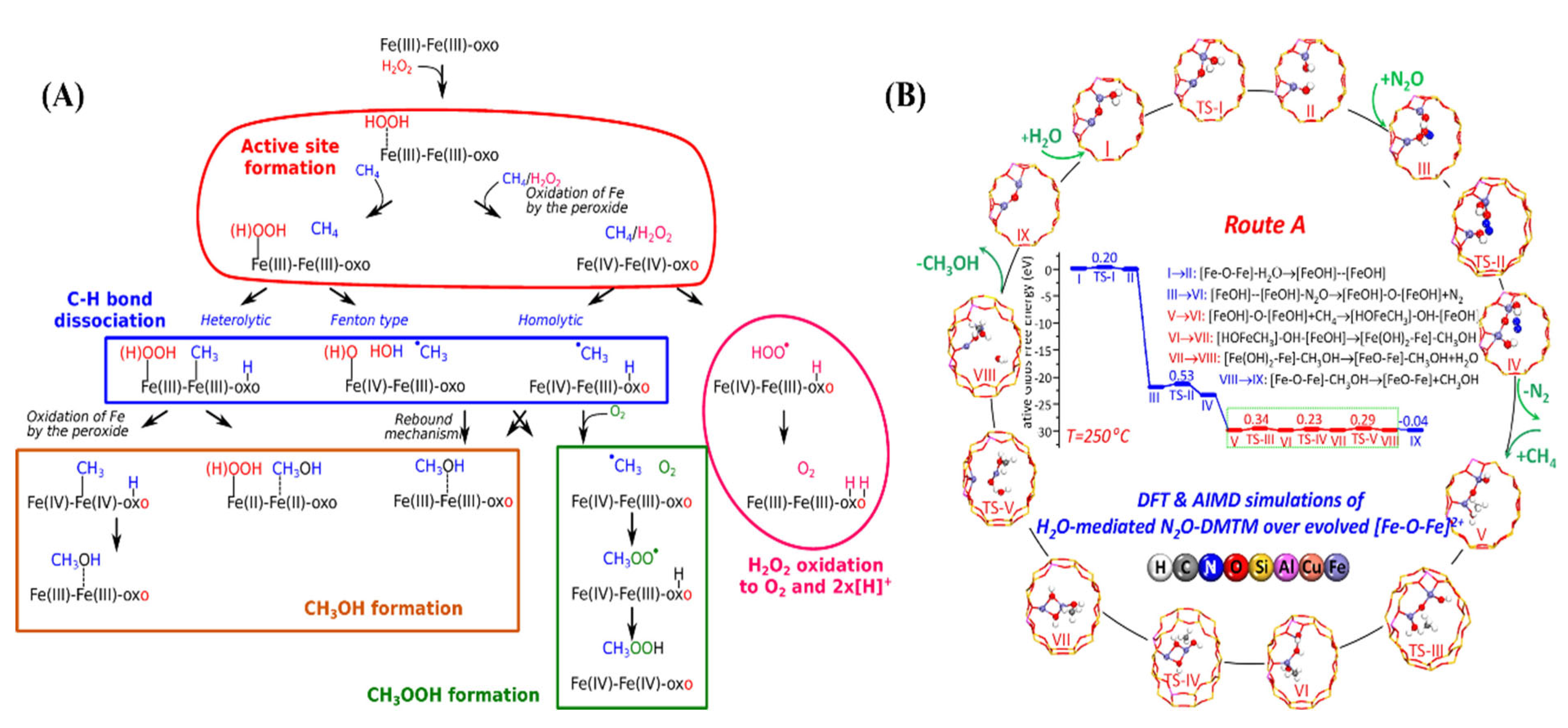
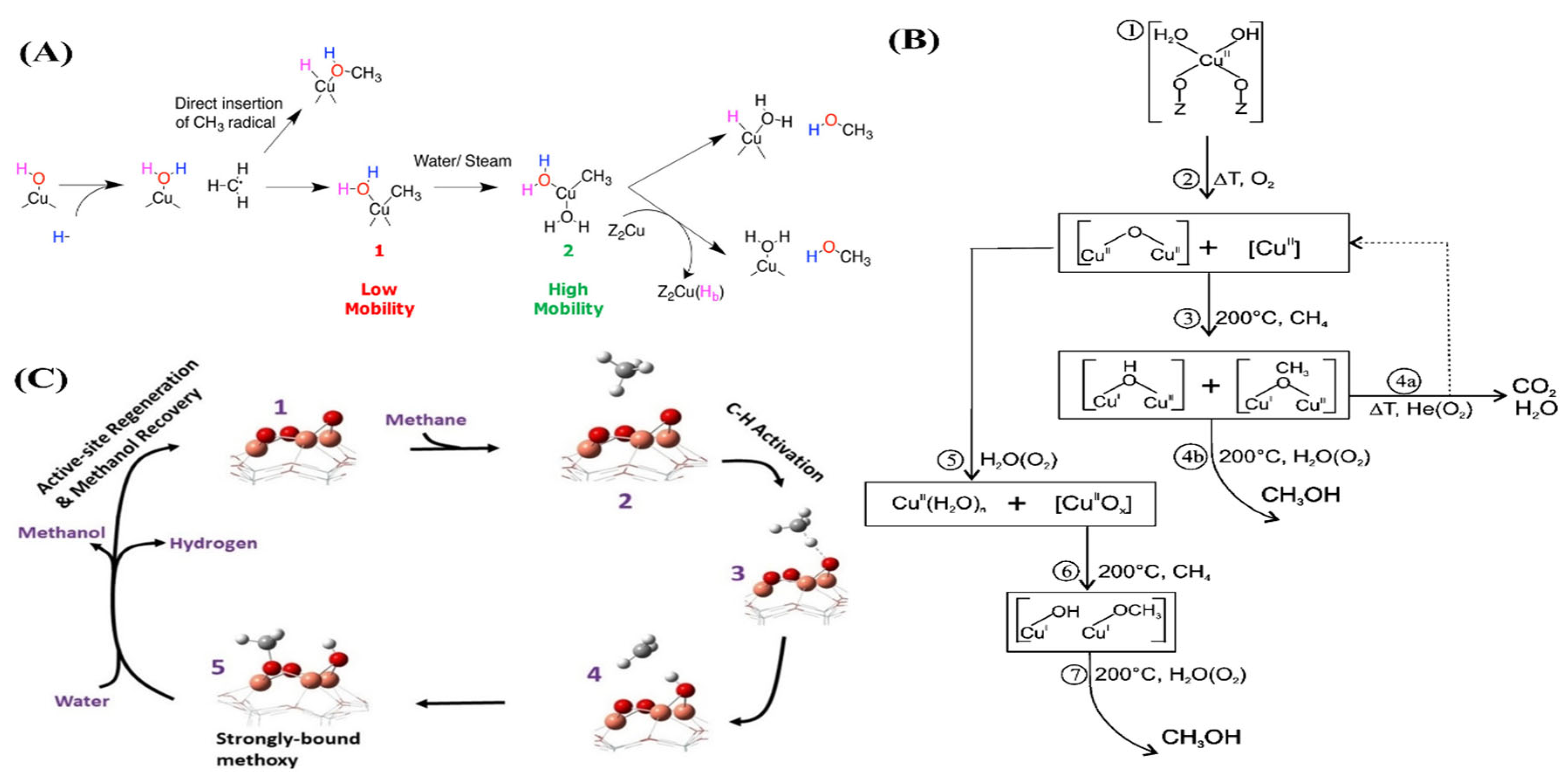
2.3. Selective Oxidation of Methane
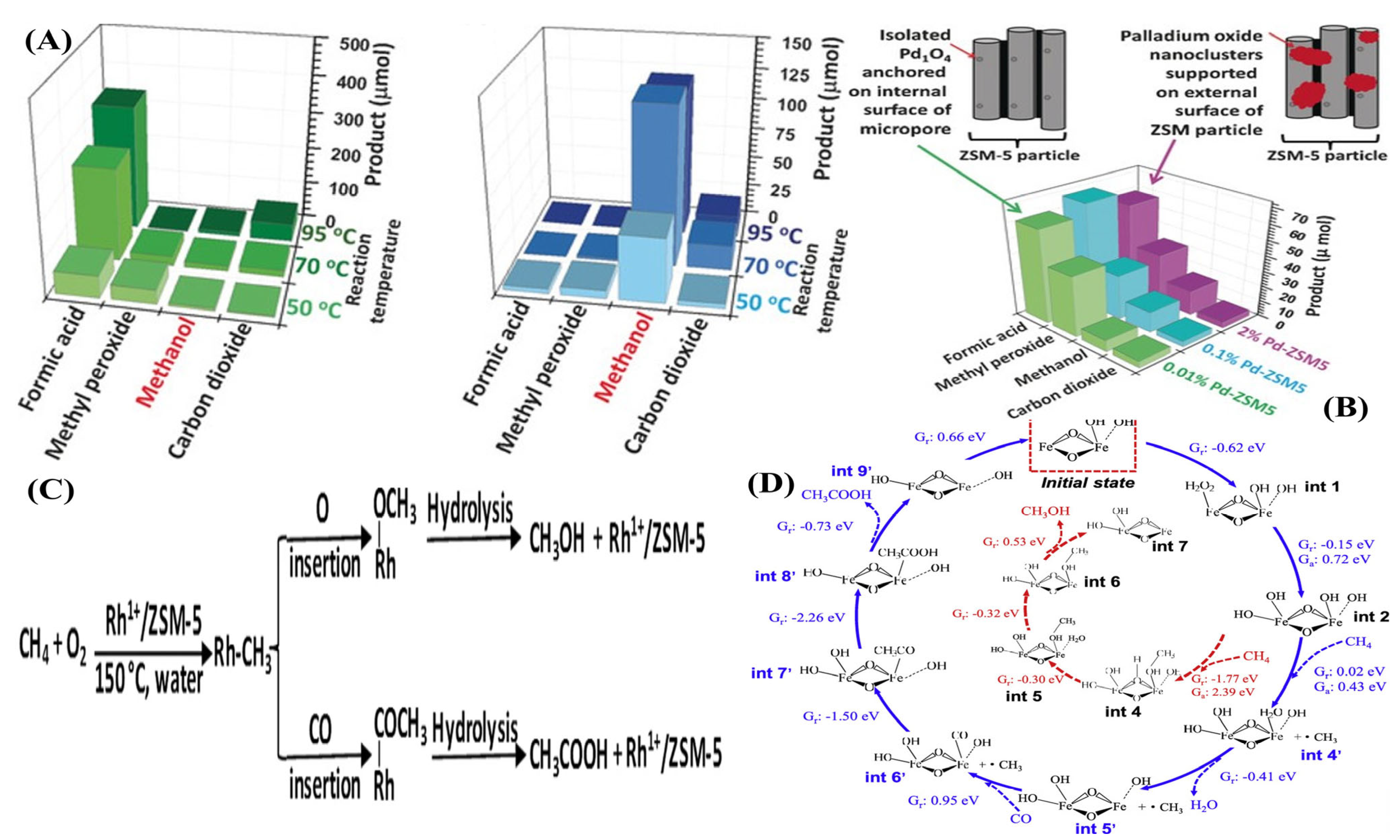
2.3.1. Selective Oxidation of Methane to Methanol
2.3.2. Selective Oxidation of Methane to Formaldehyde
2.3.3. Selective Oxidation of Methane to Formic Acid
2.3.4. Selective Oxidation of Methane to Acetic Acid
3. Conclusive Remarks and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Peng, H.; Rao, C.; Zhang, N.; Wang, X.; Liu, W.; Mao, W.; Han, L.; Zhang, P.; Dai, S. Confined ultrathin Pd-Ce nanowires with outstanding moisture and SO2 tolerance in methane combustion. Angew. Chem. Int. Ed. 2018, 57, 8953. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Grappe, H.A.; Chakraborty, A.; Orkoulas, G. Postextraction separation, on-board storage, and catalytic conversion of methane in natural gas: A review. Chem. Rev. 2016, 116, 11436–11499. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.L.; Shen, M.K.; Fan, G.Z.; Yang, A.; Meyer, J.R.; Ou, Y.N.; Yin, B.; Fortner, J.; Foston, M.; Li, Z.S.; et al. Facet-dependent enhancement in the activity of bismuth vanadate microcrystals for the photocatalytic conversion of methane to methanol. ACS Appl. Nano Mater. 2018, 1, 6683–6691. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Choudhary, V.R. Energy-efficient syngas production through catalytic oxy-methane reforming reactions. Angew. Chem. Int. Ed. 2008, 47, 1828–1847. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.L.; Kumar, V.P.; Zhu, C.J.; Wang, H.Y.; Smith, K.J.; Wolf, M.O.; Maclachlan, M.J. Bowtie-shaped NiCo2O4 catalysts for low-temperature methane combustion. Adv. Funct. Mater. 2019, 29, 1807519. [Google Scholar] [CrossRef]
- Thauer, R.K. Functionalization of methane in anaerobic microorganisms. Angew. Chem. Int. Ed. 2010, 49, 6712–6713. [Google Scholar] [CrossRef]
- Olivos-Suarez, A.I.; Szecsenyi, A.; Hensen, E.J.M.; Ruiz-Martinez, J.; Pidko, E.A.; Gascon, J. Strategies for the direct catalytic valorization of methane using heterogeneous catalysis: Challenges and opportunities. ACS Catal. 2016, 6, 2965–2981. [Google Scholar] [CrossRef]
- Han, J.Y.; Zemlyanov, D.Y.; Ribeiro, F.H. Catalytic combustion of methane on palladium single crystals. Catal. Today 2006, 117, 506–513. [Google Scholar] [CrossRef]
- McCarty, J.G. Durable catalysts for cleaner air. Nature 2000, 403, 35–36. [Google Scholar] [CrossRef]
- Ravi, M.; Sushkevich, V.L.; Knorpp, A.J.; Newton, M.A.; Palagin, D.; Pinar, A.B.; Ranocchiari, M.; Bokhoven, J.A.V. Misconceptions and challenges in methane-to-methanol over transition-metal-exchanged zeolites. Nat. Catal. 2019, 2, 485–494. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, S.; Tang, Y.; Li, Y.; Nguyen, L.; Li, Y.; Shan, J.; Xiao, D.; Gagne, R.; Frenkel, A.I.; et al. Low-temperature transformation of methane to methanol on Pd1O4 single sites anchored on the internal surface of microporous silicate. Angew. Chem. Int. Ed. 2016, 55, 13441. [Google Scholar] [CrossRef]
- Zuo, H.L.; Meynen, V.; Klemm, E. Selective oxidation of methane with hydrogen peroxide towards formic acid in a micro fixed-bed reactor. Chem. Ing. Tech. 2017, 89, 1759–1765. [Google Scholar] [CrossRef]
- Tang, Y.; Li, Y.T.; Fung, V.; Jiang, D.; Huang, W.X.; Zhang, S.; Lwasawa, Y.; Sakata, T.; Nguyen, L.; Zhang, X.Y.; et al. Single rhodium atoms anchored in micropores for efficient transformation of methane under mild conditions. Nat. Commun. 2018, 9, 1231. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.J.; Li, M.W.; Allard, L.F.; Lee, S.; Flytzani-Stephanopoulos, M. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 2017, 551, 605–608. [Google Scholar] [CrossRef]
- Shamzhy, M.; Opanasenko, M.; Concepcion, P.; Martinez, A. New trends in tailoring active sites in zeolite-based catalysts. Chem. Soc. Rev. 2019, 48, 1095–1149. [Google Scholar] [CrossRef]
- Sun, Q.M.; Wang, N.; Yu, J.H. Advances in catalytic applications of zeolite-supported metal catalysts. Adv. Mater. 2021, 33, 2104442. [Google Scholar] [CrossRef]
- Vogt, E.T.C.; Weckhuysen, B.M. Fluid catalytic cracking: Recent developments on the grand old lady of zeolite catalysis. Chem. Soc. Rev. 2015, 44, 7342–7370. [Google Scholar] [CrossRef] [Green Version]
