
Gold and Ceria Modified NiAl Hydrotalcite Materials as Catalyst Precursors for Dry Reforming of Methane

 , , , , , and
, , , , , and
Abstract
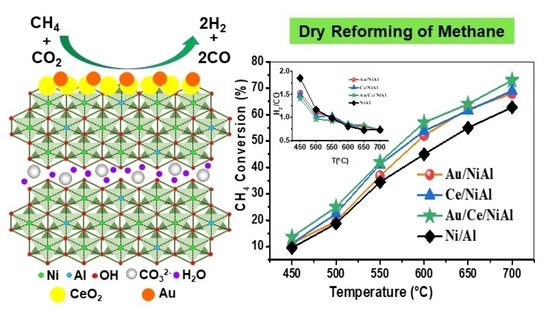
:
1. Introduction
2. Results and Discussions
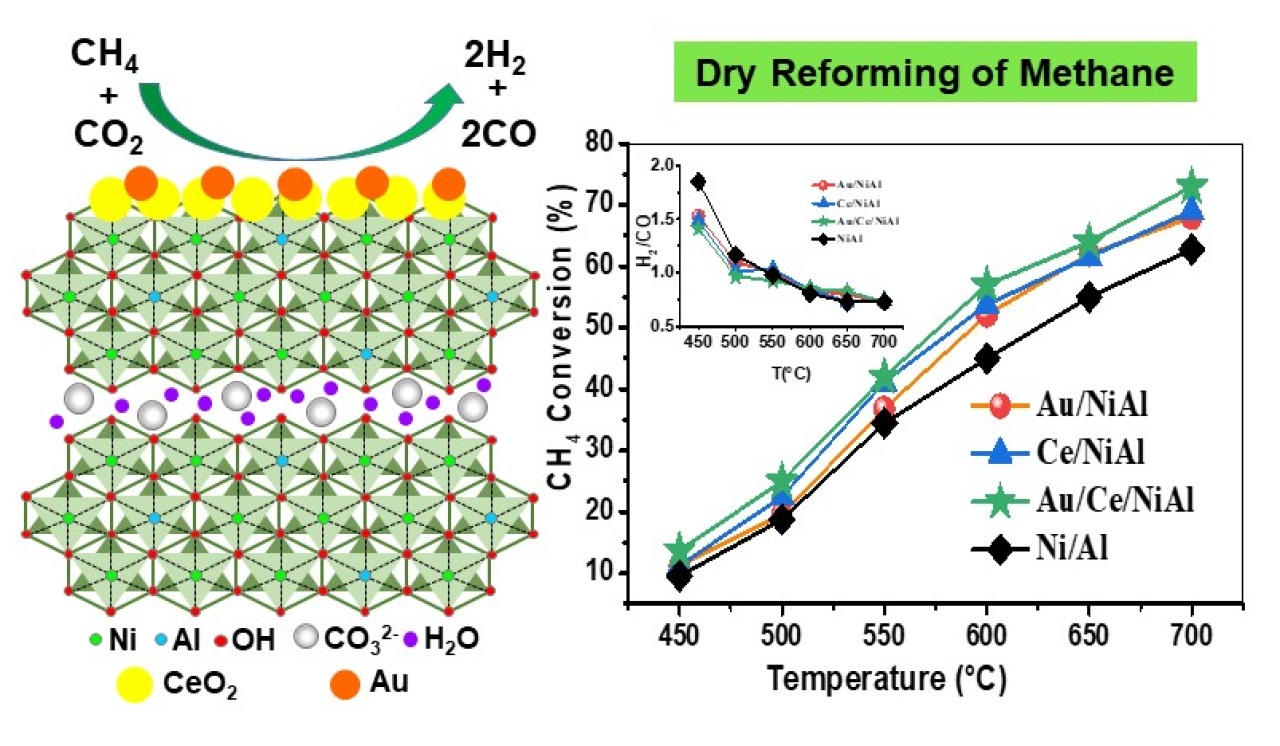
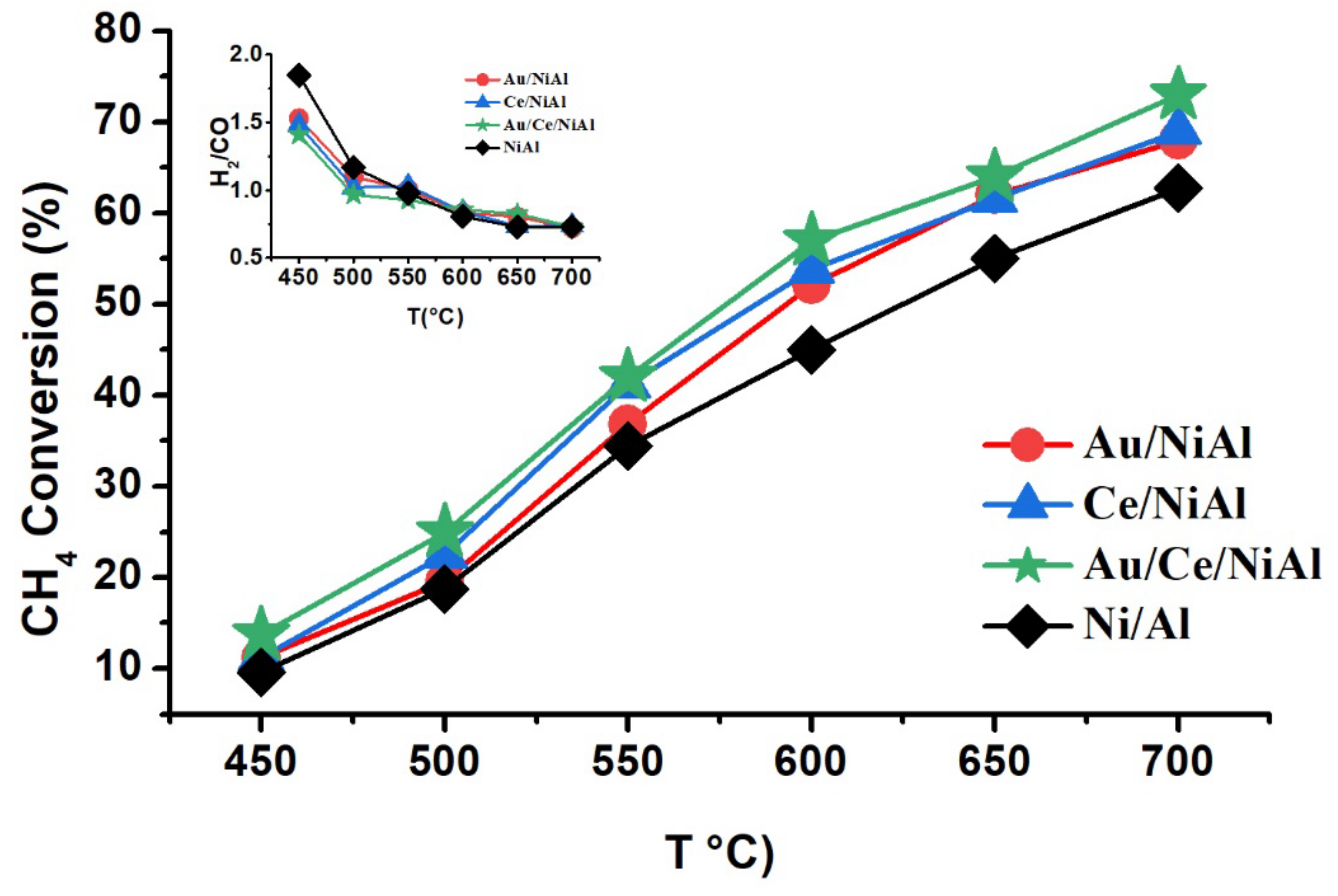
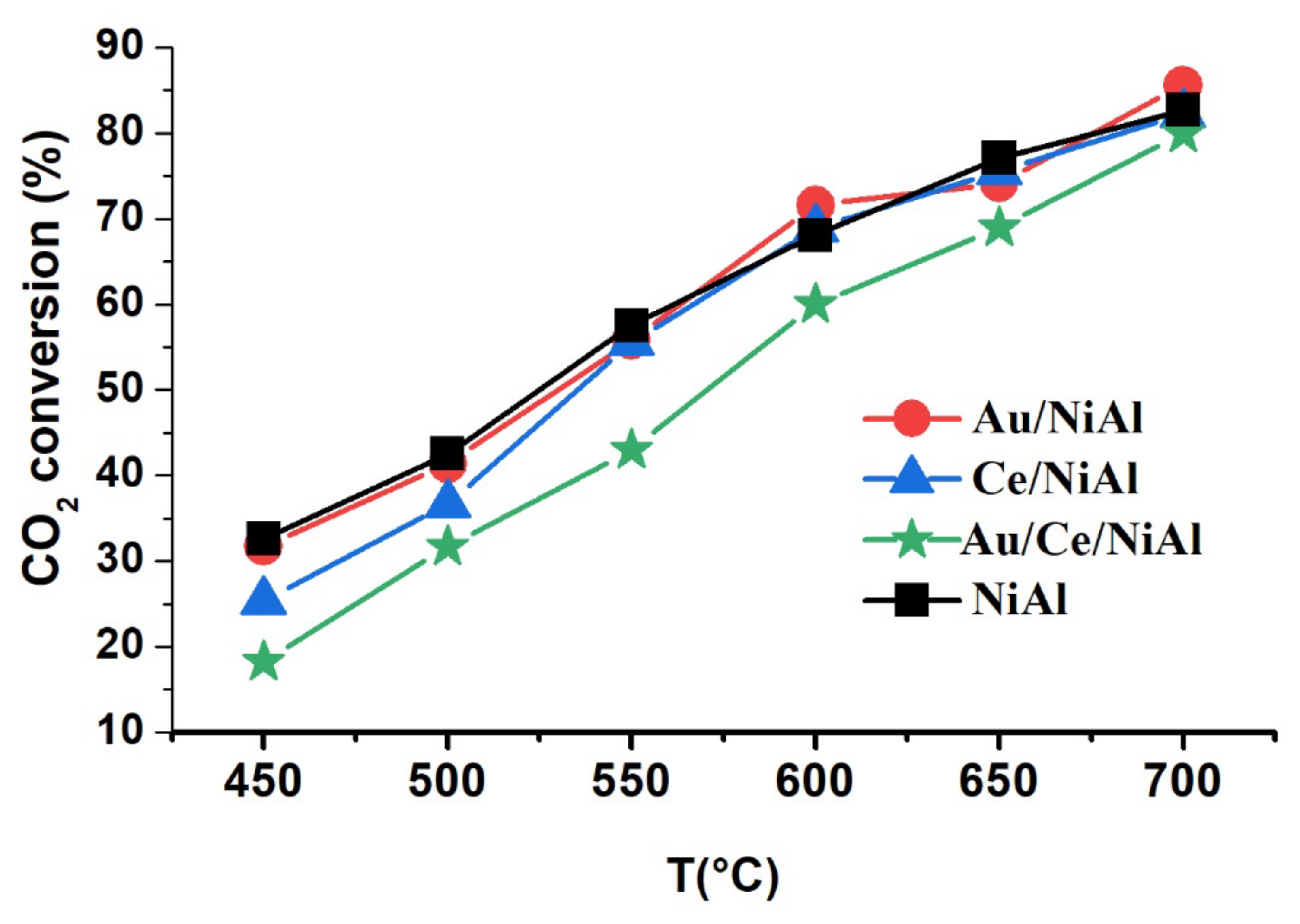
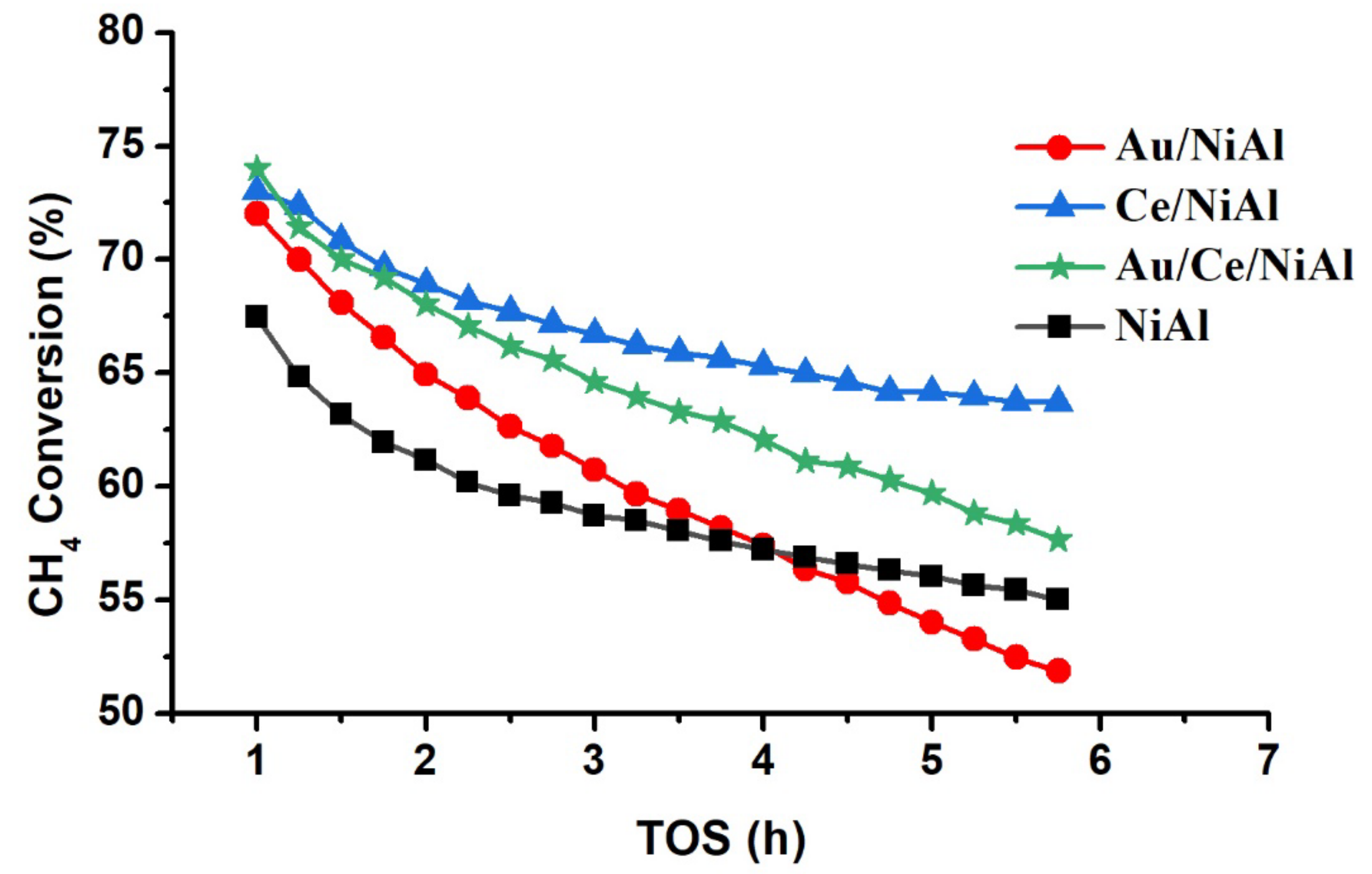
2.1. Catalytic Results
2.2. Characterization
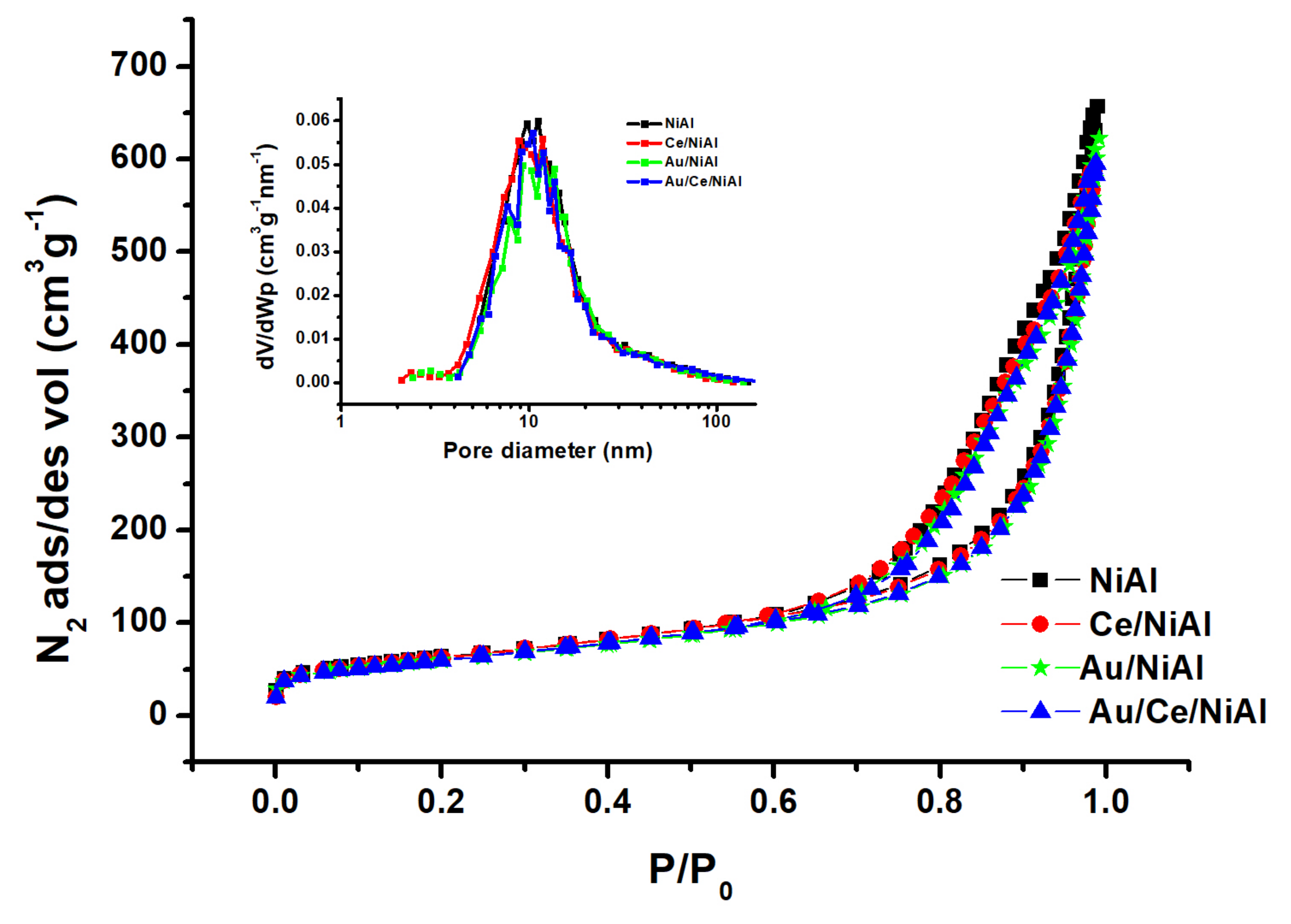
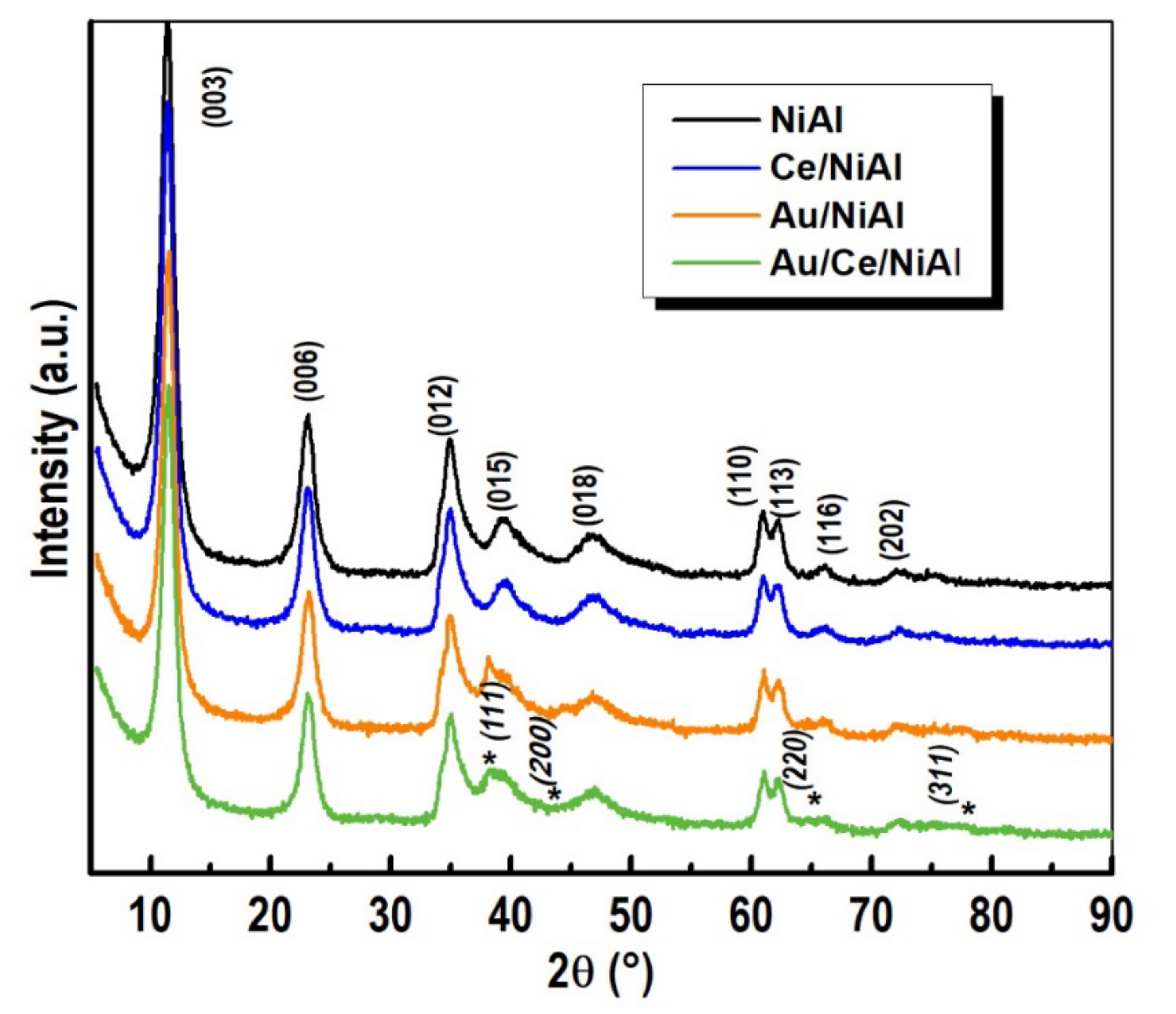
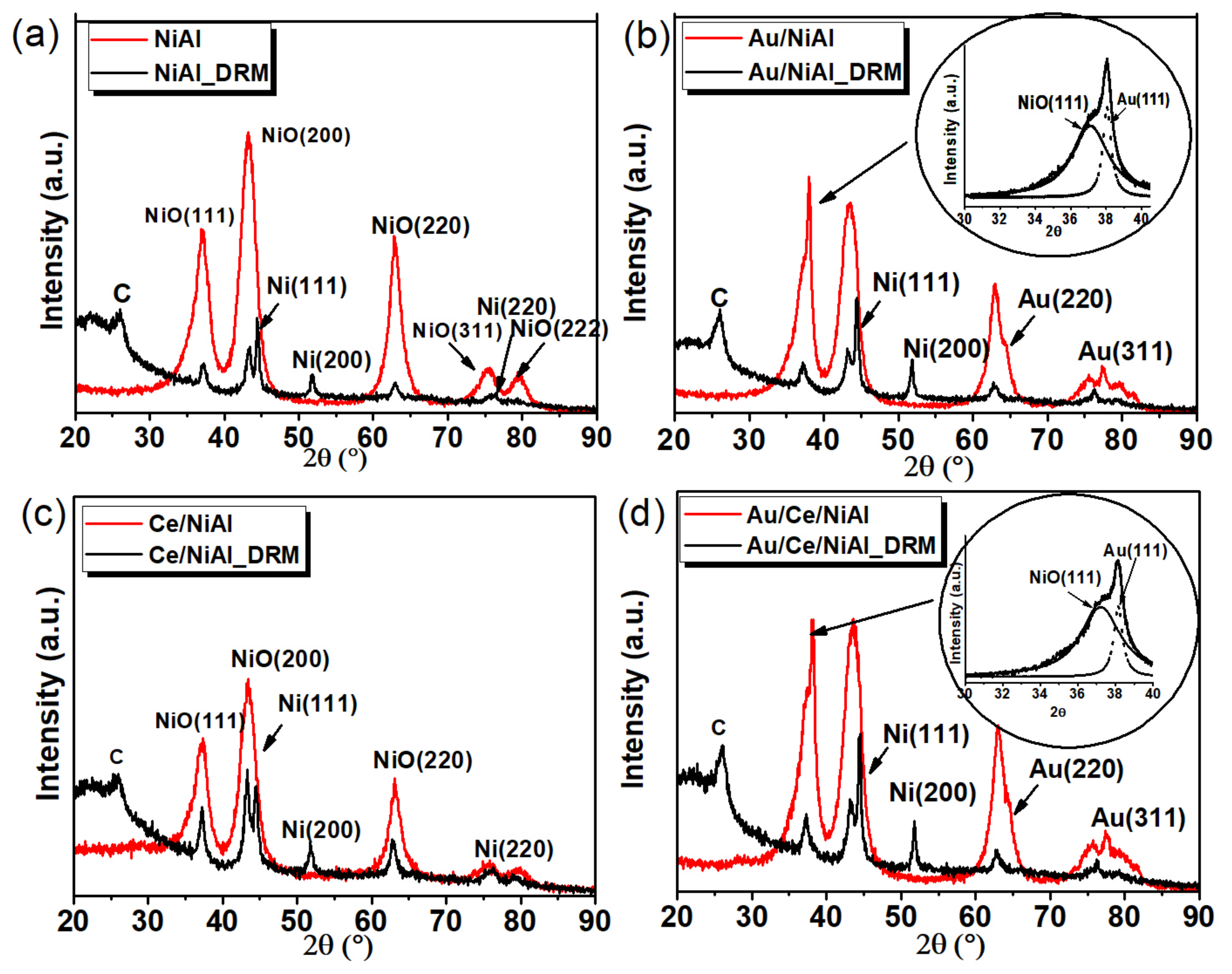
2.2.1. BET and XRD Analyses
2.2.2. TPR Analyses
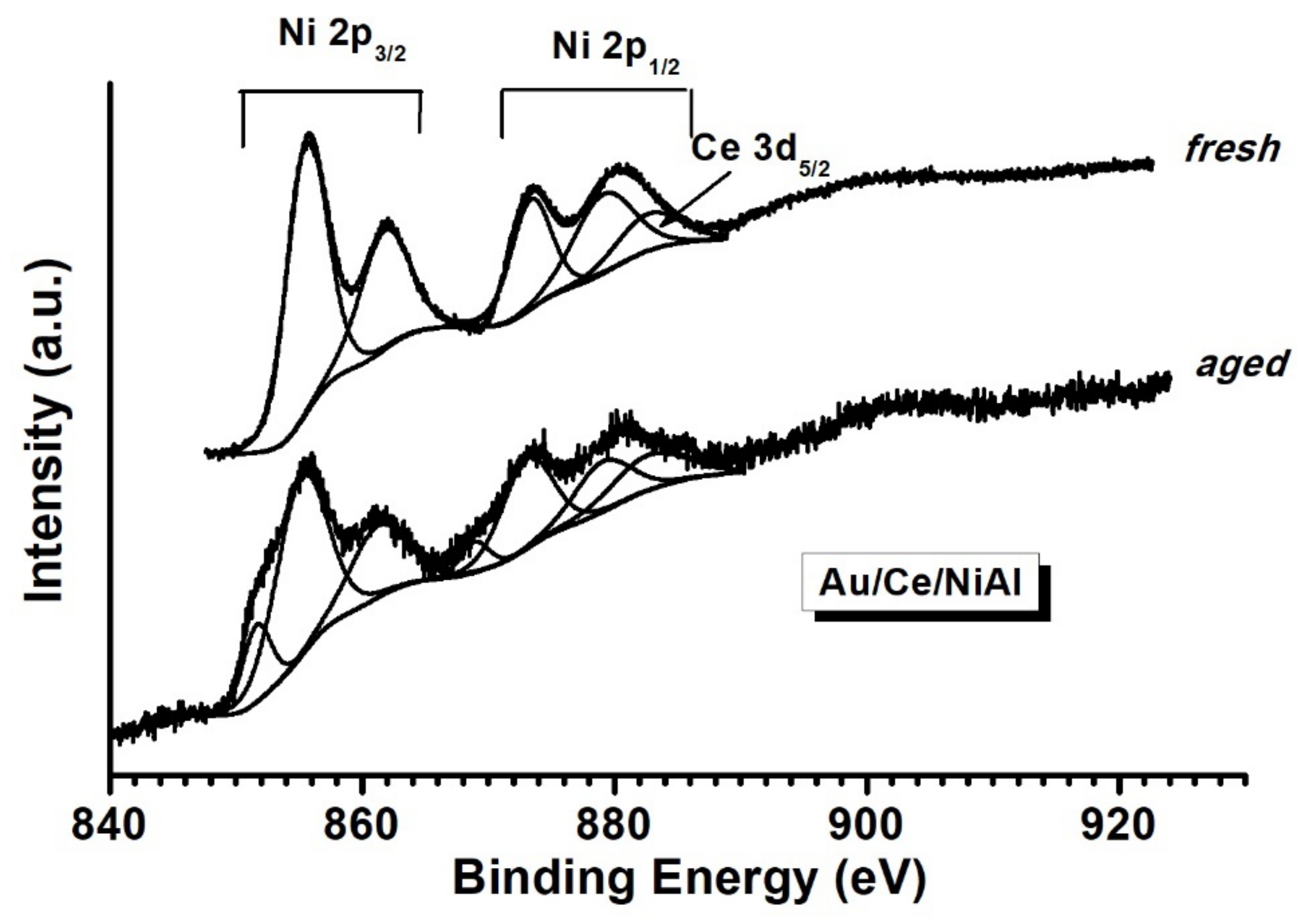
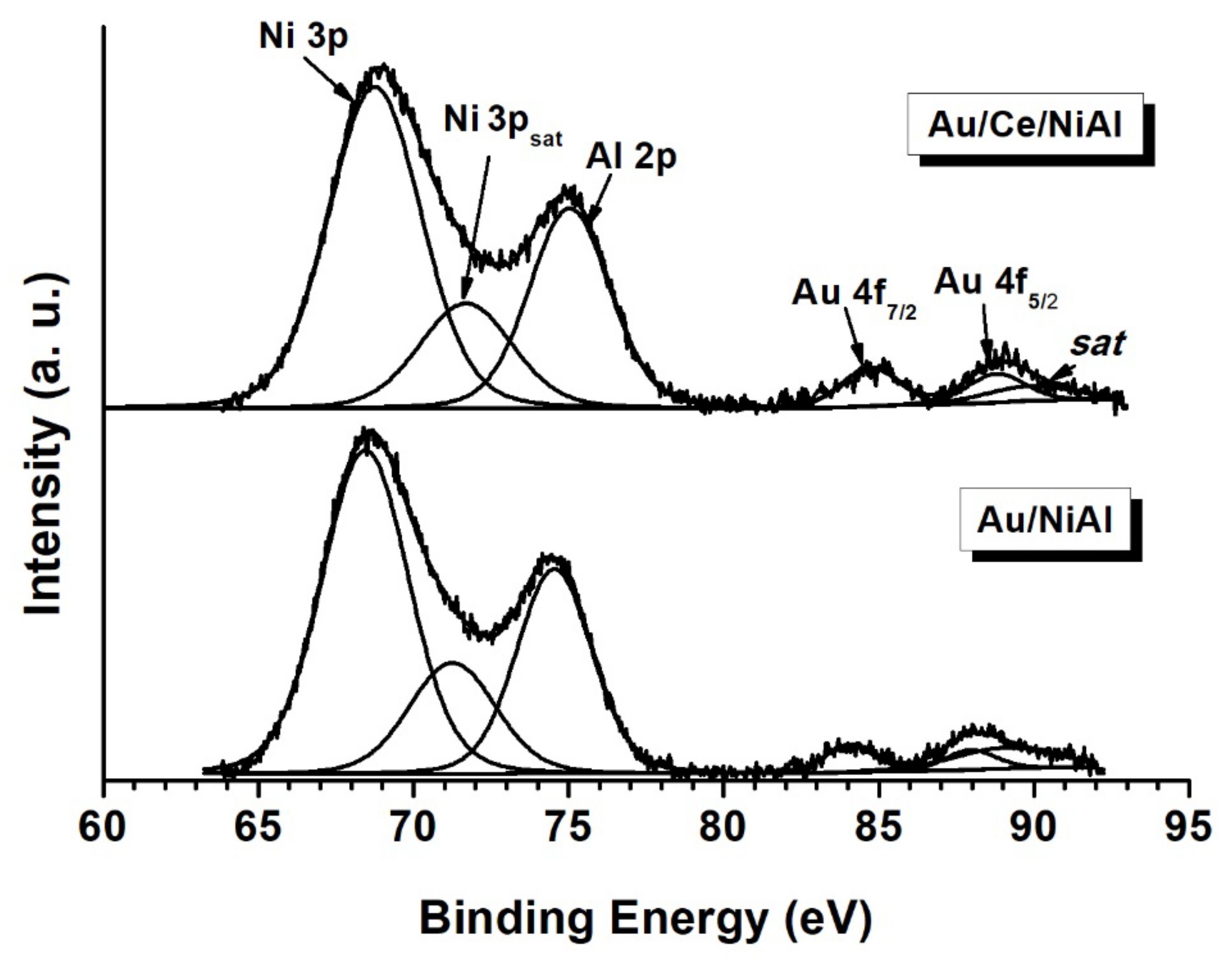
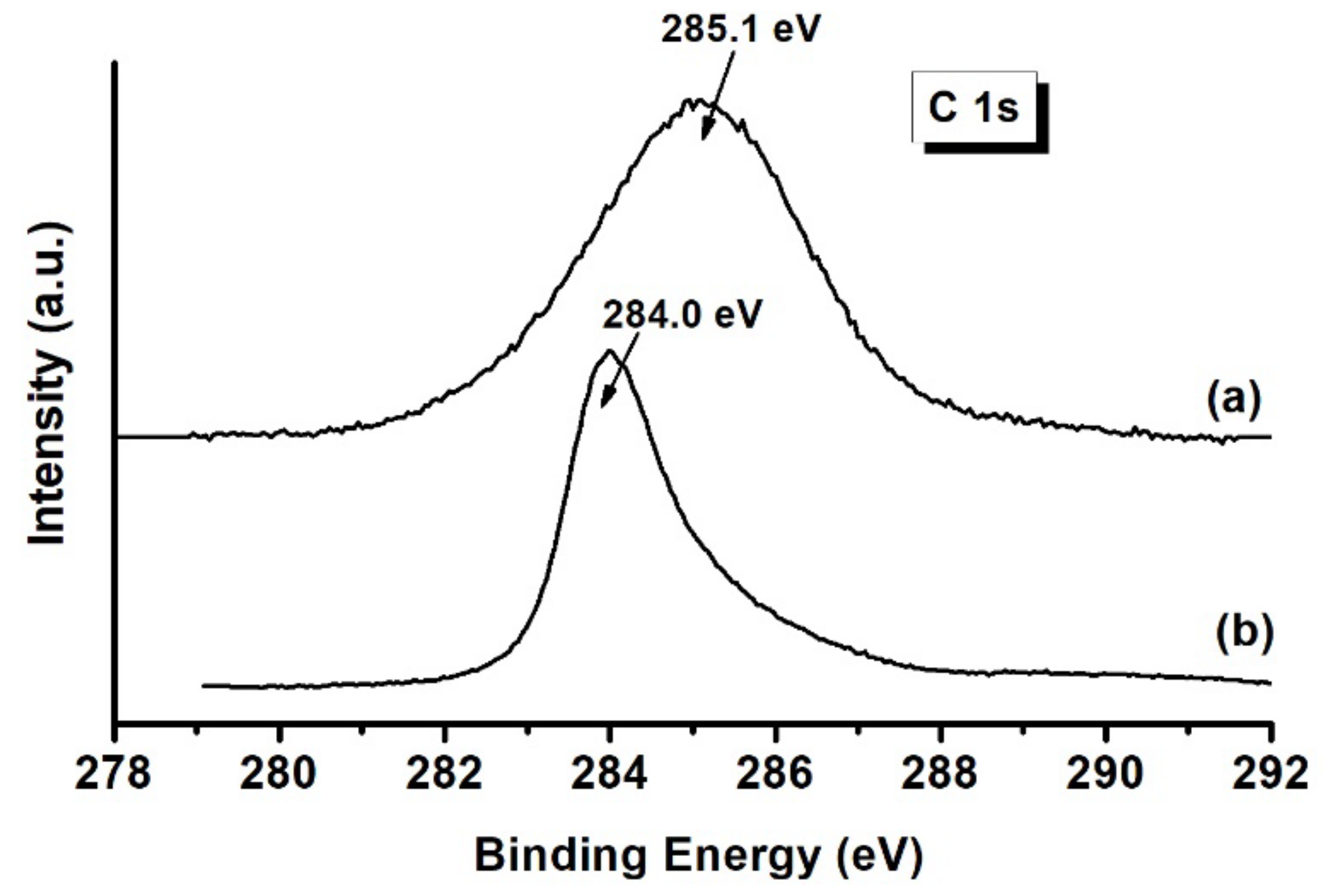
2.2.3. XPS Analyses
3. Experimental
3.1. Synthesis of Materials
3.2. Characterization
3.3. Catalytic Measurements
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wittich, K.; Krämer, M.; Bottke, N.; Schunk, S.A. Catalytic Dry Reforming of Methane: Insights from Model Systems. ChemCatChem 2020, 12, 2130–2147. [Google Scholar] [CrossRef]
- Seo, H.O. Recent Scientific Progress on Developing Supported Ni catalysts for Dry (CO2) Reforming of Methane. Catalysts 2018, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Hu, Y.H. Advances in catalytic conversion of methane and carbon dioxide to highly valuable products. Energy Sci. Eng. 2019, 7, 4–29. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Ge, Z.; Zhu, G.; Wang, Z.; Ashok, J.; Kawi, S. Anti-Coking and Anti-Sintering Ni/Al2O3 Catalysts in the dry reforming of methane: Recent Progress and Prospects. Catalysts 2021, 11, 1003. [Google Scholar] [CrossRef]