- Sudarsanam, P.; Peeters, E.; Makshina, E.V.; Parvulescu, V.I.; Sels, B.F. Advances in porous and nanoscale catalysts for viable biomass conversion. Chem. Soc. Rev. 2019, 48, 2366–2421. [Google Scholar] [CrossRef]
- Dusselier, M.; Davis, M.E. Small-pore zeolites: Synthesis and catalysis. Chem. Rev. 2018, 118, 5265–5329. [Google Scholar] [CrossRef]
- Sun, Q.; Xie, Z.K.; Yu, J.H. The state-of-the-art synthetic strategies for SAPO-34 zeolite catalysts in methanol-to-olefin conversion. Natl. Sci. Rev. 2018, 5, 542. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.H.; Sun, M.H.; Wang, Z.; Yang, W.M.; Xie, Z.K.; Su, B.L. Hierarchically structured zeolites: From design to application. Chem. Rev. 2020, 120, 11194–11294. [Google Scholar] [CrossRef] [PubMed]
- Spivey, J.J.; Hutchings, G. Catalytic aromatization of methane. Chem. Soc. Rev. 2014, 43, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, I.; Yarulina, I.; Kapteijn, F.; Gascon, J. Progress in developing a structure-activity relationship for the direct aromatization of methane. ChemCatChem 2019, 11, 39. [Google Scholar] [CrossRef]
- Yang, L.; Huang, H.M. Transition-metal-catalyzed direct addition of unactivated C–H bonds to polar unsaturated bonds. Chem. Rev. 2015, 115, 3468–3517. [Google Scholar] [CrossRef]
- Balcells, D.; Clot, E.; Eisenstein, O. C–H bond activation in transition metal species from a computational perspective. Chem. Rev. 2010, 110, 749–823. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.L.; Bao, X.H. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: Challenges and prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous functionalization of methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef]
- Yang, S.Q.; Hu, T.L. Reverse-selective metal-organic framework materials for the efficient separation and purification of light hydrocarbons. Coord. Chem. Rev. 2022, 468, 214628. [Google Scholar] [CrossRef]
- Kumar, P.; Al-Attas, T.A.; Hu, J.G.; Kibria, M.G. Single atom catalysts for selective methane oxidation to oxygenates. ACS Nano 2022, 16, 8557–8618. [Google Scholar] [CrossRef]
- Campo, P.D.; Martinez, C.; Corma, A. Activation and conversion of alkanes in the confined space of zeolite-type materials. Chem. Soc. Rev. 2021, 50, 8511–8595. [Google Scholar] [CrossRef]
- Petrov, A.W.; Ferri, D.; Krumeich, F.; Nachtegaal, M.; Bokhoven, J.A.V.; Krocher, O. Stable complete methane oxidation over palladium based zeolite catalysts. Nat. Commun. 2018, 9, 2545. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Yu, K.L.; Zhang, Y.P.; Zhu, X.L.; He, F.; Eliasson, B. Characterization of plasma treated Pd/HZSM-5 catalyst for methane combustion. Appl. Catal. B 2004, 47, 95–100. [Google Scholar] [CrossRef]
- Shi, C.K.; Yang, L.F.; Cai, J.X. Cerium promoted Pd/HZSM-5 catalyst for methane combustion. Fuel 2007, 86, 106–112. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Sun, L.W.; Hu, X.F.; Zhang, Y.B.; Tian, H.Y.; Yang, X.G. Anti-sintering Pd@silicalite-1 for methane combustion: Effects of the moisture and SO2. Appl. Surf. Sci. 2019, 494, 1044–1054. [Google Scholar] [CrossRef]
- Nguyen, T.S.; Mckeever, P.; Arredondo-Arechavala, M.; Wang, Y.C.; Slater, T.J.A.; Haigh, S.J.; Beale, A.M.; Thompson, J.M. Correlation of the ratio of metallic to oxide species with activity of PdPt catalysts for methane oxidation. Catal. Sci. Technol. 2020, 10, 1408–1421. [Google Scholar] [CrossRef]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective oxidation of methane by the bis(ì-oxo)dicopper core stabilized on ZSM-5 and mordenite zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef]
- Devos, J.; Bols, M.L.; Plessres, D.; Goethem, C.V.; Seo, J.W.; Hwang, S.J.; Sels, B.F.; Dusselier, M. Synthesis–structure–activity relations in Fe-CHA for C–H activation: Control of Al distribution by interzeolite conversion. Chem. Mater. 2020, 32, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.A.; Raynes, S.; Apperley, D.C.; Taylor, R.A. Framework effects on activation and functionalisation of methane in zinc-exchanged zeolites. ChemPhysChem 2020, 21, 673. [Google Scholar] [CrossRef]
- Beznis, N.V.; Laak, A.N.C.V.; Weckhuysen, B.M.; Bitter, J.H. Oxidation of methane to methanol and formaldehyde over Co-ZSM-5 molecular sieves: Tuning the reactivity and selectivity by alkaline and acid treatments of the zeolite ZSM-5 agglomerates. Microporous Mesoporous Mater. 2011, 138, 176–183. [Google Scholar] [CrossRef]
- Lim, J.B.; Jo, D.H.; Hong, S.B. Palladium-exchanged small-pore zeolites with different cage systems as methane combustion catalysts. Appl. Catal. B 2017, 219, 155–162. [Google Scholar] [CrossRef]
- Chen, H.Y.; Lu, J.; Fedeyko, J.M.; Raj, A. Zeolite supported Pd catalysts for the complete oxidation of methane: A critical review. Appl. Catal. A 2022, 633, 118534. [Google Scholar] [CrossRef]
- Zhang, Y.; Glarborg, P.; Andersson, M.P.; Johansen, K.; Torp, T.K.; Jensen, A.D.; Christensen, J.M. Influence of the support on rhodium speciation and catalytic activity of rhodium-based catalysts for total oxidation of methane. Catal. Sci. Technol. 2020, 10, 6035–6044. [Google Scholar] [CrossRef]
- Petrov, A.W.; Ferri, D.; Krocher, O.; Bokhoven, J.A.V. Design of stable palladium-based zeolite catalysts for complete methane oxidation by postsynthesis zeolite modification. ACS Catal. 2019, 9, 2303–2312. [Google Scholar] [CrossRef]
- Zhang, L.L.; Chen, J.F.; Yang, H.L.; Wang, X.H.; Rui, Z.B. In situ mercaptosilane-assisted confinement of Pd nanoparticles in Beta for high-efficient methane oxidation. Catal. Today 2022, 400, 124–131. [Google Scholar] [CrossRef]
- Feng, X.Q.; Wang, T.; Mu, L.; Chen, Z.L.; Liang, J.H.; Xiao, C. Excellent stability for catalytic oxidation of methane over core–shell Pd@silicalite-1 with complete zeolite shell in wet conditions. Catal. Today 2022, 400, 66–72. [Google Scholar] [CrossRef]
- Tang, X.; Lou, Y.; Zhao, R.L.; Tang, B.J.; Guo, W.Y.; Guo, Y.L.; Zhan, W.C.; Jia, Y.Y.; Wang, L.; Dai, S.; et al. Confinement of subnanometric PdCo bimetallic oxide clusters in zeolites for methane complete oxidation. Chem. Eng. J. 2021, 418, 129398. [Google Scholar] [CrossRef]
- Doyle, A.M.; Postolache, R.; Shaw, D.; Rothon, R.; Tosheva, L. Methane oxidation over zeolite catalysts prepared from geothermal fluids. Microporous Mesoporous Mater. 2019, 285, 56–60. [Google Scholar] [CrossRef]
- Friberg, I.; Clark, A.H.; Ho, P.H.; Sadokhina, N.; Smales, G.J.; Woo, J.; Auvray, X.; Ferri, D.; Nachtegaal, M.; Krocher, O.; et al. Structure and performance of zeolite supported Pd for complete methane oxidation. Catal. Today 2021, 382, 3–12. [Google Scholar] [CrossRef]
- Yabushita, M.; Yoshida, M.; Muto, F.; Horie, M.; Kunitake, Y.; Nishitoba, T.; Maki, S.; Kanie, K.; Yokoi, T.; Muramatsu, A. Hydrothermal synthesis of Ga-substituted MFI zeolites via a mechanochemical process and their catalytic activity for methane transformation. Mol. Catal. 2019, 478, 110579. [Google Scholar] [CrossRef]
- Losch, P.; Huang, W.X.; Vozniuk, O.; Goodman, E.D.; Schmidt, W.; Cargnello, M. Modular Pd/zeolite composites demonstrating the key role of support hydrophobic/hydrophilic character in methane catalytic combustion. ACS Catal. 2019, 9, 4742–4753. [Google Scholar] [CrossRef]
- Li, T.; Beck, A.; Krumeich, F.; Artiglia, L.; Ghosalya, M.K.; Roger, M.; Ferri, D.; Krocher, O.; Sushkevich, V.; Safonova, O.V.; et al. Stable palladium oxide clusters encapsulated in silicalite-1 for complete methane oxidation. ACS Catal. 2021, 11, 7371–7382. [Google Scholar] [CrossRef]
- Gao, M.Y.; Gong, Z.M.; Weng, X.F.; Shang, W.X.; Chai, Y.C.; Dai, W.L.; Wu, G.J.; Guan, N.J.; Li, L.D. Methane combustion over palladium catalyst within the confined space of MFI zeolite. Chin. J. Catal. 2021, 42, 1689–1699. [Google Scholar] [CrossRef]
- Xiao, C.; Yang, Y.; Meng, D.; Dong, L.; Luo, L.L.; Tan, Z.Y. Stable and active monolithic palladium catalyst for catalytic oxidation of methane using nanozeolite silicalite-1 coating on cordierite. Appl. Catal. A 2017, 531, 197–202. [Google Scholar] [CrossRef]
- Tang, Z.Y.; Zhang, T.; Luo, D.C.; Wang, Y.J.; Hu, Z.; Yang, R.T. Catalytic combustion of methane: From mechanism and materials properties to catalytic performance. ACS Catal. 2022, 12, 13457–13474. [Google Scholar] [CrossRef]
- He, L.; Fan, Y.L.; Bellettre, J.; Yue, J.; Luo, L.G. A review on catalytic methane combustion at low temperatures: Catalysts, mechanisms, reaction conditions and reactor designs. Renew. Sustain. Energy Rev. 2020, 119, 109589. [Google Scholar] [CrossRef]