- Sandoval-Diaz, L.E.; Schlögl, R.; Lunkenbein, T. Quo Vadis Dry Reforming of Methane? A Review on Its Chemical, Environmental, and Industrial Prospects. Catalysts 2022, 12, 465. [Google Scholar] [CrossRef]
- Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst design for dry reforming of methane: Analysis Reviews. Renew. Sustain. Energy Rev. 2018, 82, 2570–2583. [Google Scholar]
- Parsapur, R.K.; Chatterjee, S.; Huang, K.-W. The Insignificant Role of Dry Reforming of Methane in CO2 Emission Relief. ACS Energy Lett. 2020, 5, 2881–2885. [Google Scholar] [CrossRef]
- Arora, S.; Prasad, R. An overview on dry reforming of methane: Strategies to reduce carbonaceous deactivation of catalysts. RSC Adv. 2016, 6, 108688. [Google Scholar] [CrossRef]
- Muraza, O.; Galadima, A. A review on coke management during dry reforming of methane. Int. J. Energy Res. 2015, 39, 1196–1216. [Google Scholar] [CrossRef]
- Bermúdez, J.; Fidalgo, B.; Arenillas, A.; Menéndez, J. Dry reforming of coke oven gases over activated carbon to produce syngas for methanol synthesis. Fuel 2010, 89, 2897–2902. [Google Scholar] [CrossRef] [Green Version]
- Han, J.W.; Park, J.S.; Choi, M.S.; Lee, H. Uncoupling the size and support effects of Ni catalysts for dry reforming of methane. Appl. Catal. B 2017, 203, 625–632. [Google Scholar] [CrossRef]
- Akri, M.; Zhao, S.; Li, X.; Zang, K.; Lee, A.F.; Isaacs, M.A.; Xi, W.; Gangarajula, Y.; Luo, J.; Ren, Y.; et al. Atomically dispersed nickel as coke-resistant active sites for methane dry reforming. Nat. Commun. 2019, 10, 5181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, C.; Kranenborg, J.; Monti, M.; Weckhuysen, B.M. Structure Sensitivity in Steam and Dry Reforming over Nickel: Activity and Carbon formation. ACS Catal. 2020, 10, 1428–1438. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Du, X.-H.; Li, J.; Wang, P.; Zhu, J.; Ge, F.-J.; Zhou, J.; Song, M.; Zhu, W.-Y. A comparison of Al2O3 and SiO2 supported Ni-based catalysts on their performance for the dry reforming of methane. J. Fuel Chem. Technol. 2019, 47, 199–208. [Google Scholar] [CrossRef]
- Singh, R.; Dhir, A.; Mohapatra, S.K.; Mahia, S.K. Dry reforming of methane using various catalysts in the process: Review. Biomass Convers. Biorefinery 2020, 10, 567–587. [Google Scholar] [CrossRef]
- Emamdoust, A.; La Parola, V.; Pantaleo, G.; Testa, M.L.; Farjami Shayesteh, S.; Venezia, A.M. Partial oxidation of methane over SiO2 supported Ni and NiCe catalysts. J. Energy Chem. 2020, 47, 1–9. [Google Scholar] [CrossRef]
- Marinho, A.L.A.; Toniolo, F.S.; Noronha, F.B.; Epron, F.; Duprez, D.; Bion, N. Highly active and stable Ni dispersed on mesoporous CeO2-Al2O3 catalysts for production of syngas by dry reforming of methane. Appl. Catal. B 2021, 10, 1428–1438. [Google Scholar] [CrossRef]
- Yang, R.; Xing, C.; Lv, C.; Shi, L.; Tsubaki, N. Promotional effect of La2O3 and CeO2 on Ni/gAl2O3 catalysts for CO2 reforming of CH4. Appl. Catal. A 2010, 385, 92–100. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, L.; Wang, Y.; Wang, S.; Zhao, Q.; Mao, D.; Hu, C. Low -Temperature Catalytic CO2 Dry Reforming of Methane on Ni-Si/ZrO2 catalyst. ACS Catal. 2018, 8, 6495–6506. [Google Scholar] [CrossRef]