- Bunting, R.J.; Thompson, J.; Hu, P. The mechanism and ligand effects of single atom rhodium supported on ZSM-5 for the selective oxidation of methane to methanol. Phys. Chem. Chem. Phys. 2020, 22, 11686–11694. [Google Scholar] [CrossRef]
- Xue, W.J.; Mei, D.H. Mechanistic understanding of methane combustion over H-SSZ-13 zeolite encapsulated palladium nanocluster catalysts. Chem. Eng. J. 2022, 444, 136671. [Google Scholar] [CrossRef]
- Jørgensen, M.; Gronbeck, H. First-principles microkinetic modeling of methane oxidation over Pd(100) and Pd(111). ACS Catal. 2016, 6, 6730–6738. [Google Scholar] [CrossRef]
- Bu, X.Y.; Ran, J.Y.; Niu, J.T.; Ou, Z.L.; Tang, L.; Huang, X. Reaction mechanism insights into CH4 catalytic oxidation on Pt13 cluster: A DFT study. Mol. Catal. 2021, 515, 111891. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, C.; Wang, J.Y.; Li, H.Y.; Duan, Y.; Liu, Z.; Lee, J.Y.; Hu, X.; Xi, S.B.; Du, Y.H.; et al. The interplay between the suprafacial and intrafacial mechanisms for complete methane oxidation on substituted LaCoO3 perovskite oxides. J. Catal. 2020, 390, 1–11. [Google Scholar] [CrossRef]
- Saracco, G.; Geobaldo, F.; Baldi, G. Methane combustion on Mg-doped LaMnO3 perovskite catalysts. Appl. Catal. B 1999, 20, 277–288. [Google Scholar] [CrossRef]
- Ciuparu, D.; Altman, E.; Pfefferle, L. Contributions of lattice oxygen in methane combustion over PdO-based catalysts. J. Catal. 2001, 203, 64–74. [Google Scholar] [CrossRef]
- Stotz, H.; Maier, L.; Boubnov, A.; Gremminger, A.T.; Grunwaldt, J.D.; Deutschmann, O. Surface reaction kinetics of methane oxidation over PdO. J. Catal. 2019, 370, 152–175. [Google Scholar] [CrossRef]
- Hu, W.D.; Shao, Z.J.; Cao, X.M.; Hu, P. Multi sites vs single site for catalytic combustion of methane over Co3O4(110): A first-principles kinetic Monte Carlo study. Chin. J. Catal. 2020, 41, 1369–1377. [Google Scholar] [CrossRef]
- Tao, F.F.; Shan, J.J.; Nguyen, L.; Wang, Z.Y.; Zhang, S.R.; Zhang, L.; Wu, Z.L.; Huang, W.X.; Zeng, S.B.; Hu, P. Understanding complete oxidation of methane on spinel oxides at a molecular level. Nat. Commun. 2015, 6, 7798. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Sushkevich, V.L.; Knorpp, A.J.; Newton, M.A.; Mizuno, S.C.M.; Wakihara, T.; Okubo, T.; Liu, Z.D.; Bokhoven, J.A.V. Cu-erionite zeolite achieves high yield in direct oxidation of methane to methanol by isothermal chemical looping. Chem. Mater. 2020, 32, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Brezicki, G.; Zheng, J.; Paolucci, C.; Schlogl, R.; Davis, R.J. Effect of the Co-cation on Cu speciation in Cu-exchanged mordenite and ZSM-5 catalysts for the oxidation of methane to methanol. ACS Catal. 2021, 11, 4973–4987. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Jones, W.; Liu, Y.S.; He, Q.; Song, W.Y.; Du, P.F.; Yang, B.; An, H.Y.; Farmer, D.M.; et al. Identifying key mononuclear Fe species for low-temperature methane oxidation. Chem. Sci. 2021, 12, 3152–3160. [Google Scholar] [CrossRef]
- Tabor, E.; Dedecek, J.; Mlekodaj, K.; Sobalik, Z.; Andrikopoulos, P.C.; Sklenak, S. Dioxygen dissociation over man-made system at room temperature to form the active α-oxygen for methane oxidation. Sci. Adv. 2020, 6, 9776. [Google Scholar] [CrossRef]
- Kim, M.S.; Park, E.D. Aqueous-phase partial oxidation of methane with H2O2 over Fe-ZSM-5 catalysts prepared from different iron precursors. Microporous Mesoporous Mater. 2021, 324, 111278. [Google Scholar] [CrossRef]
- Sun, S.M.; Barnes, A.J.; Gong, X.X.; Lewis, R.J.; Dummer, N.F.; Bere, T.; Shaw, G.; Richards, N.; Morgan, D.J.; Hutchings, G.J. Lanthanum modified Fe-ZSM-5 zeolites for selective methane oxidation with H2O2. Catal. Sci. Technol. 2021, 11, 8052–8064. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Bols, M.L.; Rhoda, H.M.; Plessers, D.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Cage effects control the mechanism of methane hydroxylation in zeolites. Science 2021, 373, 327–331. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Narsimhan, K.; Serna, P.; Meyer, R.J.; Dinca, M.; Román-Leshkov, Y. Continuous partial oxidation of methane to methanol catalyzed by diffusion-paired copper dimers in copper-exchanged zeolites. J. Am. Chem. Soc. 2019, 141, 11641–11650. [Google Scholar] [CrossRef]