- Pantaleo, G.; La Parola, V.; Testa, M.L.; Venezia, A.M. CO2 reforming of CH4 over SiO2-Supported Ni Catalyst: Effect of Sn as Support and metal Promoter. Ind. Eng. Chem. Res. 2021, 60, 18684–18694. [Google Scholar] [CrossRef]
- Horváth, A.; Beck, A.; Maróti, B.; Sáfrán, G.; Pantaleo, G.; Liotta, L.F.; Venezia, A.M.; La Parola, V. Strong impact of indium promoter on Ni/Al2O3 and Ni/CeO2-Al2O3 catalysts used in dry reforming of methane. Appl. Catal. A 2021, 621, 18174. [Google Scholar] [CrossRef]
- Horváth, A.; Guczi, L.; Kocsonya, A.; Sáfrán, G.; La Parola, V.; Liotta, L.F.; Pantaleo, G.; Venezia, A.M. Sol-derived AuNi/MgAl2O4 catalysts: Formation, structure and activity in dry reforming of methane. Appl. Catal. A 2013, 468, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Bhatar, S.; Abedin, M.A.; Kanitkar, S.; Spivey, J.J. A review on dry reforming of methane over perovskite derived catalysts. Catal. Today 2021, 365, 2–23. [Google Scholar] [CrossRef]
- Debęk, R.; Motak, M.; Grzybek, T.; Galvez, M.E.; Da Costa, P. A short review on the Catalytic Activity of Hydrotalcite- Derived Materials for Dry Reforming of Methane. Catalysts 2017, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Marcu, I.-C.; Pavel, O.D. Layered Double Hydroxide-Based Catalytic Materials for Sustainable Processes. Catalysts 2022, 12, 816. [Google Scholar] [CrossRef]
- Abdelsadek, Z.; Holgado, J.P.; Halliche, D.; Caballero, A.; Cherifi, O.; Cortes, S.G.; Masset, P.J. Examination of the deactivation Cycle of NiAl and NiMgAl- Hydrotalcite derived catalysts in the Dry Reforming of Methane. Catal. Lett. 2021, 151, 2596–2715. [Google Scholar] [CrossRef]