- Yang, J.Y.; Du, X.R.; Qiao, B.T. Methane oxidation to methanol over copper-containing zeolite. Chem 2021, 7, 2270–2272. [Google Scholar] [CrossRef]
- Park, M.B.; Ahn, S.H.; Mansouri, A.; Ranocchiari, M.; Bokhoven, J.A.V. Comparative study of diverse copper zeolites for the conversion of methane into methanol. ChemCatChem 2017, 9, 3705. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; Bokhoven, J.A.V. The direct catalytic oxidation of methane to methanol—A critical assessment. Angew. Chem. Int. Ed. 2017, 56, 16464. [Google Scholar] [CrossRef]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.A.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; et al. Methane to methanol: Structure–activity relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef] [Green Version]
- Szecsenyi, A.; Li, G.; Gascon, J.; Pidko, E.A. Mechanistic complexity of methane oxidation with H2O2 by single-site Fe/ZSM-5 catalyst. ACS Catal. 2018, 8, 7961–7972. [Google Scholar] [CrossRef]
- Li, S.C.; Wang, Y.J.; Wu, T.; Schneider, W.F. First-principles analysis of site- and condition-dependent Fe speciation in SSZ-13 and implications for catalyst optimization. ACS Catal. 2018, 8, 10119–10130. [Google Scholar] [CrossRef]
- Alayon, E.M.C.; Nachtegaal, M.; Bodi, A.; Bokhoven, J.A.V. Reaction conditions of methane-to-methanol conversion affect the structure of active copper sites. ACS Catal. 2014, 4, 16–22. [Google Scholar] [CrossRef]
- Han, B.Z.; Yang, Y.; Xu, Y.Y.; Etim, U.J.; Qiao, K.; Xu, B.J.; Yan, Z.F. A review of the direct oxidation of methane to methanol. Chin. J. Catal. 2016, 37, 1206–1215. [Google Scholar] [CrossRef]
- Wang, X.X.; Wang, Y.; Tang, Q.H.; Guo, Q.; Zhang, Q.H.; Wan, H.L. MCM-41-supported iron phosphate catalyst for partial oxidation of methane to oxygenates with oxygen and nitrous oxide. J. Catal. 2003, 217, 457–467. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.X.; Su, Z.; Guo, Q.; Tang, Q.H.; Zhang, Q.H.; Wan, H.L. SBA-15-supported iron phosphate catalyst for partial oxidation of methane to formaldehyde. Catal. Today 2004, 93, 155–161. [Google Scholar] [CrossRef]
- Ruddy, D.A.; Ohler, N.L.; Bell, A.T.; Tilley, T.D. Thermolytic molecular precursor route to site-isolated vanadia–silica materials and their catalytic performance in methane selective oxidation. J. Catal. 2006, 238, 277–285. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Loridant, S.; Launay, H.; Pigamo, A.; Dubois, J.L.; Millet, J.M.M. Study of new catalysts based on vanadium oxide supported on mesoporous silica for the partial oxidation of methane to formaldehyde: Catalytic properties and reaction mechanism. J. Catal. 2006, 237, 38–48. [Google Scholar] [CrossRef]
- An, D.L.; Zhang, Q.H.; Wang, Y. Copper grafted on SBA-15 as efficient catalyst for the selective oxidation of methane by oxygen. Catal. Today 2010, 157, 143–148. [Google Scholar] [CrossRef]
- Li, Y.; An, D.L.; Zhang, Q.H.; Wang, Y. Copper-catalyzed selective oxidation of methane by oxygen: Studies on catalytic behavior and functioning mechanism of CuOx/SBA-15. J. Phys. Chem. C 2008, 112, 13700–13708. [Google Scholar] [CrossRef]
- Zhu, K.X.; Liang, S.X.; Cui, X.J.; Huang, R.; Wan, N.B.; Hua, L.; Li, H.Y.; Chen, H.Y.; Zhao, Z.C.; Hou, G.J.; et al. Highly efficient conversion of methane to formic acid under mild conditions at ZSM-5-confined Fe-sites. Nano Energy 2021, 82, 105718. [Google Scholar] [CrossRef]
- Wang, V.C.C.; Maji, S.; Chen, P.P.Y.; Lee, H.K.; Yu, S.S.F.; Chan, S.I. Alkane oxidation: Methane monooxygenases, related enzymes, and their biomimetics. Chem. Rev. 2017, 117, 8574–8621. [Google Scholar] [CrossRef]
- Qi, G.D.; Davies, T.E.; Nasrallah, A.; Sainna, M.A.; Howe, A.G.R.; Lewis, R.J.; Quesne, M.; Catlow, C.R.A.; Willock, D.J.; He, Q.; et al. Au-ZSM-5 catalyses the selective oxidation of CH4 to CH3OH and CH3COOH using O2. Nat. Catal. 2022, 5, 45–54. [Google Scholar] [CrossRef]
- Li, H.Y.; Fei, M.C.; Troiano, J.L.; Ma, L.; Yan, X.X.; Tieu, P.; Yuan, Y.C.; Zhang, Y.H.; Liu, T.Y.; Pan, X.Q.; et al. Selective Methane Oxidation by Heterogenized Iridium Catalysts. J. Am. Chem. Soc. 2023, 145, 769–773. [Google Scholar] [CrossRef]
- Pannov, G.I.; Sobolev, V.I.; Kharitonov, A.S. The role of iron in N2O decomposition on ZSM-5 zeolite and reactivity of the surface oxygen formed. J. Mol. Catal. 1990, 61, 85–97. [Google Scholar] [CrossRef]