- Dai, H.; Zhu, Y.; Xiong, S.; Xiao, X.; Huang, L.; Deng, J.; Zhou, C. Dry Reforming of Methane over Ni/MgO@Al Catalysts with Unique Feature of Sandwich Structure. Chem. Sel. 2021, 6, e202102788. [Google Scholar] [CrossRef]
- Touahra, F.; Sehailia, M.; Ketir, W.; Bachari, K.; Chebout, R.; Tarri, M.; Cherifi, O.; Halliche, D. Effect of the Ni/Al ratio of Hydrotalcite-type catalysts on their performance in the methane dry reforming process. Appl. Petrochem. Res. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Daza, C.E.; Gallego, J.; Mondragon, F.; Moreno, S.; Molina, R. High stability of Ce-promoted Ni/Mg-Al catalysts derived from Hydrotalcites in dry reforming of methane. Fuel 2010, 89, 592–603. [Google Scholar] [CrossRef]
- Lucredio, A.F.; Assaf, J.M.; Assaf, E.M. Reforming of a model sulfur-free biogas on Ni catalysts supported on Mg(Al)O derived from hydrotalcite precursors: Effect of La and Rh addition. Biomass Bioenergy 2014, 60, 8–17. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chang, V.W.; Schumaker, D.J. CO2 reforming of methane to syngas; I: Evaluation of Hydrotalcite clay-derived catalysts. Appl. Clay Sci. 1998, 13, 317–328. [Google Scholar] [CrossRef]
- Gabrovska, M.; Ivanov, I.; Nikolova, D.; Krstic, J.; Venezia, A.M.; Crisan, D.; Crisan, M.; Tenchev, K.; Idakiev, V.; Tabakova, T. Improved Water–Gas Shift Performance of Au/NiAl LDHs Nanostructured Catalysts via CeO2 Addition. Nanomaterials 2021, 11, 366. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Khader, M.M.; Almarri, M.; Abdelmoneim, A.G. Ni-Based Catalysts for the dry reforming of methane. Catal. Today 2020, 343, 26–37. [Google Scholar] [CrossRef]
- Nikoo, M.K.; Amin, N.A.S. Thermodynamic analysis of carbon dioxide reforming in view of solid carbon formation. Fuel Process. Technol. 2011, 92, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Cui, C.; Da Costa, P.; Hu, C. The effect of absorbed oxygen species on carbon-resistance of Ni-Zr catalyst modified by Al and Mn for dry reforming of methane. Catal. Today 2022, 384–386, 257–264. [Google Scholar]
- Debęk, R.; Motak, M.; Grzybek, T.; Galvez, M.E.; Da Costa, P. Catalytic activity of hydrotalcite-derived catalysts in the dry reforming of methane: The effect of Ce promotion and feed gas composition. React. Kinet. Mech. Catal. 2017, 121, 185–208. [Google Scholar] [CrossRef] [Green Version]
- Guil-Lopez, R.; La Parola, V.; Pena, M.A.; Fierro, J.L.G. Evolution of the Ni-active centres into ex Hydrotalcite oxide catalysts during the COx-free hydrogen production by methane decomposition. Int. J. Hydrogen Energy 2012, 37, 7042–7705. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, J.; Wu, Y.; Pan, J.; Zhang, Q.; Tan, Y.; Han, Y. Insight into the effects of the oxygen species over Ni/ZrO2 catalyst surface on methane reforming with carbon dioxide. Appl. Catal. B 2019, 244, 427–437. [Google Scholar] [CrossRef]
- Pantaleo, G.; La Parola, V.; Deganello, F.; Singha, R.K.; Bal, R.; Venezia, A.M. Ni/CeO2 catalysts for methane partial oxidation: Synthesis, driven structural and catalytic effects. Appl. Catal. B 2016, 189, 233–241. [Google Scholar] [CrossRef]