- Sobolev, V.I.; Panov, G.I.; Kharitonov, A.S.; Romannikov, V.N.; Volodin, A.M.; Ione, K.G. Catalytic properties of ZSM-5 zeolites in N2O decomposition: The role of iron. J. Catal. 1993, 139, 435–443. [Google Scholar] [CrossRef]
- Dubkov, K.A.; Sobolev, V.I.; Talsi, E.P.; Rodkin, M.A.; Watkins, N.H.; Shteinman, A.A.; Panov, G.I. Kinetic isotope effects and mechanism of biomimetic oxidation of methane and benzene on FeZSM-5 zeolite. J. Mol. Catal. A 1997, 123, 155–161. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Serna, P.; Meyer, R.J.; Dinca, M.; Roman-Leshkov, Y. Viewpoint on the partial oxidation of methane to methanol using Cu- and Fe-exchanged zeolites. ACS Catal. 2018, 8, 8306–8313. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Su, Y.; Wang, A.Q.; Weckhuysen, B.M.; Luo, W.H. Efficient synthesis of monomeric Fe species in zeolite ZSM-5 for the low-temperature oxidation of methane. ChemCatChem 2021, 13, 2766–2770. [Google Scholar] [CrossRef]
- Citek, C.; Gary, J.B.; Wasinger, E.C.; Stack, T.D.P. Chemical plausibility of Cu(III) with biological ligation in pMMO. J. Am. Chem. Soc. 2015, 137, 6991–6994. [Google Scholar] [CrossRef]
- Bols, M.L.; Hallaert, S.D.; Snyder, B.E.R.; Devos, J.; Plessers, D.; Rhoda, H.M.; Dusselier, M.; Schoonheydt, R.A.; Pierloot, K.; Solomon, E.I.; et al. Spectroscopic identification of the α-Fe/α-O active site in Fe-CHA zeolite for the low-temperature activation of the methane C–H bond. J. Am. Chem. Soc. 2018, 140, 12021–12032. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Vanelderen, P.; Bols, M.L.; Hallaert, S.D.; Bottger, L.H.; Ungur, L.; Pierloot, K.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. The active site of low-temperature methane hydroxylation in iron-containing zeolites. Nature 2016, 536, 317–321. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Tanaka, T.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Methane partial oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ active species in large-pore zeolites. ACS Catal. 2018, 8, 1500–1509. [Google Scholar] [CrossRef]
- Li, G.; Vassilev, P.; Sanchez-Sanchez, M.; Lercher, J.A.; Hensen, E.J.M.; Pidko, E.A. Stability and reactivity of copper oxo-clusters in ZSM-5 zeolite for selective methane oxidation to methanol. J. Catal. 2016, 338, 305–312. [Google Scholar] [CrossRef]
- Heyer, A.J.; Plessers, D.; Braun, A.; Rhoda, H.M.; Bols, M.L.; Hedman, B.; Hodgson, K.O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Methane activation by a mononuclear copper active site in the zeolite mordenite: Effect of metal nuclearity on reactivity. J. Am. Chem. Soc. 2022, 144, 19305–19316. [Google Scholar] [CrossRef] [PubMed]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Bokhoven, J.A.V. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Knorpp, A.J.; Sushkevich, V.L.; Palagin, D.; Bokhoven, J.A.V. Active sites and mechanisms in the direct conversion of methane to methanol using Cu in zeolitic hosts: A critical examination. Chem. Soc. Rev. 2020, 49, 1449–1486. [Google Scholar] [CrossRef] [Green Version]
- Wulfers, M.J.; Teketel, S.; Lpek, B.; Lobo, R.F. Conversion of methane to methanol on copper-containing small-pore zeolites and zeotypes. Chem. Commun. 2015, 51, 4447–4450. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Monocopper active site for partial methane oxidation in Cu-exchanged 8MR zeolites. ACS Catal. 2016, 6, 6531–6536. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Bokhoven, J.A.V. The effect of the active-site structure on the activity of copper mordenite in the aerobic and anaerobic conversion of methane into methanol. Angew. Chem. Int. Ed. 2018, 57, 8906. [Google Scholar] [CrossRef]
- Liu, N.; Li, Y.; Dai, C.N.; Xu, R.N.; Yu, G.Q.; Wang, N.; Chen, B.H. H2O in situ induced active site structure dynamics for efficient methane direct oxidation to methanol over Fe-BEA zeolite. J. Catal. 2022, 414, 302–312. [Google Scholar] [CrossRef]
- Adeyiga, O.; Odoh, S.O. Methane over-oxidation by extra-framework copper-oxo active sites of copper-exchanged zeolites: Crucial role of traps for the separated methyl group. ChemPhysChem 2021, 22, 1101. [Google Scholar] [CrossRef]