- Tabakova, T.; Gabrovska, M.; Nikolova, D.; Ivanov, I.; Venezia, A.M.; Tenchev, K. Exploring the role of promoters (Au, Cu and Re) in the performance of Ni-Al layered double hydroxides for water-gas shift reaction. Int. J. Hydrog. Energy 2022, in press. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouqueroi, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure App. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Inorganic Crystal Structure Database (ICSD); FIZ Karlsruhe, GmbH: Eggenstein-Leopoldshafen, Germany, 2014.
- Klug, H.P.; Alexander, L.E. X-Ray Diffraction Procedures for Polycrystalline and Amorphous Materials, 2nd ed.; John Wiley and Sons: New York, NY, USA, 1974. [Google Scholar]
- La Parola, V.; Liotta, L.F.; Pantaleo, G.; Testa, M.L.; Venezia, A.M. CO2 reforming of CH4 over Ni supported on SiO2 modified by TiO2 and ZrO2: Effect of the support synthesis procedure. Appl. Catal. A 2022, 642, 118704. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Ni wt% | Al wt% | Ce wt% | Au wt% | S (m2g−1) | Vtot (cm3g−1) | dNiO * (nm) | dAu (nm) | dNi ** (nm) |
|---|---|---|---|---|---|---|---|---|---|
| NiAl | 61.7 | 11.3 | 226 | 1.00 | 5(21) | 25 | |||
| Au/NiAl | 59.9 | 11.0 | 3 | 215 | 0.97 | 4(21) | 22 | 23 | |
| CeNiAl | 61.1 | 12.3 | 0.8 | 224 | 0.92 | 4(12) | 16 | ||
| Au/CeNiAl | 59.3 | 10.9 | 0.8 | 3 | 215 | 0.97 | 4(10) | 18 | 20 |
| Samples | CH4 Conversion * (%) | CO2 Conversion * (%) | H2 Yield * (%) | H2/CO | %C ** | TC *** (°C) |
|---|---|---|---|---|---|---|
| NiAl | 67 (19) | 83 (07) | 39(22) | 0.7 | 244 | 742 |
| Au/NiAl | 72 (28) | 86 (08) | 43(26) | 0.7 | 244 | 720 |
| Ce/NiAl | 73 (12) | 83 (08) | 43(10) | 0.7 | 316 | 704 |
| Au/Ce/NiAl | 74 (22) | 88 (10) | 44(20) | 0.7 | 240 | 690 |