- Lucas, A.D.; Valverde, J.L.; Rodriguez, L.; Sanchez, P.; Garcia, M.T. Partial oxidation of methane to formaldehyde over Mo/HZSM-5 catalysts. Appl. Catal. A 2000, 203, 81–90. [Google Scholar] [CrossRef]
- Hammond, C.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Rahim, M.H.; Forde, M.M.; Thetford, A.; Murphy, D.M.; Hagen, H.; Stangland, E.E.; et al. Catalytic and mechanistic insights of the low-temperature selective oxidation of methane over Cu-promoted Fe-ZSM-5. Chem. Eur. J. 2012, 18, 15735–15745. [Google Scholar] [CrossRef]
- Yang, N.T.; Ren, Z.L.; Yang, C.G.; Wu, P.; Zeng, G.F. Direct oxidation of CH4 to HCOOH over extra-framework stabilized Fe@MFI catalyst at low temperature. Fuel 2021, 305, 121624. [Google Scholar] [CrossRef]
- Shahami, M.; Shantz, D.F. Zeolite acidity strongly influences hydrogen peroxide activation and oxygenate selectivity in the partial oxidation of methane over M,Fe-MFI (M: Ga, Al, B) zeolites. Catal. Sci. Technol. 2019, 9, 2945–2951. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; Rahim, M.H.A.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Yu, X.; Wu, B.; Huang, M.; Lu, Z.X.; Li, J.; Zhong, L.S.; Sun, Y.H. IrFe/ZSM-5 synergistic catalyst for selective oxidation of methane to formic acid. Energy Fuels 2021, 35, 4418–4427. [Google Scholar] [CrossRef]
- Narsimhan, K.; Michaelis, V.K.; Mathies, G.; Gunther, W.R.; Griffin, R.G.; Roman-Leshkov, Y. Methane to acetic acid over Cu-exchanged zeolites: Mechanistic insights from a site-specific carbonylation reaction. J. Am. Chem. Soc. 2015, 137, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.M.; Qi, G.D.; Xu, J.; Li, B.J.; Wang, C.; Deng, F. NMR-spectroscopic evidence of intermediate-dependent pathways for acetic acid formation from methane and carbon monoxide over a ZnZSM-5 zeolite catalyst. Angew. Chem. Int. Ed. 2012, 51, 3850–3853. [Google Scholar] [CrossRef]
- Fang, Z.H.; Huang, M.Y.; Liu, B.; Chen, J.; Jiang, F.; Xu, Y.B.; Liu, X.H. Insights into Fe species structure-performance relationship for direct methane conversion toward oxygenates over Fe-MOR catalysts. ChemCatChem 2022, 14, e202200218. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Metal Loading (wt%) | Topology | Reaction Condition | Catalytic Activity | Stability | Ref. |
|---|---|---|---|---|---|---|
| Pd/H-MOR | 1.01 wt% Pd | MOR | 1 vol% CH4, 4 vol% O2, N2 (balance), GHSV = 70,000 h−1 | T98% = 475 °C | – | [31] |
| Pd/Na-MOR | 0.99 wt% Pd | MOR | 1 vol% CH4, 4 vol% O2, N2 (balance), GHSV = 70,000 h−1 | T98% = 425 °C | Kept stable during 90 h of on-stream reaction | [31] |
| Pd/HZSM-5 | 2.05 wt% Pd | MFI | 1 vol% CH4, 4 vol% O2, N2 (balance), GHSV = 15,000 h−1 | T98% = 450 °C | No deactivation within 10 h of reaction | [32] |
| Pd-Ce/HZSM-5 | 0.95 wt% Pd | MFI | 2 vol% CH4, 8 vol% O2, N2 (balance), GHSV = 48,000 h−1 | T98% = 375 °C | Kept stable during 30 h of on-stream reaction | [33] |
| Pd@Silicalite-1 | 0.98 wt% Pd | MFI | 1 vol% CH4, 20 vol% O2, N2 (balance), GHSV = 24,000 h−1 | T90% = 309 °C | Kept stable during 100 h of on-stream reaction | [34] |
| PdPt/TiO2/ZSM-5 | 5.31 wt% Pd 2.21 wt% Pt | MFI | 1 vol% CH4, 10 vol% O2, Ar (balance), GHSV = 24,000 h−1 | T90% = 319 °C | Kept stable during 35 h of on-stream reaction | [35] |
| Rh/ZSM-5 | 1.95 wt% Pd | MFI | 2500 ppm CH4, 10 vol% O2, N2 (balance), GHSV = 150,000 h−1 | T98% = 420 °C | Kept stable during 20 h of on-stream reaction | [42] |
| Pd/ZSM-5 | 0.93 wt% Pd | MFI | 1 vol% CH4, 4 vol% O2, N2 (balance), GHSV = 70,000 h−1 | T98% = 410 °C | Kept stable during 80 h of on-stream reaction | [43] |
| PdO/Beta | 0.7 wt% Pd | – | 1 vol% CH4, 20 vol% O2, N2 (balance), GHSV = 30,000 h−1 | T98% = 350 °C | Kept stable after 6 cycle tests | [44] |
| Pd@Silicalite-1 | 0.83 wt% Pd | MFI | 0.5 vol% CH4, 20 vol% O2, N2 (balance), GHSV = 20,000 h−1 | T98% = 360 °C | Kept stable during 200 h of on-stream reaction | [45] |
| PdCo@ZSM-5 | 0.54 wt% Pd 0.091 wt% Co | MFI | 1 vol% CH4, 20 vol% O2, N2 (balance), GHSV = 60,000 h−1 | T98% = 385 °C | Kept stable during 20 h of on-stream reaction | [46] |
| Na-FAU-Pd | 5.65 wt% Pd | FAU | 5 vol% CH4, 10 vol% O2, He (balance), GHSV = 40,000 h−1 | T98% = 245 °C | – | [47] |
| Catalyst | Method | Metal Precursor | Metal Loading (wt%) | Topology | Oxidant | Reaction Temperature (°C) | Desired Product, Yield (μmol/gcat) | Desired Product, Selectivity | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Cu-ERI | ion-exchanged | (CH3COO)2Cu | 4.2 wt% Cu | ERI | O2 | 300 | CH3OH, 147 | CH3OH, 95% | [66] |