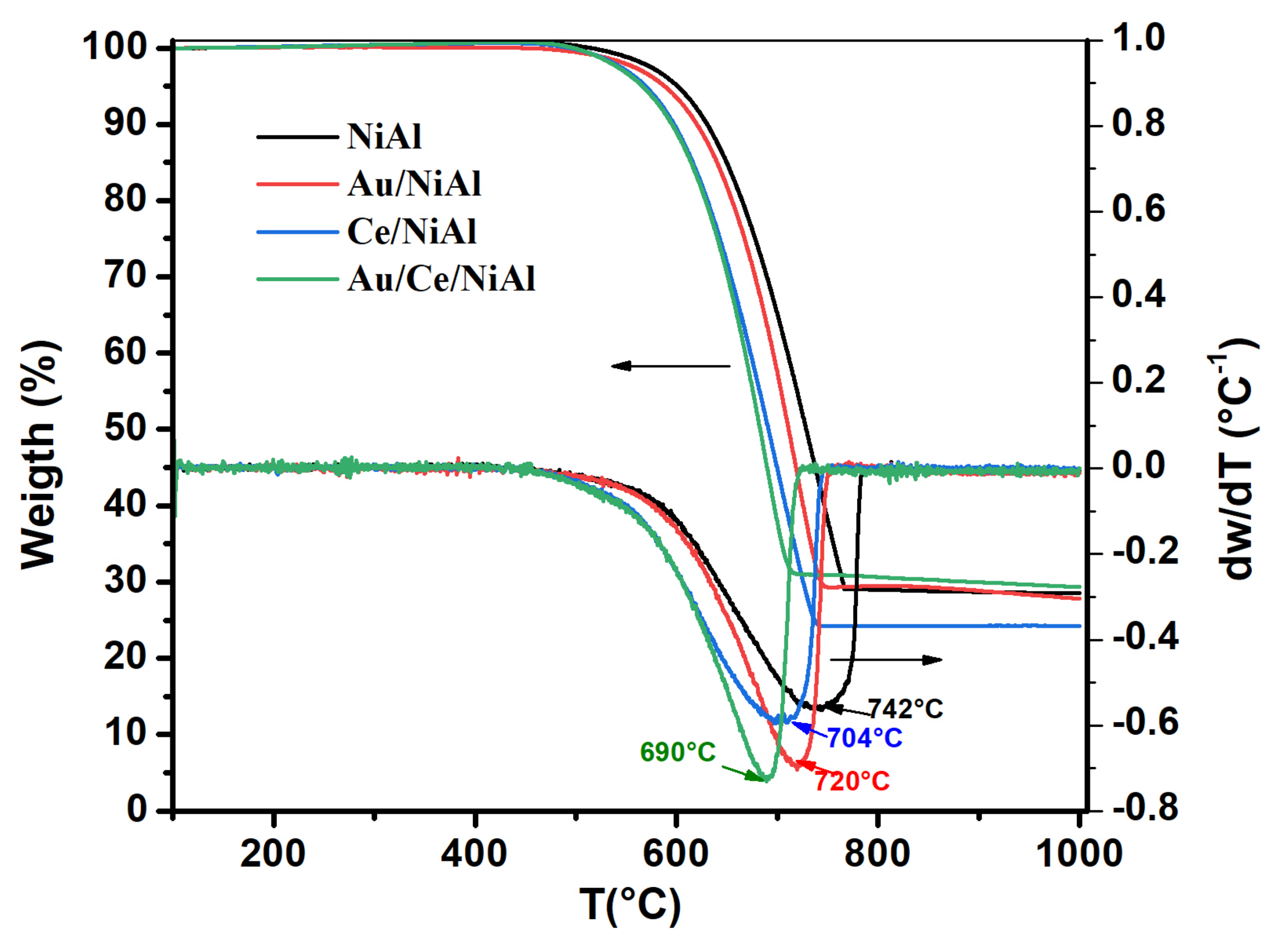
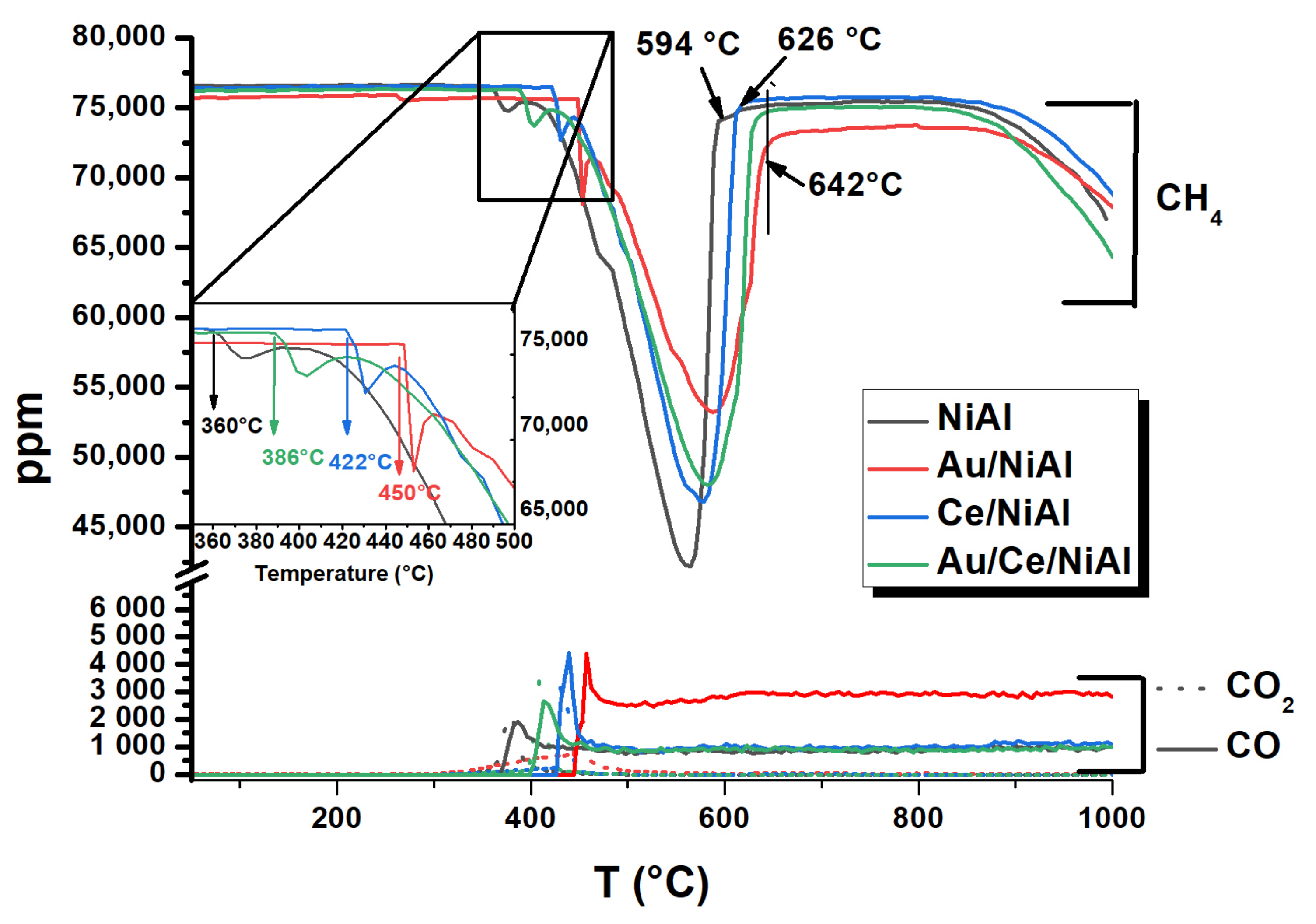
| Samples | T Light − off (C°) | T peak (°C) | Tdec range (°C) | %C * | TC ** (°C) |
|---|---|---|---|---|---|
| NiAl | 360 | 562 | 234 | 300 | 702 |
| Au/NiAl | 450 | 591 | 194 | 144 | 702 |
| Ce/NiAl | 422 | 582 | 208 | 426 | 704 |
| Au/Ce/NiAl | 386 | 584 | 257 | 566 | 732 |
| Samples | Ni 2p3/2 (eV) | Ni 3p (eV) | Au 4f7/2 (eV) | Al 2p (eV) | Ni/Al XPS * | Au/Al XPS * |
|---|---|---|---|---|---|---|
| NiAl | 855.7 | 68.6 | 74.6 | 0.5 (2.5) | - | |
| Au/NiAl | 855.8 | 68.4 | 84.0 | 74.5 | 0.6 (2.5) | 0.004 (0.036) |
| Ce/NiAl | 855.6 | 68.4 | 74.5 | 0.7 (2.5) | - | |
| Au/Ce/NiAl | 855.6 | 68.5 | 84.0 | 74.6 | 0.6 (2.5) | 0.007 (0.037) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Parola, V.; Pantaleo, G.; Liotta, L.F.; Venezia, A.M.; Gabrovska, M.; Nikolova, D.; Tabakova, T. Gold and Ceria Modified NiAl Hydrotalcite Materials as Catalyst Precursors for Dry Reforming of Methane. Catalysts 2023, 13, 606. https://doi.org/10.3390/catal13030606
La Parola V, Pantaleo G, Liotta LF, Venezia AM, Gabrovska M, Nikolova D, Tabakova T. Gold and Ceria Modified NiAl Hydrotalcite Materials as Catalyst Precursors for Dry Reforming of Methane. Catalysts. 2023; 13(3):606. https://doi.org/10.3390/catal13030606
Chicago/Turabian StyleLa Parola, Valeria, Giuseppe Pantaleo, Leonarda Francesca Liotta, Anna Maria Venezia, Margarita Gabrovska, Dimitrinka Nikolova, and Tatyana Tabakova. 2023. "Gold and Ceria Modified NiAl Hydrotalcite Materials as Catalyst Precursors for Dry Reforming of Methane" Catalysts 13, no. 3: 606. https://doi.org/10.3390/catal13030606