| Cu-H-MOR | ion-exchanged | Cu(CH3COO)2· H2O | 0.42 wt% Cu | MOR | O2 | 200 | CH3OH, 39 | CH3OH, 90% | [67] |
| Fe/ZSM-5 | wet impregnation | Fe(NO3)3·9H2O | 0.1 wt% Fe | MFI | H2O2 | 50 | CH3OH, 66 | CH3OH, 90% | [68] |
| Fe-ferrierite | ion-exchanged | Fe(C5H7O2)3 | 2.0 wt% Fe | FER | O2 | 300 | CH3OH, 75 | CH3OH, 93% | [69] |
| Fe-ZSM-5 | ion-exchanged | FeSO4 | 0.46 wt% Fe | MFI | H2O2 | 50 | CH3OH, 25 | CH3OH, 78% | [70] |
| Fe-ZSM-5 | ion-exchanged | FeCl2 | 0.45 wt% Fe | MFI | H2O2 | 50 | CH3OH, 18 | CH3OH, 70% | [70] |
| Fe-ZSM-5 | wet impregnation | FeSO4 | 0.54 wt% Fe | MFI | H2O2 | 50 | CH3OH, 13 | CH3OH, 63% | [70] |
| LaFe-ZSM-5 | wet impregnation | Fe(NO3)3·9H2O La(NO3)3·6H2O | 0.34 wt% Fe 0.18 wt% La | MFI | H2O2 | 50 | CH3OH, 114 | CH3OH, 99% | [71] |
| Fe-CHA | wet impregnation | Fe(C5H7O2)3 | 0.22 wt% Fe | CHA | N2O | 160 | CH3OH, 27 | CH3OH, 87% | [72] |
| Cu-MOR | ion-exchanged | Cu(NO3)2·3H2O | 0.95 wt% Cu | MOR | H2O | 200 | CH3OH, 20 | CH3OH, 97% | [73] |
| Cu-SSZ-13 | ion-exchanged | Cu(CH3COO)2· H2O | 0.57 wt% Cu | CHA | O2 | 270 | CH3OH, 83 | CH3OH, 98% | [74] |
| Cu/CHA | ion-exchanged | Cu(CH3COO)2· H2O | 1.05 wt% Cu | CHA | O2 | 300 | CH3OH, 54.3 | CH3OH, 91% | [75] |
| Cu-SSZ-39 | ion-exchanged | (CH3COO)2Cu | 0.256 wt% Cu | CHA | O2 | 450 | CH3OH, 36 | CH3OH, 84% | [76] |
| Cu-SPAO-34 | ion-exchanged | (CH3COO)2Cu | (0.6 wt% Cu | CHA | O2 | 450 | CH3OH, 15 | CH3OH, 71% | [76] |
| Cu-MOR | ion-exchanged | Cu(NO3)2·3H2O | 0.6 wt% Cu | MOR | O2 | 400 | CH3OH, 31.6 | CH3OH, 98% | [77] |
| Cu-SSZ-13 | ion-exchanged | (CH3COO)2Cu | 0.5 wt% Cu | CHA | O2 | 200 | CH3OH, 118 | CH3OH, 88% | [78] |
| Fe-BEA | ion-exchanged | Fe(NO3)3·9H2O | 1.04 wt% Fe | BEA | N2O | 250 | CH3OH, 227 | CH3OH, 73% | [79] |
| FePO4/MCM-41 | wet impregnation | Fe(NO3)3 | 40 wt% Fe | – | N2O | 550 | HCHO, 58 | HCHO, 79% | [39] |
| FePO4/SBA-15 | wet impregnation | Fe(NO3)3 | 5.0 wt% Fe | – | O2 | 500 | HCHO, 46 | HCHO, 81% | [80] |
| Co-ZSM-5 | wet impregnation | Co(NO3)2·6H2O | 10.0 wt% Co | MFI | O2 | 360 | HCHO, 40 | HCHO, 75% | [81] |
| Mo/ZSM-5 | wet impregnation | (NH4)6Mo7O24· 4H2O | 6.5 wt% Mo | MFI | O2 | 600 | HCHO, 22 | HCHO, 73% | [82] |
| VOx/SBA-15 | wet impregnation | NH4VO3 | 1.7 wt% V | – | O2 | 600 | HCHO, 54 | HCHO, 85% | [83] |
| VOx/MCM-41 | wet impregnation | NH4VO3 | 3.5 wt% V | – | O2 | 550 | HCHO, 28 | HCHO, 83% | [84] |
| CuOx/SBA-15 | wet impregnation | Cu(C5H7O2)2 | 0.6 wt% Cu | – | O2 | 625 | HCHO, 78 | HCHO, 71% | [85] |
| Fe/ZSM-5 | Ball-milling | Fe(NO3)3·9H2O | 0.5 wt% Fe | MFI | H2O2 | 70 | HCOOH, 115 | HCOOH, 96% | [86] |
| Fe/ZSM-5 | ion-exchanged | Fe(NO3)3 | 0.03 wt% Fe | MFI | H2O2 | 80 | HCOOH, 383 | HCOOH, 91% | [87] |
| Pd1O4/ZSM-5 | incipient wetness impregnation | Pd(NO3)2 | 0.01 wt% Pd | MFI | H2O2 | 95 | HCOOH, 323 | HCOOH, 78% | [11] |
| IrFe/ZSM-5 | wet impregnation | H2IrCl6·6H2O FeCl3·6H2O | 0.01 wt% Ir 0.6 wt% Fe | MFI | H2O2 | 50 | HCOOH, 182 | HCOOH, 71% | [88] |
| Au/ZSM-5 | deposition–precipitation | HAuCl4·3H2O | 0.5 wt% Au | MFI | O2 | 240 | CH3COOH, 13 | CH3COOH, 71% | [89] |
| Rh/Na-ZSM-5 | incipient wetness impregnation | Rh(NO3)3 | 0.5 wt% Rh | MFI | O2 | 150 | CH3COOH, 2200 | CH3COOH, 90% | [14] |
| Rh/ZSM-5 | incipient wetness impregnation | Rh(NO3)3 | 0.1 wt% Rh | MFI | O2 | 150 | CH3COOH, 820 | CH3COOH, 70% | [13] |
| Fe/ZSM-5 | wet impregnation | FeCl3·6H2O | 0.31 wt% Fe | MFI | H2O2 | 50 | CH3COOH, 925 | CH3COOH, 100% | [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.; Fan, W.; Wang, X.; Lin, H.; Tao, J.; Liu, Y.; Deng, J.; Jing, L.; Dai, H. Methane Oxidation over the Zeolites-Based Catalysts. Catalysts 2023, 13, 604. https://doi.org/10.3390/catal13030604
Wu L, Fan W, Wang X, Lin H, Tao J, Liu Y, Deng J, Jing L, Dai H. Methane Oxidation over the Zeolites-Based Catalysts. Catalysts. 2023; 13(3):604. https://doi.org/10.3390/catal13030604
Chicago/Turabian StyleWu, Linke, Wei Fan, Xun Wang, Hongxia Lin, Jinxiong Tao, Yuxi Liu, Jiguang Deng, Lin Jing, and Hongxing Dai. 2023. "Methane Oxidation over the Zeolites-Based Catalysts" Catalysts 13, no. 3: 604. https://doi.org/10.3390/catal13030604